
Abstract
Embryos utilize oocyte-donated RNAs until they become capable of producing RNAs through embryonic genome activation (EGA). The sperm’s influence over pre-EGA RNA content of embryos remains unknown. Recent studies have revealed that sperm donate non-genomic components upon fertilization. Thus, sperm may also contribute to RNA presence in pre-EGA embryos. The first objective of this study was to investigate whether male fertility status is associated with the RNAs present in the bovine embryo prior to EGA. A total of 65 RNAs were found to be differentially expressed between 2–4 cell bovine embryos derived from high and low fertility sires. Expression patterns were confirmed for protein phosphatase 1 regulatory subunit 36 (PPP1R36) and ataxin 2 like (ATXN2L) in three new biological replicates. The knockdown of ATXN2L led to a 22.9% increase in blastocyst development. The second objective of this study was to characterize the parental origin of RNAs present in pre-EGA embryos. Results revealed 472 sperm-derived RNAs, 2575 oocyte-derived RNAs, 2675 RNAs derived from both sperm and oocytes, and 663 embryo-exclusive RNAs. This study uncovers an association of male fertility with developmentally impactful RNAs in 2–4 cell embryos. This study also provides an initial characterization of paternally-contributed RNAs to pre-EGA embryos. Furthermore, a subset of 2–4 cell embryo-specific RNAs was identified.
Similar content being viewed by others


Introduction
Prior to EGA, the freshly fertilized zygote depends on maternal RNAs donated by the oocyte1,2. During EGA, the embryo degrades maternal RNAs and begins producing its own RNAs1,2. This process is termed the ‘maternal to zygotic transition’1,2. These oocyte RNAs are considered important precursors to successful embryonic development1,3. For instance, several maternal mRNAs alter cell fate. Depletion of the maternal mRNAs perilipin 2 (PLIN2)4, tripartite motif-containing 36 (TRIM36)5, and DND microRNA-mediated repression inhibitor 1 (DND1)6 disrupts the cortical rotation and microtubule formation events involved in axial patterning. Additionally, the presence of the maternal vegt protein (VegT) is instrumental for the determination of cell fate during primary germ layer formation in the blastula of Xenopus embryos7. Furthermore, proteins translated from the maternally-derived RNAs POU domain class 5 transcription factor 3 (POU5F3) and SRY-box transcription factor 3 (SOX3) mediate competence of cells prior to germ layer formation by remodeling chromatin structure just before initiation of inductive signaling in Xenopus tropicalis embryos8. The oocyte clearly influences embryonic development by contributing RNAs to the zygote at fertilization. However, sperm contributions to RNA patterns in the pre-EGA embryo are still unclear.
Older literature has suggested that the sperm only donates its chromosomes to the embryo at fertilization9,10. However, over time, studies have shown that the sperm contributes additional non-genetic components to the embryo9,11. It is now accepted that the sperm can transfer DNA methylation patterns12,13, mRNAs14,15,16,17,18, small non-coding RNAs19, and proteins20,21 to the embryo. Each of these non-genetic components is capable of regulating mRNA presence and activity22,23,24,25,26. Furthermore, sperm DNA methylation27,28, mRNAs29, small non-coding RNAs30,31, and proteins32,33,34 are all associated with male fertility status. The RNAs present in the embryo prior to EGA are important for determining cell fate and developmental success of embryos4,5,6,7,8. Previously, our lab reported that bull fertility status is associated with gene expression at the blastocyst stage27. However, the influence of male fertility over the mRNA content in pre-EGA embryos has not yet been evaluated on a whole-transcriptome scale.
Direct delivery of sperm RNA is perhaps the most straightforward influence of the sperm over pre-EGA embryo RNA content. Ostermeier et al.14 demonstrated that the sperm-specific transcripts protamine 2 (PRM2) and clusterin (CLU) could be transferred from human sperm to zona-free hamster oocytes. Further, they demonstrated that the transcripts were still present three hours post-fertilization14. A study in pigs confirmed that the paternal PRM2 and CLU transcripts were passed to zygotes16. Additionally, studies have evaluated sperm transcript stability. The transcripts pregnancy specific beta-1-glycoprotein 1 (PSG1) and major histocompatibility complex, class I, E (HLA-E), but not PRM2 were shown to remain stable for 24 hours following human sperm delivery to hamster oocytes17. Another group showed that the mouse sperm-derived forkhead box G1 (FOXG1) and Wnt family member 4 (WNT4) transcripts are transferred to the zygote15. Further, the WNT4 transcript was translated at the 1-cell stage. The WNT4 protein remained stable following the loss of the transcript at the 2-cell stage15.
The functional importance of sperm-derived RNAs during embryonic development remains largely unknown. Sperm RNA function has been criticized because there is a large difference in RNA quantity between sperm and oocytes. A single spermatozoon contains 20–30 fg of RNA35, while a single oocyte contains 0.5 ng of RNA36. However, a small number of studies have demonstrated that sperm RNA function deserves a thorough investigation. In particular, the sperm-derived factor phospholipase C zeta (PLCζ) initiates the post-fertilization calcium oscillations that are required for embryo cleavage20. PLCζ is found as both a protein and an RNA in sperm37. PLCζ knockout male mice are infertile38. However, injecting PLCζ mRNA and the sperm of PLCζ knockouts into oocytes induces calcium oscillations and leads to the production of healthy pups38. The injection of only the mRNA extracted from sperm cells also leads to the production of calcium oscillations39. This could mean that the sperm-borne PLCζ RNA is translated prior to the activation of cell division39. Another example of a functional sperm RNA is DEAD-box helicase 3 Y-linked (DDX3Y). The sperm-borne DDX3Y transcript was found in freshly fertilized mouse zygotes, but not in oocytes18. Microinjection of an antisense RNA reduced the number of male cleavage-stage embryos produced and caused a lower cleavage rate of embryos18. These studies show that select sperm-borne RNAs may be indispensable during early embryonic development. Therefore, the milieu of paternally-contributed RNAs in the pre-EGA embryo should be further understood.
The first objective of this study was to evaluate whether the fertility status of bulls was associated with transcriptomic profiles of pre-EGA embryos. We utilized high-throughput sequencing to identify differentially expressed RNAs. Following validation, the differentially expressed RNA ATXN2L was knocked down in zygotes, as a proof of principle that paternally-contributed RNAs are important for development. The second objective of this study was to characterize the parental origin of the RNAs present in pre-EGA embryos on a whole-transcriptome scale. To do this, we integrated the pre-EGA embryo RNA-seq data with RNA-seq data from sperm and oocytes. This study provides new information about the paternal impact on pre-EGA embryo’s RNA content and function.
Results
Gene expression analysis of 2–4 cell embryos
A total of 65 genes were differentially expressed between embryos derived from high and low fertility sires. There were 35 genes with decreased expression and 30 genes with increased expression in the embryos from high fertility sires compared to the embryos from low fertility sires (FDR < 0.1). The subset of genes selected for validation were ATXN2L, PPP1R36, amyloid beta precursor protein binding family A member 1 (APBA1), plakophilin 2 (PKP2), cadherin EGF LAG seven-pass G-type receptor 3 (CELSR3), ELAV like RNA binding protein 4 (ELAVL4), WD repeat domain 93 (WDR93), Drebrin Like (DBNL), and ENSBTAG00000046943 (LOC112444303 oncomodulin). The genes PPP1R36, PKP2, CELSR3, and ELAVL4 were chosen because they had previously-identified associations with reproductive development and fertility. On the other hand, ATXN2L, APBA1, WDR93, DBNL, and LOC112444303 oncomodulin were selected because their roles in fertility were still unclear. In total, nine genes were chosen for qRT-PCR validation (Table 1). All three validation replicates for ATXN2L and PPP1R36 reproduced the same trend found in RNA-Seq results. The gene ATXN2L had an average fold difference of 2.45 (P = 0.052), and PPP1R36 had an average fold difference of 4.08 (P = 0.065). The genes WDR93, ELAVL4, and LOC112444303 oncomodulin showed replication of RNA-Seq results for two out of three biological validation replicates. WDR93 had an average fold difference of 0.65 (P = 0.39), ELAVL4 had an average fold difference of 0.43 (P = 0.13), and LOC112444303 oncomodulin had an average fold difference of 0.70 (P = 0.40). The genes DBNL, CELSR3, APBA1, and PKP2 were not consistent across biological validation replicates. The 65 differentially expressed genes are found in Supplementary Table S1. Additionally, there was no significant difference in the cleavage rate between high and low fertility sire embryos (P = 0.70) (Supplementary Table S2).
Knockdown of ATXN2L with gapmer treatment
To test the possible role of ATXN2L in embryo development, knockdown experiments were performed using gapmer technology. Embryos which were treated with ATXN2L gapmer at the zygote stage showed a 22.9% increase in blastocyst rate compared to control embryos (P < 0.05). The fold change in ATXN2L was 0.44 in treated vs. control embryos (P < 0.05).
Origin of RNAs
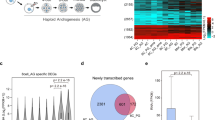
The parental origin of 6385 RNAs from 2–4 cell embryos was characterized (Fig. 1). There were 2675 RNAs derived from both the sperm and the oocyte. An additional 2575 RNAs were exclusively oocyte-derived, while 472 RNAs were exclusively sperm-derived (Supplementary Table S3). Finally, there were also 663 RNAs that were found only in 2–4 cell embryos.

Parental Origin of 2–4 Cell Embryo RNAs. The Venn diagram displays the origin of the RNAs present in 2–4 cell embryos. Detailed gene names are provided in Supplementary Table S3.
The expression levels of 24 of the differentially expressed RNAs (FDR < 0.1) in the 2–4 cell embryos was further determined. Of these, eight RNAs were downregulated and 16 RNAs were upregulated in embryos from high fertility sires versus low fertility sires. A total of four differentially expressed genes were sperm-derived. There were also two differentially expressed genes found exclusively in 2–4 cell embryos and nine that were oocyte-derived. Additionally, the sperm and oocyte co-contributed nine differentially expressed genes (Fig. 2). The parental origins of the remaining 41 differentially expressed genes were considered provisional and can be found in Supplementary Table S2.

Parental Origin of 24 Differentially Expressed RNAs between high- and low-fertility sires. The parental origin of an additional 41 genes was considered provisional and can be found in Supplementary Table S3.
Discussion
The RNAs donated by the oocyte at fertilization are known to affect the developmental competence of embryos4,5,6,7,8. It is also recognized that the sperm donates a variety of non-genetic elements12,13,14,15,16,17,18,19,20,21 during fertilization that can alter RNA expression22,23,24,25,26. This study demonstrates that male fertility is associated with RNA content in the pre-EGA embryo. The functionality of one of these RNAs was interrogated through knockdown experiments. Additionally, this study reveals a substantial number of RNAs that are directly contributed from the sperm to the embryo at fertilization. Furthermore, a suite of transcripts that are specific to the 2–4 cell embryo have also been identified.
Several of the differentially expressed genes identified through this study have been previously associated with fertility. For example, a genome-wide association study found that PKP2 and CTTNBP2NL N-terminal like (CTTNBP2NL) are associated with conception rates in Holstein cows40. Knockdown of PKP2 and overexpression of CTTNBP2NL each resulted in reduced embryo implantation rates in mice40. In this study, PKP2 was upregulated and CTTNBP2NL was downregulated in embryos from high fertility sires. Additionally, both genes were expressed in sperm. Therefore, this study demonstrates that there is paternal control over the presence of these RNAs in pre-EGA embryos. Further, the direction of differential expression for these RNAs in the pre-EGA embryos reinforces previous findings about their functions in fertility.
Additional differentially expressed genes have been associated with the regulation of spermatogenesis. For instance, the gene PPP1R36 drives autophagy during spermatogenesis41. Inhibiting autophagy in mice leads to infertile spermatozoa42. PPP1R36 was found in spermatozoa in this study. Additionally, PPP1R36 was upregulated in embryos from high fertility sires. Sperm-specific knockout mice for the autophagy regulator autophagy related 5 (ATG5) generate embryos that fail to develop beyond the 4–8 cell stage43. Thus, future studies should evaluate whether PPP1R36 is transferred to the zygote to help regulate autophagy. The mechanistic target of rapamycin kinase (mTOR) is another spermatogenesis-regulating gene that was differentially expressed in pre-EGA embryos. mTOR can shape male reproductive potential by affecting spermatogonial stem cell maintenance, Sertoli cell physiology, and blood-testis barrier architecture44. Blocking mTOR activity in mouse Sertoli cells is detrimental to sperm quality and causes infertility45. mTOR was upregulated in embryos of high fertility sires, and was also found in sperm. Future studies should evaluate whether these genes serve a dual purpose in regulating both spermatogenesis and early embryonic development.
Although many of the differentially expressed genes in this study were sperm-derived, there was a subset of these genes that were either oocyte-derived or embryo-specific. Therefore, alternative mechanisms for paternal regulation of pre-EGA RNA may be possible. Previously, our lab showed that differential DNA methylation patterns in sperm from high and low fertility sires was correlated with altered expression of the same genes in blastocysts from high and low fertility sires27. In this study, the paternally-expressed imprinted genes APBA1 and GATA binding protein 3 (GATA3)46 were both upregulated in embryos from high fertility sires. Altered DNA methylation in sperm of these genes is associated with motility47 and male fertility status48, respectively. APBA1 transcripts were found in sperm, but GATA3 was categorized as oocyte-derived in this study. Future studies should evaluate alternative sperm-derived contributions that may regulate pre-EGA embryo RNA, such as DNA methylation.
The knockdown of ATXN2L led to an increase in blastocyst development. Intriguingly, the expression of this gene was elevated in 2–4 cell embryos from high fertility sires. However, ATXN2L can have equally dynamic effects when its expression is increased or decreased49. ATXN2L is considered a regulator of stress granule and processing body (P-body) formation49. When ATXN2L is upregulated in HeLa cells, P-body numbers are reduced and stress granule numbers are increased49. Conversely, downregulation of ATXN2L in HeLa cells causes a reduction of stress granules49. P-bodies are present in non-stressed cells and contain non-translating mRNA and proteins which facilitate mRNA decay and translational repression50. Stress granules are formed when blocks of translation initiation occur in cells, and they contain various translation initiation factors and regulators of mRNA stability50. Control over RNA presence and activity is crucial to EGA51. Therefore, ATXN2L may require a careful balance during early embryonic development. Previously, a “maternal mode” and a “zygotic mode” have been proposed to initiate mRNA decay during EGA51. In the future, the use of ATXN2L as a “paternal mode” to regulate the mRNA presence should be explored. It is unclear whether the knockdown of ATXN2L is truly beneficial for embryonic development. However, our results indicate that reducing the expression of this gene can upregulate blastocyst development. Future studies should investigate the role of ATXN2L when it is overexpressed at the zygote stage. Moreover, it will be important to elucidate its possible role in regulating maternal RNAs. ATXN2L was not proven to be directly contributed by sperm in this study. Therefore, the paternal mechanism of control over ATXN2L expression should also be further assessed.
Delivery of individual sperm RNAs has been demonstrated in a variety of species14,15,16,17,18. The CLU RNA was shown to be transferred from sperm to oocytes in multiple studies14,16. CLU was found in sperm, oocytes, and 2–4 cell embryos in our dataset. This indicates it is both maternally and paternally-contributed in bovine. Additionally, calcium binding tyrosine phosphorylation regulated (CABYR) was previously identified as a sperm-specific RNA delivered to the zygote in mice52. Our study also confirmed this finding. The full subset of sperm-derived RNAs will provide a useful reference to identify fertility-related interactions between paternally-derived RNAs. For instance, this study revealed that a binding partner of CABYR called A-kinase anchoring protein 3 (AKAP3) was also sperm-derived. CABYR, AKAP3, and Ropporin form a complex in the fibrous sheath of sperm, which has been implied to function as signaling for capacitation53. Additional research should be carried out to determine whether these RNAs produce proteins that form functional complexes during early embryonic development.
Ultimately, these data also allowed additional insight into the minor EGA of the 2–4 cell embryo. Minor EGA is a pre-emptive wave of transcriptional activity which occurs at the 1 or 2–4 cell stage of bovine embryo development54. This study uniquely compiles information from both the maternally and paternally contributed RNAs. Therefore, we identified a novel subset of RNAs that are exclusively transcribed by the 2–4 cell bovine embryo during minor EGA. Conversely, a subset of the gene transcripts we identified as paternally-derived in this study is actively transcribed the 2–4 cell embryo. For example, we classified cyclin A2 (CCNA2) and cyclin dependent kinase 2 (CDK2) as maternally-derived RNAs in our study. These genes cooperate during activation of the embryonic genome55. In another study, Graf et al.49 found that CCNA2 and CDK2 belong to the category of RNAs that are maternally-derived but also transcribed by the embryo. However, several RNAs that were identified as embryo-specific through this study were also found to be present at the 4-cell stage in the study by Graf et al.49. These were deoxyribonuclease 1 (DNASE1), membrane associated ring-CH-type finger 3 (MARCH3), and ras-related C3 botulinum toxin substrate 3 (RAC3). DNASE1 can regulate DNA turnover and affects the stability of promoters56. MARCH3 mediates protein ubiquitination of Fc gamma receptor (FcγR), which affects responses to antibody-coated tumor cells57. RAC3 is a GTPase that alters cell growth by promoting cell adhesion and spreading58. These genes represent typical functions for early EGA RNAs transcribed by the embryo. However, this study also uncovered less traditional functions in the novel subset of early EGA genes.
Many of the unique embryo-specific RNAs in this study have roles in cell-cell signaling. These RNAs may serve as important regulators of embryo-mother communication during preimplantation development. For example, this study identified that sex hormone binding globulin (SHBG) expression was embryo-specific. SHBG is an androgen and estrogen transport protein which regulates plasma concentration of steroid hormones59. A deleterious mutation in SHBG is associated with prenatal death in dairy cattle60. This protein can be internalized by neurons or prostate cancer cells61,62. Further, SHBG can transport sex steroids into these target cells61,62. Another transcript found only in 2–4 cell embryos was tumor necrosis factor (TNF), a known mediator of embryo-maternal communication in mammals63. This gene modulates prostaglandin F2alpha (PGF2α) and is thought to aid in transferring the embryo from the oviduct to the uterus64. Additionally, secreted seminal-vesicle Ly-6 protein 1 (SOLD1) expression was also found to be embryo-specific in this study. SOLD1 is a signal at the fetomaternal interface that can regulate trophoblast invasiveness65. Future studies should interrogate whether these 2–4 cell embryo-specific RNAs can regulate signaling in the reproductive tract.
In conclusion, the present study demonstrated that sperm assists in regulating a substantial amount of pre-EGA RNA content in the early embryo and that male fertility is associated with RNA patterns in pre-EGA embryos. The knockdown of one of these genes, ATXN2L, resulted in substantially increased blastocyst development. This study also provided a whole-transcriptome characterization for oocyte- and sperm-contributed RNAs prior to EGA. A novel group of RNAs that are produced specifically by the 2–4 cell embryo was also identified. Future studies should evaluate whether non-RNA components contributed by the sperm to the zygote are capable of regulating pre-EGA RNA content in the embryo. Additionally, the roles of paternally-influenced RNAs in pre-EGA embryos should be further explored.
Methods
Ethics statement
This study did not require approval from the Animal Care and Use Committee. Cows used for oocyte aspirations were not cared for at the University of Wisconsin – Madison facilities. Ovaries used for oocyte aspirations were purchased from Applied Reproductive Technology, LLC (Monona, WI, USA), and we were permitted by the company to perform in vitro fertilization (IVF) using the ovaries.
Bull semen selection
Semen samples were donated by Semex, Canada and the fertility status of sires was designated by the company as either high or low fertility based on the Repromax™ system, which simultaneously accounts for Sire Conception Rate (SCR), Agri-Tech Analysis (ATA), and Canada’s Non-Return Rate (NRR) data. Permission was granted by Semex to perform IVF. A total of 12 bulls were used for embryo production, which included six high fertility and six low fertility bulls.
In vitro embryo production
In vitro production of embryos was performed as described previously66,67. Ovaries were supplied by Applied Reproductive Technology, LLC (Monona, WI, USA), from which follicles were aspirated for oocyte collection. Oocytes were washed in a TL-Hepes solution which contained 3% bovine serum albumin, sodium pyruvate, and gentamicin. Cohorts of 10 oocytes were matured for 24 h in 50 µl of M-199 maturation media which contained gonadotropins (FSH and LH), estradiol, sodium pyruvate, 10% fetal bovine serum, and gentamicin. In each of the six IVF replicates, matured oocytes were washed in the supplemented TL-Hepes solution and then split evenly into two groups for fertilization (200–380 oocytes/sire). One oocyte group was fertilized using a high fertility sire, and the second group was fertilized using a low fertility sire (Fig. 3). Overall, a total of 1737 oocytes were allocated to high fertility bulls and 1606 oocytes were allocated to low fertility bulls across six IVF replicates. New bulls were used in each replicate. In total, six high fertility bulls and six low fertility bulls were used for 2–4 cell embryo generation. Age differences of the bulls paired in each IVF replicate were checked using a paired t-test, and bull pairs used for IVF showed no substantial differences in age. Cohorts of 10 oocytes were transferred to 44 µl drops of fertilization medium, which was supplemented with fatty acid-free bovine serum albumin (FAF-BSA), sodium pyruvate, and gentamicin. Sperm preparation was performed with a Percoll gradient as previously described68. The final sperm concentration was adjusted to 1 million per mL, using 2 µl of sperm per drop. Penicillamine-hypotaurine-epinephrine and heparin (2 µl each) were added to fertilization droplets. Oocytes and sperm were co-cultured for 20 h. Then, presumptive zygotes were stripped of cumulus cells, washed with the supplemented TL-Hepes solution, and placed in 50 µl drops of CR1aa culture media69,70 which were supplemented with FAF-BSA, sodium pyruvate, amino acids, and gentamicin. Presumptive zygotes were cultured in cohorts of 20–25. At 40 h post-fertilization, embryos that had reached the 2–4 cell stage were counted and collected into 100 µl of lysis solution for RNA-Sequencing (RNA-Seq). Statistical analysis of cleavage rates was performed using a paired t-test. Three biological replicates of embryo groups containing 37–70 cleavage-stage embryos for each high and low fertility bull were used for RNA-Seq.

Schematic of experimental design for embryo generation. Each replicate included one high fertility and one low fertility sire.
RNA-Seq profiling of 2–4 cell embryos and statistical analysis
Total RNA was extracted from embryo groups using the RNAqueous™-Micro Total RNA Isolation Kit (Ambion, Austin, TX, USA). The Repli-G WTA single cell kit (Qiagen, Germantown, MD, USA) was then used for whole transcriptome amplification (WTA). Purification of cDNA was carried out using Agencourt AMPure XP (Beckman Coulter, Brea, CA, USA). Library preparation was performed using the Nextera DNA library preparation kit (Illumina, San Diego, CA, USA). Then, 150 bp paired-end sequencing was carried out on the HiSeq X Ten (Illumina). The quality of fastq files containing the raw sequence reads was evaluated using FastQC software (v 0.11.8). Adapters were trimmed from all sample fastq files using Trimmomatic software (v. 0.38) and the reads were simultaneously quality trimmed by removing reads shorter than 35 bp. All the trimmed files were re-checked for quality using FastQC software. Paired-end read files were aligned to the UMD3.1 bovine genome assembly (Bos taurusUMD3.1.dna.toplevel.fa) using Tophat (v 2.1.1)71. The number of reads per gene in each sample was calculated by HTSeq-count (v 0.6.1)72 using UMD3.1 as reference genome (Bos_taurus.UMD3.1.94.gtf). Differential expression analysis was performed on 2–4 cell samples using EdgeR (v 3.20.7). The statistical tests were corrected for multiple testing using the Benjamini-Hochberg method as implemented in EdgeR73. A false discovery rate (FDR) cutoff of 10% was used to identify significant genes.
Evaluation of gene expression in biological replicates
Three additional biological replicates of in vitro-produced 2–4 cell embryos were generated for validation purposes. New high- and low-fertility sires were used for each replicate. RNA was extracted from groups of 180–280 2–4 cell embryos for each sire. Whole transcriptome amplification was performed as described above. The iTaq Universal SYBR Green Supermix (Bio-Rad) was used for quantitative real-time PCR (qRT-PCR). Cycling was performed with a CFX-Connect Real-Time PCR Detection System (Bio-Rad), under the following conditions: 95 °C for 30 seconds, followed by 40 cycles of 95 °C for 5 seconds, and 60 °C for 30 seconds. To select a reference gene, the genes beta-actin (ACTB), ATP synthase F1 subunit beta (ATP5B), beta-2-microglobulin (B2M), and calnexin (CANX) were compared for expression stability across 2–4 cell embryo samples. The most stable gene, ACTB, was selected as the internal control74. All primers were designed to span an intron, in order to avoid DNA amplification (Supplementary Table S4). Fold change in gene expression was determined using the 2−ΔΔCT method as described75. If a sample CT was greater than 33, it was considered beyond the threshold for quantification. Validation within individual biological replicates was considered, and those replicates with >1.5-fold difference in accordance with sequencing results were categorized as individually validated. For determination of significance across all three replicates, a paired t-test of normalized gene expression values (ΔCT) was used.
Knockdown of ATXN2L using gapmer supplementation
ATXN2L was selected for further functional analysis based on its significance in validation, and because its role in fertility had not yet been explored. Knockdown was performed with a locked nucleic acid gapmer specific to ATXN2L, which was custom designed by Qiagen. Both 1 µM and 3 µM concentrations of the gapmer were evaluated, and the optimal concentration of 1 µM was selected. Each gapmer was supplemented to culture media 24 hours post-fertilization. Simultaneously, a control group of embryos was cultured, where an equivalent volume of water (vehicle of gapmer) was supplemented. Embryos remained in culture until blastocysts were graded at eight days post-fertilization. For grading, embryos were categorized as either blastocyst or degenerate. The blastocyst rate was calculated as the percentage of cleaved embryos that developed to the blastocyst stage. Three IVF replicates of ATXN2L gapmer knockdowns were carried out. Additionally, a cohort of embryos was supplemented with a bovine negative control gapmer, which is a scramble sequence designed so that it does not align to any known bovine sequences. Immediately after blastocyst grading, embryos were collected in lysis buffer, and RNA was extracted with the same method described above for the 2–4 cell embryos. Gene expression analysis was performed in order to confirm gapmer activity, and fold change in gene expression was calculated using the 2−ΔΔCT method75. The significance of both blastocyst rate and gene expression was determined using a paired t-test.
Identification of RNAs expressed in sperm
Sperm RNA was extracted from the two sires (one high fertility, one low) from one of the sequenced IVF replicates. One straw of cryopreserved semen per sire was subjected to RNA extraction. Each straw was thawed for one minute at 35–37 °C. Thawed semen was transferred to a 1.5 mL tube and centrifuged for four minutes at 4000 RPM. The supernatant was removed, and cells were suspended in a somatic cell lysis buffer76 for four minutes on ice. Sperm was then centrifuged four minutes at 4000 RPM and lysis supernatant was aspirated. Somatic cell lysis buffer suspension was repeated once. Following somatic cell lysis, sperm were suspended in 0.75 mL of TRIzol (Invitrogen, Carlsbad, CA, USA), and RNA extraction was carried out according to manufacturer instructions. Then, RNA was amplified through WTA, purified, and sequenced. Reads were processed as described for 2–4 cell embryo samples. RNAs with a read count greater than five in either sample were considered to be expressed in sperm.
Identification of RNAs expressed in oocytes
Studies that evaluated bovine oocyte RNA expression in multiple replicates were identified using NCBI Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo). Two studies provided freely available data on bovine oocyte RNA expression in multiple replicates77,78, and were thus used for comparison to our sequencing results. Data from Xie et al.77 is under accession number GSE18290 and data from Graf et al.49 is under accession number GSE52415. The averages across oocyte replicates was used for comparison. The data from Xie et al.77 was generated using the Affymetrix GeneChip® Bovine Genome Array, which includes all high quality and publicly identified bovine transcripts. The Affymetrix-gene-code was converted to gene ID through DAVID79,80, and a minimum probe threshold of 40 raw intensity units81 was used as a cutoff to classify whether or not a gene was expressed. The data from Graf et al.49 were generated through RNA-Seq and aligned to UMD 3.1, and a cutoff of greater than five reads78 was used to classify whether a gene was expressed in oocytes for this dataset.
Characterization of RNA origin
To classify the parental origin of RNAs, the reads from 2–4 cell embryo samples were merged with sequencing results from sperm samples and literature-derived oocyte RNA expression data. Any gene with greater than five reads in at least one of the six replicates of 2–4 cell embryos was considered expressed, and the thresholds stated above were used to determine sperm and oocyte expression. Genes that did not meet the threshold for any of the 2–4 cell embryo samples were omitted from the analysis. The parental origin of RNAs was determined based on RNA expression in different sample types. Ultimately, the RNAs were sorted into four categories: (1) both oocyte- and sperm-derived (2) oocyte-derived; (3) sperm-derived; (4) only 2–4 cell embryo. If a gene was only found in one gamete-specific dataset (rather than both) it was labeled as provisional. Provisionally-labeled genes were not included in final counts of parental origin, but all genes and parental origin labels can be found in Supplementary Table S3.
Accession codes
Oocyte RNA expression data from Xie et al.77 is under accession number GSE18290, and oocyte RNA expression data from Graf et al.49 is under accession number GSE52415.
Data availability
The majority of data generated is published in the current study (and its Supplementary Information files). The datasets generated during the current study are available from the corresponding author on reasonable request.
References
Schultz, R. M. The molecular foundations of the maternal to zygotic transition in the preimplantation embryo. Hum. Reprod. Update 8, 323–331 (2002).
Schier, A. F. The Maternal-Zygotic Transition: Death and Birth of RNAs. Science 316, 406–407 (2007).
Svoboda, P., Franke, V. & Schultz, R. M. Sculpting the Transcriptome During the Oocyte-to-Embryo Transition in Mouse. Current Topics in Developmental Biology 113, (Elsevier Inc. 2015).
Chan, A. P., Kloc, M., Larabell, C. A., LeGros, M. & Etkin, L. D. The maternally localized RNA fatvg is required for cortical rotation and germ cell formation. Mech. Dev. 124, 350–363 (2007).
Cuykendall, T. N. & Houston, D. W. Vegetally localized Xenopus trim36 regulates cortical rotation and dorsal axis formation. Development 136, 3057–3065 (2009).
Mei, W. et al. Maternal Dead-End1 is required for vegetal cortical microtubule assembly during Xenopus axis specification. Dev. 140, 2334–2344 (2013).
Zhang, J. et al. The role of maternal VegT in establishing the primary germ layers in Xenopus embryos. Cell 94, 515–524 (1998).
Gentsch, G. E., Spruce, T., Owens, N. D. L. & Smith, J. C. Maternal pluripotency factors initiate extensive chromatin remodelling to predefine first response to inductive signals. Nat. Commun. 10 (2019).
Krawetz, S. A. Paternal contribution: new insights and future challenges. Nat. Rev. Genet. 6, 633–642 (2005).
Galeraud-Denis, I., Lambard, S. & Carreau, S. Relationship between chromatin organization, mRNAs profile and human male gamete quality. Asian J. Androl. 9, 587–592 (2007).
Immler, S. The sperm factor: paternal impact beyond genes. Heredity (Edinb). 121, 239–247 (2018).
Rakyan, V. K. et al. Transgenerational inheritance of epigenetic states at the murine AxinFu allele occurs after maternal and paternal transmission. Proc. Natl. Acad. Sci. USA 100, 2538–2543 (2003).
Edwards, C. A. & Ferguson-Smith, A. C. Mechanisms regulating imprinted genes in clusters. Curr. Opin. Cell Biol. 19, 281–289 (2007).
Ostermeier, C. G., Miller, D., Huntriss, J. D., Diamond, M. P. & Krawetz, S. A. Delivering spermatozoan RNA to the oocyte. Nature 429, 154 (2004).
Fang, P. et al. Estimated Diversity of Messenger RNAs in Each Murine Spermatozoa and Their Potential Function During Early Zygotic Development. Biol. Reprod. 90, 1–11 (2014).
Kempisty, B. et al. Analysis of selected transcript levels in porcine spermatozoa, oocytes, zygotes and two-cell stage embryos. Reprod. Fertil. Dev. 20, 513–518 (2008).
Avendaño, C., Franchi, A., Jones, E. & Oehninger, S. Pregnancy-specific β-1-glycoprotein 1 and human leukocyte antigen-E mRNA in human sperm: Differential expression in fertile and infertile men and evidence of a possible functional role during early development. Hum. Reprod. 24, 270–277 (2009).
Yao, C.-J. et al. The role of Dby mRNA in early development of male mouse zygotes. Asian J. Androl. 12, 567–577 (2010).
Kawano, M., Kawaji, H., Grandjean, V., Kiani, J. & Rassoulzadegan, M. Novel Small Noncoding RNAs in Mouse Spermatozoa, Zygotes and Early Embryos. PLoS One 7, e44542 (2012).
Saunders, C. M. et al. PLC zeta: a sperm-specific trigger of Ca(2+) oscillations in eggs and embryo development. Development 129, 3533–3544 (2002).
MacLeod, G. & Varmuza, S. The application of proteomic approaches to the study of mammalian spermatogenesis and sperm function. FEBS J. 280, 5635–5651 (2013).
Orang, A. V., Safaralizadeh, R. & Kazemzadeh-Bavili, M. Mechanisms of miRNA-mediated gene regulation from common downregulation to mRNA-specific upregulation. Int. J. Genomics 2014 (2014).
Klose, R. J. & Bird, A. P. Genomic DNA methylation: The mark and its mediators. Trends Biochem. Sci. 31, 89–97 (2006).
Wei, J. W., Huang, K., Yang, C. & Kang, C. S. Non-coding RNAs as regulators in epigenetics (Review). Oncol. Rep. 37, 3–9 (2017).
Hentze, M. W., Castello, A., Schwarzl, T. & Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 19, 327–341 (2018).
Roux, P. P. & Topisirovic, I. Signaling Pathways Involved in the Regulation of mRNA Translation. Mol. Cell. Biol. 38, 1–26 (2018).
Kropp, J. et al. Male fertility status is associated with DNA methylation signatures in sperm and transcriptomic profiles of bovine preimplantation embryos. BMC Genomics 18, 1–15 (2017).
Fang, L. et al. Integrating Signals from Sperm Methylome Analysis and Genome-Wide Association Study for a Better Understanding of Male Fertility in Cattle. Epigenomes 3, 10 (2019).
Card, C. J., Krieger, K. E., Kaproth, M. & Sartini, B. L. Oligo-dT selected spermatozoal transcript profiles differ among higher and lower fertility dairy sires. Anim. Reprod. Sci. 177, 105–123 (2017).
Yuan, S. et al. Sperm-borne miRNAs and endo-siRNAs are important for fertilization and preimplantation embryonic development. Development 143, 635–647 (2016).
Fagerlind, M., Stålhammar, H., Olsson, B. & Klinga-Levan, K. Expression of miRNAs in Bull Spermatozoa Correlates with Fertility Rates. Reprod. Domest. Anim. 50 (2015).
Rahman, M. S., Kwon, W. S. & Pang, M. G. Prediction of male fertility using capacitation-associated proteins in spermatozoa. Mol. Reprod. Dev. 84, 749–759 (2017).
Park, Y. J., Kwon, W. S., Oh, S. A. & Pang, M. G. Fertility-related proteomic profiling bull spermatozoa separated by percoll. J. Proteome Res. 11, 4162–4168 (2012).
Peddinti, D. et al. Comprehensive proteomic analysis of bovine spermatozoa of varying fertility rates and identification of biomarkers associated with fertility. BMC Syst. Biol. 2, 1–13 (2008).
Parthipan, S. et al. Spermatozoa input concentrations and RNA isolation methods on RNA yield and quality in bull (Bos taurus). Anal. Biochem. 482, 32–39 (2015).
Olszanska, B. & Borgul, A. Quantitation of nanogram amounts of nucleic acids in the presence of proteins by the ethidium bromide staining technique. in. Acta Biochimica Polonica 37, 59–63 (1990).
Kashir, J., Nomikos, M. & Lai, F. A. Phospholipase C zeta and calcium oscillations at fertilisation: The evidence, applications, and further questions. Adv. Biol. Regul. 67, 148–162 (2018).
Nozawa, K., Satouh, Y., Fujimoto, T., Oji, A. & Ikawa, M. Sperm-borne phospholipase C zeta-1 ensures monospermic fertilization in mice. Sci. Rep. 8, 1–10 (2018).
Parrington, J., Anthony Lai, F. & Swann, K. The soluble mammalian sperm factor protein that triggers Ca2+ oscillations in eggs: Evidence for expression of mRNA(s) coding for sperm factor protein(s) in spermatogenic cells. Biol. Cell 92, 267–275 (2000).
Sugimoto, M. et al. Genetic variants related to gap junctions and hormone secretion influence conception rates in cows. Proc. Natl. Acad. Sci. 110, 19495–19500 (2013).
Zhang, Q. et al. The germline-enriched Ppp1r36 promotes autophagy. Sci. Rep. 6, 1–9 (2016).
Wang, H. et al. Atg7 is required for acrosome biogenesis during spermatogenesis in mice. Cell Res. 24, 852–869 (2014).
Tsukamoto, S. et al. Autophagy is essential for preimplantation development of mouse embryos. Science 321, 117–120 (2008).
Oliveira, P., Cheng, C. Y. & Alves, M. G. Emerging Role for Mammalian Target of Rapamycin in Male Fertility. Trends Endocrinol Metab. 28, 165–267 (2017).
Boyer, A. et al. mTOR Regulates Gap Junction Alpha-1 Protein Trafficking in Sertoli Cells and Is Required for the Maintenance of Spermatogenesis in Mice. Biol. Reprod. 95, 1–11 (2016).
Luedi, P. P. et al. Computational and experimental identification of novel human imprinted genes. Cold Spring Harb. Lab. Press 1723–1730, https://doi.org/10.1101/gr.6584707 (2007).
Pacheco, S. E. et al. Integrative DNA methylation and gene expression analyses identify DNA packaging and epigenetic regulatory genes associated with low motility sperm. PLoS One 6, 1–10 (2011).
Sujit, K. M. et al. Genome-wide differential methylation analyses identifies methylation signatures of male infertility. Hum. Reprod. 33, 2256–2267 (2018).
Kaehler, C. et al. Ataxin-2-Like Is a Regulator of Stress Granules and Processing Bodies. PLoS One 7, 1–12 (2012).
Decker, C. J. & Parker, R. P-Bodies and Stress Granules: Possible Roles in the Control of Translation and mRNA Degradation. Cold Spring Harb. Perspect. Biol. 1–16, https://doi.org/10.1101/cshperspect.a012286 (2012).
Lee, M. T., Bonneau, A. R. & Giraldez, A. J. Zygotic genome activation during the maternal-to-zygotic transition. Annu Rev Cell Dev Biol. 581–613, https://doi.org/10.1146/annurev-cellbio-100913-013027 (2014).
Johnson, G. D., Mackie, P., Jodar, M., Moskovtsev, S. & Krawetz, S. A. Chromatin and extracellular vesicle associated sperm RNAs. Nucleic Acids Res. 43, 6847–6859 (2015).
Li, Y. F. et al. CABYR binds to AKAP3 and Ropporin in the human sperm fibrous sheath. Asian J. Androl. 13, 266–274 (2011).
Memili, E. & First, N. L. Control of Gene Expression at the Onset of Bovine Embryonic Development. Biol. Reprod. 61, 1198–1207 (1999).
Kanka, J. et al. Association of the transcription profile of bovine oocytes and embryos with developmental potential. Anim. Reprod. Sci. 134, 29–35 (2012).
Martínez-Balbás, M. A., Dey, A., Rabindran, S. K., Ozato, K. & Wu, C. Displacement of sequence-specific transcription factors from mitotic chromatin. Cell 83, 29–38 (1995).
Fatehchand, K. et al. Toll-like receptor 4 ligands down-regulate Fcγ receptor IIb (FcγRIIb) via MARCH3 protein-mediated ubiquitination. J. Biol. Chem. 291, 3895–3904 (2016).
Haataja, L., Kaartinen, V., Groffen, J. & Heisterkamp, N. The small GTPase Rac3 interacts with the integrin-binding protein CIB and promotes integrin αIIbβ3-mediated adhesion and spreading. J. Biol. Chem. 277, 8321–8328 (2002).
Hammond, G. L. Plasma steroid-binding proteins: Primary gatekeepers of steroid hormone action. J. Endocrinol. 230, R13–R25 (2016).
Fritz, S. et al. Detection of Haplotypes Associated with Prenatal Death in Dairy Cattle and Identification of Deleterious Mutations in GART, SHBG and SLC37A2. PLoS One 8, 2–9 (2013).
Caldwell, J. D., Shapiro, R. A., Jirikowski, G. F. & Suleman, F. Internalization of sex hormone-binding globulin into neurons and brain cells in vitro and in vivo. Neuroendocrinology 86, 84–93 (2007).
Cunningham, M. & Gilkeson, G. Estrogen receptors in immunity and autoimmunity. Clin. Rev. Allergy Immunol. 40, 66–73 (2011).
Correia-Álvarez, E. et al. Early embryonic and endometrial regulation of tumor necrosis factor and tumor necrosis factor receptor 2 in the cattle uterus. Theriogenology 83, 1028–1037 (2015).
Siemieniuch, M. J., Woclawek-Potocka, I., Deptula, K., Okuda, K. & Skarzynski, D. J. Effects of Tumor Necrosis Factor-α and Nitric Oxide on Prostaglandins Secretion by the Bovine Oviduct Differ in the Isthmus and Ampulla and Depend on the Phase of the Estrous Cycle. Exp. Biol. Med. 234, 1056–1066 (2009).
Awad, M., Koshi, K., Kizaki, K., Takahashi, T. & Hashizume, K. SOLD1 is expressed in bovine trophoblast cell lines and regulates cell invasiveness. Reprod. Biol. Endocrinol. 12, 1–10 (2014).
Kropp, J. & Khatib, H. Characterization of microRNA in bovine in vitro culture media associated with embryo quality and development. J. Dairy Sci. 98, 6552–6563 (2015).
Gross, N., Kropp, J. & Khatib, H. Sexual Dimorphism of miRNAs Secreted by Bovine In vitro-produced Embryos. Front. Genet. 8, 1–10 (2017).
Parrish, J. J., Krogenaes, A. & Susko-Parrish, J. L. Effect of bovine sperm separation by either swim-up or Percoll method on success of in vitro fertilization and early embryonic development. Theriogenology 44, 859–869 (1995).
Rosenkrans, C. F., Zeng, G. Q., MCNamara, G. T., Schoff, P. K. & First, N. L. Development of bovine embryos in vitro as affected by energy substrates. Biol. Reprod. 49, 459–62 (1993).
Sagirkaya, H. et al. Developmental and molecular correlates of bovine preimplantation embryos. Reproduction 131, 895–904 (2006).
Daehwan, K. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, 1–13 (2013).
Anders, S., Pyl, P. T. & Huber, W. HTSeq-A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2009).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 3, 1–12 (2002).
Livak, K. J. & Schmittgen, T. D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 25, 402–408 (2001).
Goodrich, R., Johnson, G. & Krawetz, Sa The preparation of human spermatozoal RNA for clinical analysis. Arch. Androl. J. Reprod. Syst. 53, 161–167 (2007).
Xie, D. et al. Rewirable gene regulatory networks in the preimplantation embryonic development of three mammalian species. Genome Res. 20, 804–815 (2010).
Graf, A. et al. Fine mapping of genome activation in bovine embryos by RNA sequencing. Proc. Natl. Acad. Sci. USA 111, 4139–4144 (2014).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Huang, D. W., Sherman, B. T. & Lempicki, R. A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13 (2009).
Mach, N. et al. Pleiotropic effects of polymorphism of the gene diacylglycerol-O-transferase 1 (DGAT1) in the mammary gland tissue of dairy cows. J. Dairy Sci. 95, 4989–5000 (2012).
Acknowledgements
The authors would like to thank John Parrish and Rick Monson for mentorship on the production of embryos. This research was supported by the College of Agriculture and Life Sciences, University of Wisconsin-Madison (HATCH MULTI-STATE RESEARCH FORMULA FUND, award number MSN168911).
Author information
Authors and Affiliations
Contributions
N.G. conducted the experiments, M.G.S. and F.P. analyzed data, and H.K. conceived the experiments. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gross, N., Strillacci, M.G., Peñagaricano, F. et al. Characterization and functional roles of paternal RNAs in 2–4 cell bovine embryos. Sci Rep 9, 20347 (2019). https://doi.org/10.1038/s41598-019-55868-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-55868-3
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
