
Abstract
Expanding our capabilities to unambiguously identify ancient traces of life in ancient rocks requires laboratory experiments to better constrain the evolution of biomolecules during advanced fossilization processes. Here, we submitted RNA to hydrothermal conditions in the presence of a gel of Al-smectite stoichiometry at 200 °C for 20 days. NMR and STXM-XANES investigations revealed that the organic fraction of the residues is no longer RNA, nor the quite homogeneous aromatic-rich residue obtained in the absence of clays, but rather consists of particles of various chemical composition including amide-rich compounds. Rather than the pure clays obtained in the absence of RNA, electron microscopy (SEM and TEM) and diffraction (XRD) data showed that the mineralogy of the experimental residues includes amorphous silica and aluminosilicates mixed together with nanoscales phosphates and clay minerals. In addition to the influence of clay minerals on the degradation of organic compounds, these results evidence the influence of the presence of organic compounds on the nature of the mineral assemblage, highlighting the importance of fine-scale mineralogical investigations when discussing the nature/origin of organo-mineral microstructures found in ancient rocks.
Similar content being viewed by others

Introduction
The ancient fossil record carries key information on the first steps of the history of life on Earth1,2,3. Yet, although ancient microfossils may still comprise (partially preserved) biogenic organic material4,5,6,7,8,9,10, the identification of traces of life in ancient rocks has always been fraught with difficulties pertaining to fossilization and burial-induced degradation processes11,12,13,14. As a result, the interpretation of the Archean palaeobiological record is full of controversies, among which the most famous one is centered on filamentous microstructures found in black chert veins within the 3.46 Ga Apex Basalt (Pilbara Craton, Western Australia)15,16. For more than 30 years now, these microstructures have been considered by some authors as the most ancient remains of life on Earth, based on their morphologies and isotopic signatures17,18. Yet, besides contamination issues19,20, it has recently been argued, based on nanoscale chemical imaging, that these filamentous microstructures are not microfossils (i.e. true body fossils of microorganisms), but rather abiotic stacks of clay minerals fortuitously arranged in roughly filamentous patterns and interleaved with nanoscale mineral phases and organic particles21,22,23.
Laboratory experiments may provide clues regarding the potential impact that hydrothermal/diagenetic processes have had on these microstructures, thereby potentially shedding new light on their origin14,24. Although pioneered experimental studies investigated the interactions between biogenic organic compounds and pure silica25,26,27,28, phosphates29,30,31 or oxides32,33, the chemical evolution and mineralogical evolution that may undergo a mixture of reactive biomolecules (i.e. prone to degradation) and clay minerals during advanced fossilization processes remain poorly constrained. In the present study, we experimentally submitted a mixture of RNA (i.e. the most emblematic organic macromolecule of the living world and the central molecule of the ‘RNA world’ hypothesis34,35) and a gel of Al-smectite stoichiometry (i.e. a clay mineral ubiquitously distributed in the continental crust36) to experimental hydrothermal conditions at 200 °C for 20 days within initially pure water. Additional experiments were conducted in the absence of the gel or in the absence of RNA to serve as controls.
Results and Discussion
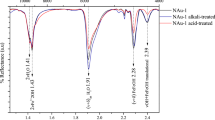
Results evidence that RNA underwent significant transformations during the experiments (Fig. 1A). While the CP MAS-NMR signature of RNA is dominated by the signals of ribose (signal between 60 and 105 ppm) and nucleobases (signal between 140 and 170 ppm), the CP MAS-NMR signatures of the residues of experiments conducted either in the presence or absence of a gel of Al-smectite stoichiometry are dominated by aromatic/heterocyclic and aliphatic carbons (signals between 100 and 150 ppm and between 55 and 70 ppm, respectively). In absence of RNA, the gel of Al-smectite stoichiometry evolved into a crystallized montmorillonite36 (Figs. 1B and 2A). In contrast, only a small amount of crystalline phyllosilicates is detected together with mainly amorphous phases in residues of experiments conducted in the presence of RNA (Fig. 1B). Surprisingly, the phyllosilicate that crystallizes is not montmorillonite: instead of exhibiting the 060 reflection of dioctahedral montmorillonite at 1.498 Å, this phase exhibits a 060 reflection at 1.532 Å, i.e. a value more typical of 2:1 trioctahedral Mg-rich smectites36 (Fig. 1B).

NMR and XRD characterization of the experimental residues. (A) CP MAS-13C-NMR spectra of the residues of experiments conducted in the presence or absence of a gel of Al-smectite stoichiometry. The CP MAS-13C-NMR spectrum of RNA is also shown for comparison. (B) Powder XRD patterns of the residues of experiments conducted in the presence or absence of RNA. Values correspond to hkl reflections of clay minerals. Stars correspond to peaks attributed to phosphates.

SEM characterization of the experimental residues. (A) SEM image of the montmorillonite produced during the experiments conducted in the absence of RNA. (B) SEM image of a fraction of the residues of experiments conducted in the presence of RNA, showing a cluster/pile of carbon-free micrometric spheres of pure amorphous silica (left) and a pluri-decimicrometric heterogeneous aggregate (right).
SEM investigations reveal that, rather than pure clay minerals (Fig. 2A), the presence of RNA leads to complex organo-mineral residues composed of (i) clusters of carbon-free micrometric spheres of pure amorphous silica, and (ii) heterogeneous aluminosilicate-rich aggregates containing Al, Mg, P, C and N in addition to Si (Fig. 2B). The heterogeneous nature of these aggregates is highlighted at the sub-micrometer scale by TEM imaging and EDX mapping of cryo-ultramicrotome sections (Fig. 3A–D): these aggregates are composed of submicrometric particles of more or less N-rich amorphous aluminosilicates and (Ca,Mg)-phosphates associated with relatively rare Mg-smectites that appear to be in close association with organic carbon (Fig. 3C). This organic material might be either adsorbed onto or bonded to surfaces (lateral or basal) of these Mg-smectites or trapped within their interlayer spaces37.

STEM and STXM characterization of the experimental residues. (A–D) STEM-HAADF images of fractions of the residues of experiments conducted in the presence of RNA and corresponding maps (C,D). Clay minerals more or less associated with organic carbon are clearly visible (pink arrows). (E) XANES spectra at the C and N K-edges of organic compounds encountered in the residues. At the C K-edge, energy values (in eV) correspond to typical absorptions of quinones or conjugated cyclic amides (284.7), alkenes and aromatics (285.0), conjugated cycles (285.3), heterocycles (285.9), imines and nitriles (286.7), phenols and ketones (286.7/287.4), aliphatics (287.9), amides (288.2), carboxylics (288.4) and hydroxyls (289.3/289.5)42,44. Due to overlapping energies of several resonances, absorption peaks at the N K-edge cannot be univocally assigned to given functional groups, but absorption features below 400 eV generally indicate the presence of imine, nitrile and pyridinic N while absorption features above 400 eV generally indicate the presence of amide, nitro and pyrrolic N44,45. The spectra of RNA and of the residue of the experiments conducted in the absence of the gel of Al-smectite stoichiometry are shown for comparison.
Organic carbon also occurs within these residues as submicrometric particles of various chemical compositions as indicated by STXM-based XANES spectroscopy (Fig. 3E): (i) aromatic-rich organic particles (N/C ~ 0.15) containing a significant proportion of conjugated cycles and heterocycles (peaks at 285, 285.4 & 285.9 eV, respectively) and traces of carboxylics (peak at 288.4 eV), and (ii) N-rich organic particles (N/C ~ 1) dominated by amide and hydroxyl groups (absorption peaks at 288.2 & 289.5 eV, respectively). These N-rich organic particles are absent from the residues of experiments conducted in the absence of the gel of Al-smectite stoichiometry. In fact, these very homogeneous residues (N/C ~ 0.15) exhibit a XANES spectrum very similar to that of the heterocyclic, (poly)aromatic organic particles.
Although the information related to the original chemical structure of RNA was lost during the experiments, amide-rich organic compounds were produced in experiments conducted in the presence of a gel of Al-smectite stoichiometry. Similar compounds have been reported within ancient microfossils and interpreted as products of the degradation of peptidic compounds6,8. This is not the case here as RNA does not contain peptidic moieties but rather conjugated cyclic amides. Such preservation of amide moieties occurred only in the presence of the gel of Al-smectite stoichiometry, suggesting that the presence of this initially amorphous inorganic phase limited the molecular degradation of RNA, consistently with results of experiments conducted with silica27.
In addition to the influence of a mineral phase on the degradation of organic compounds, the present results evidence the influence of the presence of organic compounds on the final mineral assemblage. Instead of pure clay minerals as in the absence of RNA, the residues of experiments conducted in the presence of RNA comprise amorphous silica, aluminosilicates and nanophosphates associated with rare nanoscale clay minerals. As illustrated here, although in some cases the presence and the nature of organic materials composing ancient putative microfossils may not be informative enough to discuss their possible biogenicity, the nature of the organo-mineral assemblage may be quite rich in information. Although additional experiments would be required to properly constrain the origin of the Apex microstructures, the present study highlights the importance fine-scale mineralogical investigations, as suggested earlier21,22,23, when discussing the nature/origin of organo-mineral microstructures found in ancient rocks.
Materials and Methods
Starting materials
Pure powders of yeast RNA (SIGMA-ALDRICH) and a hydrogel of Al-smectite stoichiometry synthetized in the lab were used for the present experiments. The hydrogel was obtained by adding 25.1 mL of a solution of aluminium nitrate (81.2 g/L) and 4.01 mL of a solution of magnesium nitrate (100.45 g/L) into a 15 mL solution of ethanol containing 0.1 mol of TEOS (tetraethylorthosilicate) and 0.005 mol of CaCO3. An ammoniacal solution (33%) was then added until gelification. The gel was stored 1 night at room temperature and heated 1 day at 200 °C and 1 day at 650 °C.
Fossilization experiments
Experiments were conducted in 25 mL PARR Reactors under hydrothermal conditions using 50 mg of RNA, 100 mg of the gel of Al-smectite stoichiometry and 5 mL of water. The Ti reactor were placed at 200 °C for 20 days before being cooled at room temperature. Residues were dried overnight at 60 °C. Additional experiments were also conducted with either no mineral or no RNA to serve as controls. For TEM and STXM measurements, residues were washed 3 times with water and dichloromethane/methanol (1:1) to remove the soluble organic fraction. All experiments were triplicated to ensure reproducibility.
X-Ray diffraction
X-Ray diffraction patterns were acquired using a PANALYTICAL X’PERT PRO diffractometer (IMPMC, Paris) operating at 40 kV and 40 mA with a Co anode (Kα at 1.79 Å). Analyses were performed on finely ground powders deposited on a silicium sample holder. The angular range in 2θ was 5–80° with a step size of 0.017° for a total counting time per sample of 2 hours.
Scanning electron microscopy
SEM investigations were performed on powders deposited on carbon tape using SEM-FEG ZEISS ULTRA 55 (IMPMC, Paris).
CP MAS-13C-NMR
Cross polarization (CP) 13C nuclear magnetic resonance (13C-NMR) experiments were performed with a magic-angle spinning (MAS) probe 1 H/X at 14000 kHz in a BRUCKER AVANCE 3 500 MHz (IMPC, Paris) operating at 125.77 MHz. Samples were sealed in a 4.0 mm zircon rotor. Chemical shifts were calibrated using the carboxyl signal of adamantane (38.52 ppm). The 13C Cross-Polarization spectra were acquired with a ramp-CP contact time of 1 ms and a 1.5 s of recycle delay.
Sample preparation for TEM and STXM
Cryo-ultramicrotome sections were prepared for STXM and TEM characterization using the LEICA ultramicrotome available at UMET (Lille, France). Experimental residues were mixed with 0.1 ml of water-ethanol (50/50%Vol) before being frozen in liquid nitrogen at −160 °C. After cutting, the ultrathin slices of residues were deposited on holey carbon film TEM grids before being exposed to ambient temperature.
Transmission electron microscopy
Scanning transmission electron microscopy (STEM) and EDS mapping were performed using a THERMOFISHER TITAN THEMIS 300 microscope operated at 300 kV (CCM – Lille). Hyperspectral EDS data were obtained using the super-X detector system equipped with four windowless silicon drift detectors. These detectors have a high sensitivity for light elements and allow a high counting rate of the carbon, nitrogen and oxygen X-rays. The probe current was set at 600 pA with a dwell time at 10 µs per pixel. A key aspect of this work is the post-processing of the hyperspectral data, performed using the HYPERSPY python-based package38. The signal was first denoised using principal component analysis. Then, the EDS spectra at each pixel were fitted by a series of Gaussian functions and a physical model for background/bremsstrahlung. The integrated intensities of the Gaussian functions were used to quantify the compositions thanks to the Cliff-Lorimer method, using experimentally determined k-factors. Absorption correction was taken into account using an experimental thickness map obtained by integration of plasmon absorption bands in EELS. This step was mandatory to correct for the re-absorption within the sample of the carbon, nitrogen and oxygen X-rays. Finally, end-member phases (smectites, phosphates, amorphous silica, organic compounds) were identified and their spectra used as inputs for linear combination fitting (multiple linear least square fits). Pixels of similar composition were given the same colors scaled as a function of the proportion of each phase.
Scanning transmission X-ray microscopy
STXM/XANES investigations were conducted using the HERMES STXM beamline39,40 at the synchrotron SOLEIL (Gif-sur-Yvette, France). Carbon contamination on beamline optics is constantly removed thanks to a continuous flow of pure O2. The well-resolved 3p Rydberg peak of gaseous CO2 at 294.96 eV and the 1 s → π* photoabsorption resonance of gaseous N2 at 400.89 eV were used for energy calibration. Collecting image stacks at energy increments of 0.1 eV with a dwell time of ≤ 1 ms per pixel prevented irradiation damage41. The C- and N-XANES spectra shown here were normalized to the carbon and nitrogen quantities using QUANTORXS42. N/C values were estimated at +/− 0.0243 using QUANTORXS42.
References
Summons, R. E. & Hallmann, C. Organic Geochemical Signatures of Early Life on Earth. In Treatise on Geochemistry 33–46, https://doi.org/10.1016/B978-0-08-095975-7.01005-6 (Elsevier, 2014).
Briggs, D. E. G. & Summons, R. E. Ancient biomolecules: Their origins, fossilization, and role in revealing the history of life: Prospects & Overviews. BioEssays 36, 482–490 (2014).
Knoll, A. H., Bergmann, K. D. & Strauss, J. V. Life: the first two billion years. Philosophical Transactions of the Royal Society B: Biological Sciences 371, 20150493 (2016).
Bernard, S. et al. Exceptional preservation of fossil plant spores in high-pressure metamorphic rocks. Earth and Planetary Science Letters 262, 257–272 (2007).
Galvez, M. E. et al. Morphological preservation of carbonaceous plant fossils in blueschist metamorphic rocks from New Zealand: Fossil preservation in metamorphic rocks. Geobiology 10, 118–129 (2012).
Alleon, J. et al. Molecular preservation of 1.88 Ga Gunflint organic microfossils as a function of temperature and mineralogy. Nature Communications 7, 11977 (2016).
Delarue, F. et al. Investigation of the Geochemical Preservation of ca. 3.0 Ga Permineralized and Encapsulated Microfossils by Nanoscale Secondary Ion Mass Spectrometry. Astrobiology 17, 1192–1202 (2017).
Alleon, J. et al. Chemical nature of the 3.4 Ga Strelley Pool microfossils. Geochemical Perspectives Letters 37–42, https://doi.org/10.7185/geochemlet.1817 (2018).
Delarue, F. et al. Nitrogen isotope signatures of microfossils suggest aerobic metabolism 3.0 Gyr ago. Geochemical Perspectives Letters 32–36, https://doi.org/10.7185/geochemlet.1816 (2018).
Delarue, F. et al. Out of rock: A new look at the morphological and geochemical preservation of microfossils from the 3.46 Gyr-old Strelley Pool Formation. Precambrian Research 336, 105472 (2020).
Schopf, J. W. Precambrian Paleobiology: Problems and Perspectives. Annual Review of Earth and Planetary Sciences 3, 213–249 (1975).
Schiffbauer, J. D. et al. Ultrastructural and Geochemical Characterization of Archean–Paleoproterozoic Graphite Particles: Implications for Recognizing Traces of Life in Highly Metamorphosed Rocks. Astrobiology 7, 684–704 (2007).
Bernard, S. & Papineau, D. Graphitic Carbons and Biosignatures. Elements 10, 435–440 (2014).
Javaux, E. J. Challenges in evidencing the earliest traces of life. Nature 572, 451–460 (2019).
Awramik, S. M., Schopf, J. W. & Walter, M. R. Filamentous fossil bacteria from the Archean of Western Australia. Precambrian Research 20, 357–374 (1983).
Schopf, J. & Packer, B. Early Archean (3.3-billion to 3.5-billion-year-old) microfossils from Warrawoona Group, Australia. Science 237, 70–73 (1987).
Schopf, J. W. & Kudryavtsev, A. B. Biogenicity of Earth’s earliest fossils: A resolution of the controversy. Gondwana Research 22, 761–771 (2012).
Schopf, J. W., Kitajima, K., Spicuzza, M. J., Kudryavtsev, A. B. & Valley, J. W. SIMS analyses of the oldest known assemblage of microfossils document their taxon-correlated carbon isotope compositions. Proceedings of the National Academy of Sciences 115, 53–58 (2018).
Westall, F. Stephen Jay Gould, les procaryotes et leur évolution dans le contexte géologique. Comptes Rendus Palevol 2, 485–501 (2003).
Westall, F. Early Life on Earth: The Ancient Fossil Record. In Astrobiology: Future Perspectives (eds. Ehrenfreund, P. et al.) 287–316, https://doi.org/10.1007/1-4020-2305-7_12, (Springer Netherlands, 2005).
Brasier, M. D., Antcliffe, J., Saunders, M. & Wacey, D. Changing the picture of Earth’s earliest fossils (3.5–1.9 Ga) with new approaches and new discoveries. Proceedings of the National Academy of Sciences 112, 4859–4864 (2015).
Wacey, D., Saunders, M., Kong, C., Brasier, A. & Brasier, M. 3.46 Ga Apex chert ‘microfossils’ reinterpreted as mineral artefacts produced during phyllosilicate exfoliation. Gondwana Research 36, 296–313 (2016).
Wacey, D., Eiloart, K. & Saunders, M. Comparative multi-scale analysis of filamentous microfossils from the c. 850 Ma Bitter Springs Group and filaments from the c. 3460 Ma Apex chert. Journal of the Geological Society jgs2019–053, https://doi.org/10.1144/jgs2019-053 (2019).
Alleon, J. & Summons, R. E. Organic geochemical approaches to understanding early life. Free Radical Biology and Medicine 140, 103–112 (2019).
Oehler, J. H. & Schopf, J. W. Artificial Microfossils: Experimental Studies of Permineralization of Blue-Green Algae in Silica. Science 174, 1229–1231 (1971).
Orange, F. et al. Preservation and Evolution of Organic Matter During Experimental Fossilisation of the Hyperthermophilic Archaea Methanocaldococcus jannaschii. Origins of Life and Evolution of Biospheres 42, 587–609 (2012).
Alleon, J. et al. Early entombment within silica minimizes the molecular degradation of microorganisms during advanced diagenesis. Chemical Geology 437, 98–108 (2016).
Igisu, M. et al. Changes of aliphatic C-H bonds in cyanobacteria during experimental thermal maturation in the presence or absence of silica as evaluated by FTIR microspectroscopy. Geobiology 16, 412–428 (2018).
Li, J., Benzerara, K., Bernard, S. & Beyssac, O. The link between biomineralization and fossilization of bacteria: Insights from field and experimental studies. Chemical Geology 359, 49–69 (2013).
Li, J. et al. Impact of biomineralization on the preservation of microorganisms during fossilization: An experimental perspective. Earth and Planetary Science Letters 400, 113–122 (2014).
Miot, J., Bernard, S., Bourreau, M., Guyot, F. & Kish, A. Experimental maturation of Archaea encrusted by Fe-phosphates. Scientific Reports 7 (2017).
Picard, A., Kappler, A., Schmid, G., Quaroni, L. & Obst, M. Experimental diagenesis of organo-mineral structures formed by microaerophilic Fe(II)-oxidizing bacteria. Nature Communications 6, 6277 (2015).
Picard, A., Obst, M., Schmid, G., Zeitvogel, F. & Kappler, A. Limited influence of Si on the preservation of Fe mineral-encrusted microbial cells during experimental diagenesis. Geobiology 14, 276–292 (2016).
Orgel, L. E. The origin of life–a review of facts and speculations. Trends Biochem. Sci. 23, 491–495 (1998).
Higgs, P. G. & Lehman, N. The RNA World: molecular cooperation at the origins of life. Nature Reviews Genetics 16, 7–17 (2015).
Meunier, A. Clays. (Springer Science & Business Media, 2005).
Lagaly, G., Ogawa, M. & Dékány, I. Clay Mineral–Organic Interactions. In Developments in Clay Science vol. 5 435–505 (Elsevier, 2013).
de la Pena, F. et al. Electron Microscopy (Big and Small) Data Analysis With the Open Source Software Package HyperSpy. Microscopy and Microanalysis 23, 214–215 (2017).
Belkhou, R. et al. HERMES: a soft X-ray beamline dedicated to X-ray microscopy. Journal of Synchrotron Radiation 22, 968–979 (2015).
Swaraj, S. et al. Performance of the HERMES beamline at the carbon K-edge. Journal of Physics: Conference Series 849, 012046 (2017).
Wang, J. et al. Radiation damage in soft X-ray microscopy. Journal of Electron Spectroscopy and Related Phenomena 170, 25–36 (2009).
Le Guillou, C., Bernard, S., De la Pena, F. & Le Brech, Y. XANES-Based Quantification of Carbon Functional Group Concentrations. Analytical Chemistry 90, 8379–8386 (2018).
Alleon, J., Bernard, S., Remusat, L. & Robert, F. Estimation of nitrogen-to-carbon ratios of organics and carbon materials at the submicrometer scale. Carbon 84, 290–298 (2015).
Alleon, J. et al. Organic molecular heterogeneities can withstand diagenesis. Scientific Reports 7: 1508, https://doi.org/10.1038/s41598-017-01612-8 (2017).
Leinweber, P. et al. Nitrogen K -edge XANES – an overview of reference compounds used to identify ‘unknown’ organic nitrogen in environmental samples. Journal of Synchrotron Radiation 14, 500–511 (2007).
Acknowledgements
We gratefully acknowledge financial support from OPUS, from the DIM-MAP (project FossilEx [PI: M. Jaber] and BombEx [PI: JC. Viennet]) and Sorbonne Université (Project MarsAtLab [PI: S. Bernard]). The TEM facility in Lille (France) is supported by the Conseil Régional du Nord-Pas de Calais, and the European Regional Development Fund (ERDF). Special thanks go to Stefan Stanescu and Sufal Swaraj for their expert support with the HERMES STXM beamline at SOLEIL, and to Anne-Marie Blanchenet for her help during the Cryo-ultramicrotome sections preparations. The HERMES beamline (SOLEIL) is supported by the CNRS, the CEA, the Region Ile de France, the Departmental Council of Essonne and the Region Centre.
Author information
Authors and Affiliations
Contributions
S.B. and M.J. designed the present study. P.J. and J.C.V. conducted the fossilization experiments. P.J., J.C.V. and L.D. performed the XRD analyses. P.J., M.J. and B.R. performed the NMR analyses. P.J. performed the MEB analyses. P.J., J.C.V. and C.L.G. prepared the cryo-ultramicrotome sections. P.J. and C.L.G. performed the TEM analyses. P.J., J.C.V., S.B. and C.L.G. performed the STXM analyses. All authors contributed to the interpretation of the data and discussed their implications. P.J., S.B. and M.J. wrote the manuscript, with critical inputs from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Jacquemot, P., Viennet, JC., Bernard, S. et al. The degradation of organic compounds impacts the crystallization of clay minerals and vice versa. Sci Rep 9, 20251 (2019). https://doi.org/10.1038/s41598-019-56756-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-56756-6
This article is cited by
-
Influence of pH on the Hydrothermal Synthesis of Al-Substituted Smectites (Saponite, Beidellite, and Nontronite)
Clays and Clay Minerals (2023)
-
Exploring the link between molecular cloud ices and chondritic organic matter in laboratory
Nature Communications (2021)
-
Inherited geochemical diversity of 3.4 Ga organic films from the Buck Reef Chert, South Africa
Communications Earth & Environment (2021)
-
Constraining the preservation of organic compounds in Mars analog nontronites after exposure to acid and alkaline fluids
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
