
Abstract
A novel cerium doped compounds Mn3Gd7–xCex(SiO4)6O1.5 with an apatite structure was found and used to achieve high-efficiency degradation of tetracycline in aqueous solution. The catalysts were characterized by XRD, XPS, EDS and other techniques. The characteristic results indicated that the catalytic activity of the compound was improved due to the introduction of Ce in the structure, because Ce3+ which was stably present in the apatite structure can serve as an active site for the reaction, and in addition, there was a high presence between Ce4+ and Ce3+ on the surface of the catalyst. The redox potential and high oxygen storage capacity were also beneficial for the catalytic reaction. The results of free radical capture indicated that both superoxide radicals and hydroxyl radicals participated in the catalytic oxidation process and played an important role in the reaction. The decomposition of tetracycline followed the pseudo second-order reaction kinetics. In addition, the catalyst exhibited long-term stability and low metal leaching during the reaction, which indicated that the novel cerium-doped apatite structure material could be a promising wastewater treatment material.
Similar content being viewed by others

Introduction
Antibiotics are extensively used in the treatment of human diseases and in livestock and aquaculture, due to their broad-spectrum and extremely strong antibacterial activity1. In recent years, with the extensive use of antibiotics in human and veterinary medicine, trace antibiotics have been detected the in drinking water, surface water, groundwater, soil, and aquatic organisms2. Due to their low metabolism rate, some antibiotics remain in the natural environment for a long time, which not only damage the balance of the ecological environment, but also causes detrimental to humans, due to their enrichment in food chains or pollution of drinking water4,5. Therefore, alongside the plans of reasonable use of antibiotics in medicines, it is important also to develop effective treatment technologies to remove antibiotics from polluted water or soil3.
Tetracycline (TC) exists in trace levels in natural waters and is difficult to decompose, and the toxicity of its primary decomposition products is comparable to or even higher than that of the parent compound4. Therefore, traditional techniques including adsorption separation5 and biodegradation6 have been investigated to remove antibiotics and other antimicrobials from water. However, the adsorption method does not destroy the structure of tetracycline and cannot achieve the effect of thorough removal. On the other hand, TC inhibits metabolism during microbial degradation. Thus, rapid and efficient treatment processes must be developed for TC degradation1.
Advanced oxidation processes (AOPs) is based on highly potent chemical species7. It has the characteristics of high efficiency8, bottom measurement, the absence of secondary pollution9 and short residence time for the treatment of refractory organic matter in water and sewage. Compared with the traditional method of wastewater treatment, it has obvious advantages such as strong oxidizing ability, non-selective simple reaction conditions, and no requirements for high temperature and pressure. It can be used not only for advanced treatment of sewage, but also in combination with other treatment technologies10. In recent years, heterogeneously catalyzed ozone oxidation technology has received extensive attention in the large-scale water treatment11. Meanwhile, in order to achieve more efficient processing efficiency, catalysts for heterogeneous catalytic ozonation technology include transition metal oxide such as manganese dioxide, iron oxide, copper oxide, rare earth oxides, such as cerium oxide and their composites, have been reported as solid supports for the catalytic degradation. However, regardless if they are single metal oxide or compounds, there will be a certain amount of metal dissolution12,13, causing catalyst deactivation and secondary pollution, wasting of resources. Therefore, increasingly researches are focused on the way to low the amount of metal dissolution, in which introducing catalytically active metal ions into the crystal lattice is a good approach. In the context of lowing the active ion dissolution. doping of the crystal lattice with active metal ions, zhu et al.14 studied the A-position of the perovskite compound has 12-fold coordinated positions, and the high coordination number of the active ions can effectively improve the stability of the active metal in the compound, otherwise, Wang et al.15 also reported relationship between leaching ratio and doping content. And further, by using the active metal doping into the crystal lattice can also better disperse the active elements into the crystal lattice to better improve the catalytic performance of the sample. S.I. Suárez-Váquez et al. also reported the phenomenon16, the addition of Mn resulted in the incorporation of Mn4+ in Ti4+ sites present into the structure of the perovskite. This incorporation also enhances the relation Oads/Olatt and the catalytic properties. Finally, the catalyst doped by Mn presented the highest catalytic activity17. In addition, CNTs attached by means of CH-π, π-π stacking and Van der Waals forces could make them disperse in liquid media and leave the polyaromatic pattern unaltered, and reduce catalyst deactivation.
The compounds with an apatite-type structure A10[MO4]6O2 have great flexibility in their crystal lattice to accommodate a big number of substitutions18. The cations in a position can be substituted by foreign cations having different oxidation states or radii, thus increasing the number of the cation vacancies in the structure. Meanwhile, [PO4]3− ions can also be substituted by [SiO4]4−, [GeO4]4− anion groups under different conditions. Obviously, the component adjustment will bring a bit of active sites, to improve catalytic effect. So, it is a kind of potential candidate for catalyst designing technology.
Among the different reported catalysts, transition metal ions such as Mn(II), has demonstrated high efficiencies in catalytic ozonation for homogeneous degradation of various organic pollutants19. Manganese ion with the lowest state has a significant advantages as a redox medium for the removal of organic pollutants20. Besides, via the introduction of MnOx, large amount of surface hydroxyl groups are generated on the surface of catalyst, it play a key role in degradation adsorption and ·OH initiation, Higher multivalent MnOx (Mn(III)/Mn(IV)) enhances electron transfer, which also benefits degradants removal17. To improve the catalytic activity, cerium (Ce) with a high oxygen storage capacity has been commonly used21. As such, a redox cycle between the +3 and +4 states can be manipulated to create efficient catalysts process3. Interestingly, the doped Ce cations can enter the apatite-type lattice with equivalent substitution, and maintain the stability of the structure. Inclusion of foreign metals in the structure could lead to increases in defects inside the catalyst surface thereby creating more number of active sites22,23.
In this work, we prepared a kind of Ce-doped apatite-type compounds Mn3Gd7−xCex(SiO4)6O1.5 using traditional high temperature solid phase method and applied it in the catalytic ozonation of TC. The results showed that strong interactions between Ce atoms in apatite-type structure were established and promoted the regeneration of the catalyst and extended its lifecycle. Also, it was shown that the new composite had a high removal efficiency of TC in ozone catalytic degradation. In addition, the pathways and mechanism of TC degradation were proposed, and had a good stability performance after the reaction.
Experimental and Methods
A traditional high temperature solid-state reaction was used to prepare the Mn3Gd7−xCex(SiO4)6O1.5 compounds. The raw materials including MnCO3 (Aldrich, 99.9%), SiO2 (Aldrich, 99.9%) and Gd2O3 (Aldrich, 99.99%) and CeO2 (Aldrich, 99.99%) used for the syntheses of Mn3Gd7−xCex(SiO4)6O1.5 were purchased from the the Sinopharm Chemical Reagent Co., Ltd. Firstly, calculating the amount of each raw material required according to the stoichiometric ratio, after weighting and thoroughly mixing in the agate mortar, the mixtures were placed into little corundum crucibles, and covered with activated carbon to prevent oxidation. Finally, the samples were sintered at 1200 °C for 4 h in muffle furnace to produce the final products.
The crystal structures of the synthesized samples were examined by the X-ray powder diffractometer (XRD; D8 Advance diffractometer, Germany) with CuKα radiation (λ = 1.5418 Å) from 10° to 70° (2θ). The valence of manganese and cerium in the structure of Mn3Gd7−xCex(SiO4)6O2 was confirmed by X-ray Photoelectron Spectroscopy (XPS, Thermo Scientific) with monochromatic AlKa irradiation (150 W). The binding energy (BE) scale was calibrated in reference23 to the energy of the adventitious carbon (C 1 s) core level assigned at 284.6 eV. The visible spectra of as-prepared samples were performed on ultraviolet-visible spectroscopy (Beijing North Temple Instrument Technology Co., Ltd.) to collect the wavelength range from 500 to 700 nm at an interval of 1 nm. The molecular weight of the obtained samples was identified by methods of HPLC-mass spectrometer (LC-MS. Thermo Scientific) with high resolution search (Thermo Scientific)24. Catalytic ozonation processes were carried out in a semi-batch reactor containing 0.4 g/L of the target pollutant. The experimental device is composed of an ozone generating device, a vapor-liquid reaction device, a stirring device and an exhausting gas treating device. Ozone was produced by an ozone generator and passed into the reaction unit at 500 cm3•min−1, and the concentration of ozone is 40 mg/L. The reaction vessel was a 500 mL three-necked flask, the temperature in the vessel was maintained at 20 °C, and the agitation rate was set at 500 rpm•min−1. The exhaust gas treatment unit is a 500 mL 25 g/L Na2S2O3 solution. At certain time intervals, water samples were withdrawn from the reactor with a syringe and collected to measure the TC concentration. To test the stability and recyclability of Mn3Gd7−xCex(SiO4)6O1.5, the catalyst was filtered, centrifuged at 10,000 rpm, dried at 80 °C, and used again in another cycles.
Results and Discussion
Characterization of the materials
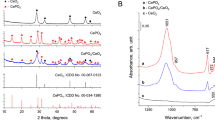
Figure 1 shows the XRD patterns of the structure of Mn3Gd7–xCex(SiO4)6O1.5 solid solution along the c axis. Two cationic sites exist in the structure: 9-fold coordinated 4f sites with C3 point symmetry and 7-fold coordinated 6 h sites with Cs point symmetry18. To verify the phase purity and structure, Mn3Gd7–xCex(SiO4)6O1.5 with different amount cerium doping were characterized by XRD. The pure phases of the solid solution were obtain, and no peaks of other impurity phases were found. All the characterized peaks are in a good agreement with the standard Ca2Gd8(SiO4)6O2 (JCPDS No.28-0212). Although the substitution amount of Ce element is increased, the structure of the compound remains isostructure with Ca2Gd8(SiO4)6O2. Crucially, the proportion of positive ions was greater than that of negative ions in the sample, which provides direct evidence of the existence of vacancies25. Moreover, XRD patterns of the Mn3Gd7−xCex(SiO4)6O1.5 samples after every cycles of TC degradation were shown in Fig. 2(b), and will be discussed in the following sections.

XRD patterns of Mn3Gd7−x(SiO4)6O1.5: xCe particles, and the standard data for Gd2Gd8(SiO4)6O2 (JCPDS card No. 28-0212) is shown as a comparison.

Catalyst on the degradation of TC, cycle stability test: (a) five times cycles, (b) XRD test of catalyst after every times using; leaching amount test: (c) leaching amount of Ce before and after recycle.
The Energy dispersive spectroscopy (EDS) were carried out for the chemical composition of the compounds prepared in the study. The result of a scanning electron microscope (SEM) image of a Mn3Gd5.5Ce1.5(SiO4)6O1.5 sample was shown in Fig. 3(a). The particles of the sample are not uniform and the particles are relatively large due to agglomeration during heating. Figure 3(b) shows an EDS elemental analysis of surface points in a rectangular region, the proportion of positive-charged ions was greater than that of negative ions in the sample, which provides direct evidence of the existence of vacancies25. In addition, Fig. 3(c–g) shows the elemental distributions of O, Si, Mn, Gd, and Ce in Mn3Gd5.5Ce1.5(SiO4)6O1.5 particles. The Mn element is less homogeneous than the remaining elements, we think this might be due to the morphology of the samples and an increased amount near the surface of the particles.

(a) EDS spectrum, (b) SEM image, (c–g)Elemental analysis of Mn3Gd5.5Ce1.5(SiO4)6O1.5 particles about O, Si, Mn, Gd, and Ce respectively.
To investigate the chemical states of the variable elements and oxygen in the sample, a wide survey scan of XPS spectra was carried out. All peaks have been corrected by C1s peaks position (284.8 eV)26. Figure 4(a) shows the XPS survey spectrum of Mn3Gd7–xCex(SiO4)6O1.5. The presence of Mn, Gd, Ce, Si, O, and C in Fig. 4(a), with no other impurity was detected. The high resolution XPS scan of the Mn2p doublet with the peak deconvolution is shown in Fig. 4(c). Both the peaks of Mn could well attach to the Mn 2p3/2 and Mn 2p1/2 at the binding energies (BE) of 642.5 eV and 653.4 eV. There was no any noticeable shoulder peaks observed in Mn2p spectra of Mn3Gd7−xCex(SiO4)6O1.5, revealing that Mn ions are in the formal chemical valance state of 2+. Analysis of the Ce 3d spectra showed that 903.5 eV(U0), 898.8 eV(V′), and 884.9 eV(V0)were ascribed to Ce3+ species while 907.5 eV(U) and 888.2 eV(V) were attributed to Ce4+ species (Fig. 4(d)). The surface concentration of Ce4+ can be determined by Ce4+ = Ce4+/(Ce4+ + Ce3+); Ce3+ = Ce3+/(Ce4+ + Ce3+), where Ce3+ = U0 + V′ + V0 and Ce4+ = U + V. Inherent challenges are present for Ce 3d XPS spectrum analysis because of the difficulty in deconvolution of individual peaks in Ce 3d3/2 and Ce 3d5/2 envelopes27. So the proportion of Ce4+ and Ce3+ was 55% and 45% in the composite28. In order to confirm the oxygen vacancies in the prepared samples, we analyzed the XPS spectra of O1s. The result of O1s BE peaks of Mn3Gd5.5Ce1.5(SiO4)6O1.5 have been showed in Fig. 4(b). The O 1s XPS spectra were deconvoluted with three major peaks, located at 529.9 eV, 531.8 eV, and 532.6 eV, which correspond to lattice oxygen species (O2−) named OI and adsorbed oxygen (e.g., O22− and O−), and hydroxyl groups (OH−), respectively29 named OII. The atom ratio of OI to OII was 4.02 for Mn3Gd5.5Ce1.5(SiO4)6O1.5, which is higher than that of Mn3Gd7(SiO4)6O1.5 to 3.17, respectively, indicating that the doping of Ce increased the oxygen defects of Mn3Gd7(SiO4)6O1.5, which is the most active oxygen, and has been reported to play an important role in the oxidation reaction30. Otherwise, the EPR comparative experiment of Mn3Gd7(SiO4)6O1.5 and Mn3Gd5.5Ce1.5(SiO4)6O1.5 is shown in Fig. 4(e). Obviously, the peaks of Mn3Gd5.5Ce1.5(SiO4)6O1.5 is higher than that of Mn3Gd7(SiO4)6O1.5, also verifing that the doping of Ce increases the oxygen vacancies, increases the activity of the catalyst.

(a) XPS spectra of Mn3Gd7−xCex(SiO4)6O1.5 synthesized in present work. (b) high resolution XPS spectra at O 1 s position of compounds. (c) Mn 2p. position of Mn3Gd7−xCex(SiO4)6O1.5 (d) Ce 3d. position of Mn3Gd7−xCex(SiO4)6O1.5; (e) Comparative EPR spectra of Mn3Gd7(SiO4)6O1.5 and Mn3Gd5.5Ce1.5(SiO4)6O1.5 particles.
Catalytic degradation of TC in Mn3Gd7−xCex(SiO4)6O1.5 system
Degradation processes of TC using Mn3Gd7−xCex(SiO4)6O1.5 as a catalyst were investigated in the presence of ozone, (Experimental conditions: Catalyst loading 2 g L−1, pH = 3.4 working volume of 200 mL, [TC]0 = 400 mg L−1, the concentration of ozone is 40 mg/L). Figure 5(a) shows the comparisons of time-dependent reaction yields between the ozonation and the six catalysts (the situation under which ozone alone as a control is also included). First of all, when only the Mn3Gd5.5Ce1.5(SiO4)6O1.5 compound was added into the TC solution, there was no adsorption of TC. It is obvious that there are substantially different performances in the catalytic oxidation of TC. The composite Mn3Gd5.5Ce1.5(SiO4)6O1.5 has the greater catalytic capability for TC degradation in comparison with pure Mn3Gd7(SiO4)6O1.5. So, Mn3Gd7(SiO4)6O1.5 was mixed with CeO2, and MnO2. It could be observed, that the introduction of Ce enhanced the catalytic degradation efficiency. To further investigate the impact of Ce, different amounts of Ce doped Mn3Gd7–xCex(SiO4)6O1.5 samples was prepared, and used in the catalytic degradation of TC. As shown in Fig. 5(b), increasing of the Ce doping concentration from 0% to 15% led to a significant increase of TC removal, implying that the cerium doping plays an important role in catalytic ozonation as an active species. When the amount of cerium doping continuously increased up to 20%, the efficiency reduces. That can be explain that when the doping amount is 15%, (211) crystal plane of the compound has the lowest crystal strength, resulting in increased defects and active sites. So the most suitable proportion of Ce doped amount in Mn3Gd7–xCex(SiO4)6O1.5 is x = 1.5 for catalytic ozonation of TC.

The catalytic catalystic degradation behavior of TC (Experimental conditions: Catalyst loading 2.5 g, pH = 3.4), [TC]0 = 400 mg L−1, working volume = 200 mL, and its degradation behavior under O3/Catalyst system, the concentration of ozone is 40 mg/L.) (a)different forms of manganese and cerium (b) different doping amount of cerium in the catalytic system (c)different initial concentrations in catalystic system (d) Pseudo-second order kinetic behavior under different tetracycline concentration in degradation process.
For ozonation catalyst process, initial TC concentration is a worth considering parameter. The experiments of different TC concentration were conducted, the result in Fig. 5(c) shows that the degradation efficiency of TC dropped with increasing of initial concentrations. When the concentration rises from 200 mg/L to 600 mg/L, the degradation efficiency dropped from 99.8% to 86.2%. The excess of TC (up to 600 mg/L) or its degradation intermediates may need to consume more active radicals, so the catalytic capability became slight low. Additionally, the mechanism of TC degradation under the catalysis of Mn3Gd5.5Ce1.5(SiO4)6O1.5 was explored under different initial TC concentration (200, 400 and 600 mg/L) according to the pseudo-first order and pseudo-second order kinetics31.
where C0 is the initial concentration of TC, Ct is the concentration of the TC in the solution after treatment at time t, and k1 and k2 are pseudo-first and pseudo-second order rate constants. Clearly, the degradation of TC was better described by the pseudo-second order kinetic model17 judged by its regression coefficient (R2 > 0.96). The fitting result are shown in Fig. 5(d), as the concentration increased from 200 to 600 mg/L the reaction rate constants k2 were 0.0036, 0.0028 and 0.0004 Lmg–1 min–1, respectively.
Possible degradation mechanism in the presence of Mn3Gd5.5Ce1.5(SiO4)6O1.5 catalyst
During TC degradation with catalysts under ozone as oxidant, several kinds of reactive radicals, such as ·OH, and·O3, could be generated and have a great influence under the activation of transition metals. In order to investigate the effect of the two reactive radicals, benzoquinone and IPA were added into the TC solutions to scavenge these radicals. Benzoquinone is widely used to quench O2−, and IPA usually used to scavenge ·OH. After 5 min of reaction, 1 mmol IPA was added to the solution, the amount of TC degradation declined from 289 mg/L to 241 mg/L (Fig. 6(a)). When the same excess amount of benzoquinone was added, the TC removal dropped to 164 mg/L. The competitive radical tests suggest superoxide is the dominating active radicals in the degradation of TC using Mn3Gd5.5Ce1.5(SiO4)6O1.5 as a catalyst. Nevertheless, the results of the scavenger experiments proved that ·OH also participated in the catalytic ozonation, the combination of the two reactive radicals leads to the efficient reaction.

(a) The effects of different scavengers on the degradation of TC and with Mn3Gd5.5Ce1.5(SiO4)6O1.5 catalyst under O3, (b) XRD patterns of the white precipitation formed in Ba(OH)2 solutions, Insert: picture of Ba(OH)2 solutions after treatment. Gas produced by TC degradation with O3 (A) and gas produced by catalytic degradation passed through the Ba(OH)2 solutions.(B), several 2 mol/L HCl was added(C), High-performance liquid chromatography of TC (c), the degradation process for 7 min(d), 15 min(e), and 60 min(f).
To confirm the catalytic effect of the Ce3+-doped composite, a couple of comparable experiments have been made. The catalytic degradation of TC using Mn3Gd5.5Ce1.5(SiO4)6O1.5 had a great efficiency than any others (Fig. 3(b)). Obviously, Ce made the difference in the process, as the XPS test showed Ce was made by a mixture of positive tri- and tetra-valent cations. Ce3+/Ce4+ is a good indicator for the redox reaction and development of superoxide free radicals32.
According to the above result, degradation reaction should occur on the catalyst surface via Ce due to its redox capability11. First, S ≡ Ce3+ could react with O3, with the electron transferring, superoxide and oxygen were produced32,33 (Eq. 3). Then the obtained O2– continues to react with another O3 to produce a peroxide (O22–) molecule and a dioxygen molecule (Eq. 4). O22– molecule would react with S ≡ Ce4+ in turn to produce Ce3+ (Eq. 5). The oxygen-containing reactive intermediate reacts with tetracycline which is also adsorbed on the surface of the catalyst33.
As reported by Jia et al.34, the react active site would be made up of the oxygen vacancy site on the catalyst surface, when catalyst was attacked by ozone, one of O atom of O3 could insert into oxygen vacancy site. Electrons transfer will easy occur in oxygen vacancies to transfer electrons to an ozone molecule, the result is obtaining a new surface bound oxygen species (O2−) and dioxygen molecule at original oxygen vacancy site, which leads to be a gas phase (Eq. 6). The following step is that another ozone molecule will react with the surface bounded O2− to form a dioxygen and the second peroxide (O22-) molecule (Eq. 7). Finally, the peroxide species was unstable and decomposes to a dioxygen and then recover the initial oxygen vacancy to join the following ozonolysis cycle33. The key point of catalysis process is to decompose the peroxide in time to make sure the oxygen vacancies recover that the peroxide can easy transfer the lattice oxygen to improve the catalyst activity33,34.
In order to further investigate the degradation process and intermediate compounds produced during reaction the HPLC-MS technology was applied. The results of the TC solution (400 mg/L) chromatography after different reaction time with the presence of the catalyst are illustrated in Fig. 6. The identification was based on mass fragmentation values and by comparing the mass spectra to a database. It is apparent, that the relative intensity of the ion [1 + H]+ of m/z 445 decreases with the reaction proceeds, whereas two new and intense ions with 461 m/z and 477 m/z are clearly detected. This means that the degradation reaction of TC solution continuously occurred. As the reaction proceeds three kinds of functional groups (double bond, amine group, and phenolic groups) of TC will compete for the ozone.
The results of the HPLC-MS indicate that fragmentations of TC yielded ions with an m/z value of 427 on the loss of NO3−, which further fragmented to the ions with the value 410 m/z on the H2O loss35. After 60 minutes of ozone catalytic degradation, there was still a small amount of compounds present (molecules with 114 m/z value were detected) (Fig. 6(d)).
However, the exact structures of these compounds could not be identified in the present study and further work still required for a more detailed structures analysis. In addition to these results, we also detected the exhausted gas generated during the degradation process, and the exhaust gas was introduced into the clarified saturated Ba(OH)2 solution as shown in the insert (A) in Fig. 6(b). It can be clearly observed that the transparent solution inset (A) Fig. 6(b) became cloudy inset (B) Fig. 6(b), and a certain amount of white precipitate was formed at the bottom of the bottle. When the excess dilute hydrochloric acid was added dropwise to the turbid solution, the cloudy disappeared and the solution became transparent again as shown in the insert (A) in Fig. 6(c). To identify the gained from the experience precipitate, it was collected from the bottom of the bottle and subjected to the XRD test. The XRD results are shown in the Fig. 6(b). The comparison with the card JCPDS no. 45–174 (Ba(CO3)2) revealed that the obtained precipitate is exactly Ba(CO3)2. This result confirmed that the reaction to produce CO2 gas as the final product of the degradation reaction progress of the TC took place in the experiment. The above experimental phenomena follows the following two reaction equations:
The degradation process proceeds through the formation of a series of mineralized intermediates, which then further oxidize to water, inorganic ions and carbon dioxide36. Based on these results. So, it can be concluded that TC is completely decomposed through the oxidation by (•OH) and (•O2−) after less than 30 min the presence of ozone. The overall degraded reaction can be supposed by the following equation:
Reusability and stability of Mn3Gd5.5Ce1.5(SiO4)6O1.5 catalyst
For economic reasons, the capability of recycling Mn3Gd5.5Ce1.5(SiO4)6O1.5 catalyst was evaluated. Figure 2(a) shows the results of catalyst after being used for five times (After each experiment, we used 10,000 rpm high speed centrifugation, and filtered the samples, then collected the samples, and dried at 80 °C in an air oven). Comparing with the first use, the degradation rate decreased slightly during the first and the fifth reactions, but TC removal was almost the same. This indicates that the catalyst still remains active after consecutive runs. XRD results shows that there is no impurity in the structure after each reaction, so the catalyst can keep a great reusability (Fig. 2(b)).
The amount of metal leaching is also an important factor in measuring the stability of the catalyst. The excessive metal leaching can lead to deactivation of the catalyst. So, an ICP measurement was carried out to determine the concentrations of dissolved active Ce after the reaction (Fig. 2(c)). When the amount of the added catalyst was 0.5 g per 200 mg/L TC solution at pH 3.4, the equilibrium concentrations of Mn, Gd, and Ce were 0.051, 0.050, and 0.086 mg/L, respectively, after the reaction was completed for 60 minutes. The amount of leached metal did not affect the activity of the catalyst, since the atoms are stable present in the apatite structure. So, a stable apatite structure can be applied as a potential structural design unit to heterogeneously catalyzed oxidative degradation.
In recent years, there are many kinds of ways have been intensively studied to completely settle an issue of tetracycline pollution in water. A preliminary and brief comparison of the degradation effects of different treating methods37,38,39,40,41 to remove tetracycline is summarized in Table 1. Compared with the mentioned works, the as-synthesized Mn3Gd5.5Ce1.5(SiO4)6O1.5 catalyst investigated in this study shows excellent performance for the degradation of tetracycline under ozone, and also shows outstanding reusability and stability.
Conclusion
In this paper, the Mn3Gd5.5Ce1.5(SiO4)6O1.5 catalyst with the apatite-type structure was successfully prepared by traditional high temperature solid phase method. The catalyst exhibited good catalytic activity and stability for the degradation of TC at room temperature. This result could attribute to the synergistic effect between the different valence of the cerium ion. Free radical scavenging experiments proved that superoxide radicals were the most active substances in the reaction process, and, thus, suggested possible degradation pathways of TC in the reaction process. In summary, Mn3Gd5.5Ce1.5(SiO4)6O1.5 is a promising catalyst for removal of TC in wastewater.
References
Wu, T. et al. Mechanistic insight into interactions between tetracycline and two iron oxide minerals with different crystal structures. Chemical Engineering Journal 366, 577–586, https://doi.org/10.1016/j.cej.2019.02.128 (2019).
Bai, C. X., Shen, F. & Qi, X. H. Preparation of porous carbon directly from hydrothermal carbonization of fructose and phloroglucinol for adsorption of tetracycline. Chinese Chemical Letters 28, 960–962 (2017).
Montini, T., Melchionna, M., Monai, M. & Fornasiero, P. Fundamentals and Catalytic Applications of CeO2-Based Materials. Chemical reviews 116, 5987–6041, https://doi.org/10.1021/acs.chemrev.5b00603 (2016).
Segura, P. A., François, M., Gagnon, C. & Sauvé, S. Review of the Occurrence of Anti-infectives in Contaminated Wastewaters and Natural and Drinking Waters. Environmental Health Perspectives 117, 675–684 (2009).
Ismadji, S., Soetaredjo, F. E. & Ayucitra, A. Natural Clay Minerals as Environmental Cleaning Agents. (Springer International Publishing, (2015).
Song, C. et al. Fate of tetracycline at high concentrations in enriched mixed culture system: biodegradation and behavior. Journal of Chemical Technology & Biotechnology 91, 1562–1568 (2016).
Xiong, Z. Degradation of p-nitrophenol (PNP) in aqueous solution by a micro-size Fe0/O3 process (mFe0/O3): Optimization, kinetic, performance and mechanism. Chemical Engineering Journal 302, 137–145 (2016).
Jie Lee, W., Bao, Y., Hu, X. & Lim, T.-T. Hybrid Catalytic Ozonation-Membrane Filtration Process with CeOx and MnOx Impregnated Catalytic Ceramic Membranes for Micropollutants Degradation. Chemical Engineering Journal, https://doi.org/10.1016/j.cej.2019.05.031 (2019).
An, T. et al. Kinetics and mechanism of advanced oxidation processes (AOPs) in degradation of ciprofloxacin in water. Applied. Catalysis B: Environmental 94, 288–294, https://doi.org/10.1016/j.apcatb.2009.12.002 (2010).
Deng, Y. & Zhao, R. Advanced Oxidation Processes (AOPs) in Wastewater Treatment. Current Pollution Reports 1, 167–176 (2015).
Zhao, L., Ma, J., Sun, Z. & Liu, H. Mechanism of heterogeneous catalytic ozonation of nitrobenzene in aqueous solution with modified ceramic honeycomb. Applied Catalysis B: Environmental 89, 326–334, https://doi.org/10.1016/j.apcatb.2008.12.009 (2009).
Bai, Z., Yang, Q. & Wang, J. Catalytic ozonation of sulfamethazine antibiotics using Ce0.1Fe0.9OOH: Catalyst preparation and performance. Chemosphere 161, 174–180, https://doi.org/10.1016/j.chemosphere.2016.07.012 (2016).
Kurian, M., Kunjachan, C. & Sreevalsan, A. Catalytic degradation of chlorinated organic pollutants over CexFe1−xO2(x: 0, 0.25, 0.5, 0.75, 1) nanocomposites at mild conditions. Chemical Engineering Journal 308, 67–77, https://doi.org/10.1016/j.cej.2016.09.039 (2017).
Zhu, J. et al. Perovskite Oxides: Preparation, Characterizations, and Applications in Heterogeneous Catalysis. ACS Catalysis 4, 2917–2940, https://doi.org/10.1021/cs500606g (2014).
Wang, B., Cao, Q. & Zhang, S. Effects of the incorporation of Fe on the electromagnetic and microwave absorption performance of La0.7Sr0.3MnO3±δ. Materials Science in Semiconductor Processing 19, 101–106, https://doi.org/10.1016/j.mssp.2013.12.010 (2014).
Suárez-Vázquez, S. I., Gil, S., García-Vargas, J. M., Cruz-López, A. & Giroir-Fendler, A. Catalytic oxidation of toluene by SrTi1−XBXO3 (B = Cu and Mn) with dendritic morphology synthesized by one pot hydrothermal route. Applied Catalysis B: Environmental 223, 201–208, https://doi.org/10.1016/j.apcatb.2017.04.042 (2018).
Sun, Q. et al. Influence of the surface hydroxyl groups of MnOx/SBA-15 on heterogeneous catalytic ozonation of oxalic acid. Chemical Engineering Journal 242, 348–356, https://doi.org/10.1016/j.cej.2013.12.097 (2014).
Liu, H. et al. Structure refinement and luminescence properties of a novel apatite-type compound Mn2Gd8(SiO4)6O2. Dyes and Pigments 140, 87–91, https://doi.org/10.1016/j.dyepig.2017.01.033 (2017).
Chen, J. et al. Magnetically Separable and Durable MnFe2O4 for Efficient Catalytic Ozonation of Organic Pollutants. Industrial & Engineering Chemistry Research 53, 6297–6306, https://doi.org/10.1021/ie403914r (2014).
Martins, R. C. & Quinta-Ferreira, R. M. Catalytic ozonation of phenolic acids over a Mn–Ce–O catalyst. Applied Catalysis B: Environmental 90, 268–277, https://doi.org/10.1016/j.apcatb.2009.03.023 (2009).
Hammouda, S. B. et al. Reactivity of novel Ceria–Perovskite composites CeO2 - LaMO3 (MCu, Fe) in the catalytic wet peroxidative oxidation of the new emergent pollutant ‘Bisphenol F’: Characterization, kinetic and mechanism studies. Applied Catalysis B: Environmental 218, 119–136, https://doi.org/10.1016/j.apcatb.2017.06.047 (2017).
Zhang, L., Tu, J., Lyu, L. & Hu, C. Enhanced catalytic degradation of ciprofloxacin over Ce-doped OMS-2 microspheres. Applied Catalysis B: Environmental 181, 561–569, https://doi.org/10.1016/j.apcatb.2015.08.029 (2016).
Zhang, J. L. et al. Catalytic ozonation of penicillin G using cerium-loaded natural zeolite (CZ): Efficacy, mechanisms, pathways and toxicity assessment. Chemical Engineering Journal 123144, 1385–8947, https://doi.org/10.1016/j.cej.2019.123144 (2019).
Min, J. W. et al. Simple, robust metal fluoride coating on layered Li1.23Ni0.13Co0.14Mn0.56O2 and its effects on enhanced electrochemical properties. Electrochimica Acta 100, 10–17, https://doi.org/10.1016/j.electacta.2013.03.085 (2013).
Wang, X. et al. Mechanism and process of methylene blue degradation by manganese oxides under microwave irradiation. Applied Catalysis B: Environmental 160-161, 211–216, https://doi.org/10.1016/j.apcatb.2014.05.009 (2014).
Wang, X. et al. Oxygen-assisted preparation of mechanoluminescent ZnS:Mn for dynamic pressure mapping. Nano. Research 11, 1967–1976, https://doi.org/10.1007/s12274-017-1813-y (2018).
Liu, W.-T., Tsai, S.-C., Tsai, T.-L., Lee, C.-P. & Lee, C.-H. Characteristic study for the uranium and cesium sorption on bentonite by using XPS and XANES. Journal of Radioanalytical and Nuclear Chemistry 314, 2237–2241, https://doi.org/10.1007/s10967-017-5584-4 (2017).
Chang, L. H., Sasirekha, N., Chen, Y. W. & Wang, W. J. Preferential Oxidation of CO in H2 Stream over Au/MnO2 −CeO2. Catalysts. Industrial & Engineering Chemistry Research 45, 4927–4935 (2006).
Zou, Z. Q., Meng, M. & Zha, Y. Q. Surfactant-Assisted Synthesis, Characterizations, and Catalytic Oxidation Mechanisms of the Mesoporous MnOx−CeO2 and Pd/MnOx−CeO2 Catalysts Used for CO and C3H8 Oxidation. J.phys.chem.c 114, 468–477 (2014).
Lim, J., Yang, Y. & Hoffmann, M. R. Activation of Peroxymonosulfate by Oxygen Vacancies-Enriched Cobalt-Doped Black TiO2 Nanotubes for the Removal of Organic Pollutants. Environmental science & technology 53, 6972–6980, https://doi.org/10.1021/acs.est.9b01449 (2019).
Yang, Y. et al. Cobalt-Doped Black TiO2 Nanotube Array as a Stable Anode for Oxygen Evolution and Electrochemical Wastewater Treatment. ACS Catal 8, 4278–4287, https://doi.org/10.1021/acscatal.7b04340 (2018).
Gonçalves, A. G., Figueiredo, J. L., Órfão, J. J. M. & Pereira, M. F. R. Influence of the surface chemistry of multi-walled carbon nanotubes on their activity as ozonation catalysts. Carbon 48, 4369–4381, https://doi.org/10.1016/j.carbon.2010.07.051 (2010).
Afzal, S., Quan, X. & Lu, S. Catalytic performance and an insight into the mechanism of CeO2 nanocrystals with different exposed facets in catalytic ozonation of p-nitrophenol. Applied Catalysis B: Environmental 248, 526–537, https://doi.org/10.1016/j.apcatb.2019.02.010 (2019).
Xiong, Z., Lai, B. & Yang, P. Insight into a highly efficient electrolysis-ozone process for N,N-dimethylacetamide degradation: Quantitative analysis of the role of catalytic ozonation, fenton-like and peroxone reactions. Water research 140, 12–23, https://doi.org/10.1016/j.watres.2018.04.030 (2018).
Jia, J., Zhang, P. & Chen, L. The effect of morphology of α-MnO2 on catalytic decomposition of gaseous ozone. Catalysis Science &. Technology 6, 5841–5847, https://doi.org/10.1039/c6cy00301j (2016).
Cao, J. et al. Degradation of tetracycline by peroxymonosulfate activated with zero-valent iron: Performance, intermediates, toxicity and mechanism. Chemical Engineering Journal 364, 45–56, https://doi.org/10.1016/j.cej.2019.01.113 (2019).
Su, C. et al. Mixed Conducting Perovskite Materials as Superior Catalysts for Fast Aqueous-Phase Advanced Oxidation: A Mechanistic Study. ACS Catalysis 7, 388–397, https://doi.org/10.1021/acscatal.6b02303 (2016).
Hao, R., Xiao, X., Zuo, X., Nan, J. & Zhang, W. Efficient adsorption and visible-light photocatalytic degradation of tetracycline hydrochloride using mesoporous BiOI microspheres. Journal of hazardous materials 209-210, 137–145, https://doi.org/10.1016/j.jhazmat.2012.01.006 (2012).
Hou, L., Wang, L., Royer, S. & Zhang, H. Ultrasound-assisted heterogeneous Fenton-like degradation of tetracycline over a magnetite catalyst. Journal of hazardous materials 302, 458–467, https://doi.org/10.1016/j.jhazmat.2015.09.033 (2016).
Zhu, X.-D., Wang, Y.-J., Sun, R.-J. & Zhou, D.-M. Photocatalytic degradation of tetracycline in aqueous solution by nanosized TiO2. Chemosphere 92, 925–932, https://doi.org/10.1016/j.chemosphere.2013.02.066 (2013).
Lv, G. et al. Synthesis of birnessite with adjustable electron spin magnetic moments for the degradation of tetracycline under microwave induction. Chemical Engineering Journal 326, 329–338, https://doi.org/10.1016/j.cej.2017.05.123 (2017).
Acknowledgements
This present work was supported by the National Key R&D Program of China (Grant no. 2017YFE0133100), the National Natural Science Foundations of China (Grant Nos 41831288 and 51672257), the Fundamental Research Funds for the Central Universities (Grant Nos 2652018305 and 2652017335). D.V.D is grateful for Russian Science Foundation (Grant 19-77-10013).
Author information
Authors and Affiliations
Contributions
J.F., N.L. and L.M. conceived the project. N.L designed and performed the experiments. J.F., D.D.,Y.B. and J.W. analyzed the data. L.M., L.L., D.D. and G.L. wrote the manuscript. All the authors discussed the results and commented on the manuscript at all stages.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Fu, J., Liu, N., Mei, L. et al. Synthesis of Ce-doped Mn3Gd7−xCex(SiO4)6O1.5 for the enhanced catalytic ozonation of tetracycline. Sci Rep 9, 18734 (2019). https://doi.org/10.1038/s41598-019-55230-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-55230-7
This article is cited by
-
Environmental fate of tetracycline antibiotics: degradation pathway mechanisms, challenges, and perspectives
Environmental Sciences Europe (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
