
Abstract
Human norovirus is the leading cause of acute gastroenteritis worldwide, affecting every year 685 million people. In about one third of cases, this virus affects children under five years of age, causing each year up to 200,000 child deaths, mainly in the developing countries. Norovirus outbreaks are associated with very significant economic losses, with an estimated societal cost of 60 billion dollars per year. Despite the marked socio-economic consequences associated, no therapeutic options or vaccines are currently available to treat or prevent this infection. One promising target to identify new antiviral agents for norovirus is the viral polymerase, which has a pivotal role for the viral replication and lacks closely homologous structures in the host. Starting from the scaffold of a novel class of norovirus polymerase inhibitors recently discovered in our research group with a computer-aided method, different new chemical modifications were designed and carried out, with the aim to identify improved agents effective against norovirus replication in cell-based assays. While different new inhibitors of the viral polymerase were found, a further computer-aided ligand optimisation approach led to the identification of a new antiviral scaffold for norovirus, which inhibits human norovirus replication at low-micromolar concentrations.
Similar content being viewed by others

Introduction
Human norovirus (HuNoV) is the main cause of acute gastroenteritis worldwide, leading to approximately 200,000 deaths every year, with an associated societal cost of 60 billion USD1. While infection with this virus is responsible for a self-limiting disease in healthy individuals, the condition often becomes severe or life-threatening in immune-compromised patients, children and the elderly2,3. Despite the serious health and economic consequences associated, no therapeutic options or vaccines are currently available4, attracting significant research efforts into the identification of antiviral candidates, to be used to treat the infection and as a prophylactic measure in the case of outbreaks. Research into much needed anti-norovirus agents has been hampered by several factors, including the inaccessibility of a cell-culture human norovirus propagation system until recently. Despite this, there are ways to study the antiviral effect of molecules by using murine norovirus (MNV), which represents a good surrogate, particularly regarding the viral enzymes involved in replication, and a HuNoV replicon cell line5,6. Currently, only a few antiviral agents are under evaluation in pre-clinical and clinical stages. In particular, two small-molecules are currently under clinical investigations: Nitazoxanide, an antiprotozoal agent which reduces the duration and alleviates the symptoms of norovirus infection with a still unknown mechanism of action7,8, and CMX521, a nucleoside analogue which shows efficacy against multiple viral genotypes9. Other therapeutic candidates in pre-clinical development include different agents targeting mainly the viral polymerase or protease, due to their essential roles in the viral replication and the absence of homologous host enzymes10,11,12,13,14,15,16,17.
As a member of the Caliciviridae family, norovirus is characterised by a single-stranded positive-sense RNA genome, which is replicated by the viral RNA-dependent RNA-polymerase (RdRp) function located in the viral non-structural protein NS718. As revealed by crystallographic data, norovirus polymerase structure highly resembles the one of other positive-strand RNA viruses19, and its activity of RNA synthesis can be initiated de novo RNA or via a VPg-primed mechanism20. Due to its essential role in the viral replication, and to the repeatedly proven success of targeting viral polymerases in antiviral drug discovery21, norovirus RdRp has been previously chosen in our research group as a promising target for the identification of new anti-norovirus agents, focussing in particular on the identification of novel non-nucleoside inhibitors (NNIs) of this enzyme. A limited number of inhibitors of this type has been reported so far for norovirus RdRp, but the majority of these compounds lack any activity against the viral replication in cellular systems, possibly due to poor cell permeability and drug-like properties22. As several crystal structures are available for human and murine norovirus polymerase, including ternary complexes with nucleotide analogues and with allosteric non-nucleoside inhibitors23,24,25,26,27,28, the study of these structures has been the starting point for a structure-based virtual screening study that led to the identification of our broad-spectrum Caliciviridae RdRp inhibitor 1 (Fig. 1)29. As is the case for other reported NNIs of norovirus RdRp, despite showing an enzyme inhibition in the low micromolar range, 1 was associated with a very mild effect against norovirus replication in cell-based systems, possibly due to its poor aqueous solubility. Moreover, this compound showed some cytotoxicity at relatively low concentrations, with a CC50 of ~64 μM, possibly due, at least in part, to its low solubility and precipitation from the assay medium.

Structural features of previous hit 1 and strategies for the rational/computer-aided modification of its scaffold.
In the present study, the structure of 1 has been rationally modified in order to improve its drug-like properties and achieve an antiviral effect against norovirus replication in cell-systems. The novel structural modifications carried out have allowed a better understanding of the functional groups required for enzymatic and antiviral activity, and the successful identification of a new anti-norovirus scaffold with antiviral EC50 values in the low micromolar range. This new scaffold represents a promising starting point for further optimisations and for the potential development of a viable treatment for norovirus infections.
Results and Discussion
Rational modifications on compound 1
1 is characterised by a central 5-phenylfuran-2-ylmethylene-pyrazolidine-3,5-dione core (planar central linker in Fig. 1), substituted at position 1 of the pyrazolidine with a benzene ring (terminal hydrophobic ring 1), and at position 4 of the phenyl ring with a N-phenylsulfonamide (terminal hydrophobic ring 2). These structural features render 1 relatively hydrophobic (calculated logP (o/w) 4.3) and poorly soluble, limiting its potential as a drug.
As described by Hashimoto et al.30, decreasing the logP of a molecule by introducing a hydrophilic group is a classical and general strategy for improving its aqueous solubility. Following this rationale, two main structural modifications have been planned for 1 to achieve this result: 1) the introduction of a carboxylic acid group at different positions of terminal hydrophobic ring 1 (compounds 2 and 3 in Fig. 1), which should also provide the potential for extra interactions with the RdRp active site; 2) the replacement of the terminal hydrophobic ring 2 with different heteroaromatics (compounds 4–6 in Fig. 1). In particular, this second approach was designed as a consequence of our previously published work29, in which a thiazole derivative of 1 showed a better antiviral activity in a cell-based assay (40% inhibition of murine norovirus (MNV)-induced plaque formation at 10 μM), without showing significant solubility issues, but was found to be relatively cytotoxic, with a low selectivity index. Both types of modifications increase the polarity of the original molecule, which could influence in a positive manner their antiviral activity against norovirus replication in cell-based systems. Moreover, as suggested above, the new modifications may improve the binding to the viral polymerase, better mimicking the polar phosphate groups present in the RNA natural substrate of this enzyme.
Another feature that is recognised to influence compound solubility in a negative fashion is molecular planarity, which enhances crystal packing: poorly soluble compounds are often found to form very stable crystals, which are difficult to be broken by the solvent molecules31. Disruption of molecular planarity is usually reflected in improved solubility. As the structure of 1 is predominantly planar, an alternative strategy to reduce its solubility issues was to attempt to disrupt its planarity, by applying a scaffold replacement procedure using Schrödinger Core Hopping tool32. During this analysis, the two terminal hydrophobic rings were kept as fixed, while searching for alternative groups to replace the central planar core in 1. Among the different modifications generated by the program, a N-2-diphenylacetamide group was chosen as a more flexible linker to replace the phenylfuran-2-ylmethylene-pyrazolidine-3,5-dione core, and the new compound 7 (Fig. 1) was designed. The new linker selection was driven by an overall molecular length preservation and synthetic accessibility. Despite penalising the new compound from a logP point of view (calculated logP (o/w) 5.3), the effect of the new central core on disrupting the planarity of 1 appears to be significant, as shown in Fig. 2.

Three-dimensional superimposition, calculated with the Alignment tool in MOE 201837, of the energetically minimised structures of 1, with carbon atoms in lilac, and 7, with carbon atoms in green-uniform. The new linker inserted in 7 significantly reduces the rigidity and planarity of the linker in 1.
Chemistry
Synthesis of compounds with modifications on the two terminal hydrophobic rings
A seven-step synthetic pathway previously developed and optimised by our research group has been used for the preparation of a small family of seven new derivatives bearing modifications on the terminal hydrophobic ring 1 or on ring 229.
As shown in Fig. 3, differently substituted phenylhydrazines (10–12, 37) were reacted with ethyl-3-chloro-3-oxopropanoate 13 in THF and triethylamine (NEt3), giving the desired hydrazides 14–16, 38. Ester-susbtituted hydrazines 10–11 were prepared in a good yield by converting the differently substituted hydrazineylbenzoic acids 8–9 into the corresponding ethyl esters via Fisher esterification using hydrochloric acid (HCl) and ethanol (EtOH). An alternative approach was required for the preparation of the ortho ethyl ester 37. In fact, under Fisher reaction conditions, an intramolecular reaction between the carboxylic acid and the hydrazine group occurs, leading to the formation of 3-indanzolinone. The desired ethyl 2-hydrazineylbenzoate 37 was obtained by reacting the ethyl 2-aminobenzoate with sodium nitrite (NaNO2) in HCl, and then reducing the intermediate diazonium salt using tin chloride (SnCl2). Hydrazides 14–16 were converted into the corresponding 1-arylpyrazolidine-3,5-diones 17–19 through an ester displacement reaction in the presence of sodium hydroxide (NaOH) and EtOH. Unfortunately, the synthesis of ortho substituted compound 39 could not be achieved, potentially due to steric hindrance that impedes the cyclization reaction. Treatment of 4-bromobenzene-1-sulfonyl chloride (24) with the appropriate aniline (20–23) in pyridine produced sulfonamides 25–28, which were then converted into the aldehyde intermediates 30–33 by Suzuky coupling with (5-formylfuran-2-yl)boronic acid 29. In particular, for compound 30, bearing the original phenyl group, our formerly reported conditions using potassium phosphate (K3PO4) as base, Pd(dppf) as catalyst, water/DMF as solvent and heating under microwave irradiation for 75 min at 130 °C25, gave the desired product, whereas no product could be obtained for derivatives 31–33 in these reaction conditions. After exploring various alternative procedures, the best reaction conditions were found using sodium carbonate (Na2CO3) as base, Pd(OAc)2 and PPh3 as catalyst, water/DME as solvent, and heating the reaction mixture under microwave irradiation for 10 min at 85 °C, which gave the desired products in moderate yields (34–51%). The two portions were then linked together by reacting aldehydes 30–33 with the appropriate arylpyrazolidine-3,5-dione 17–19 according to a Knoevenagel condensation in acetic acid at 130 °C for 3 h, giving the desired products 5, 34–35. The derivatives showing a pyrimidine (4) and a 1H-benzoimidazole ring (6) appeared to degrade under these reaction conditions, therefore 4 was obtained using EtOH under reflux overnight, while 6 was prepared using methanol under reflux for 2 hours. In the last step, hydrolysis of the ethyl ester group was performed for 34 and 35. The para carboxylic acid derivative 2 was obtained by treating the starting ethyl ester with lithium hydroxide (LiOH) overnight at room temperature, while for the meta derivative 3 sodium hydroxide (NaOH) and dioxane were used. In fact, using LiOH for the hydrolysis of 35 led to the formation of a hydroxyl amine on the -NH group of the pyrazolidine-3,5-dione ring, with this hydroxylamine side product in a 50:50 ratio with the desired compound 3. The use of NaOH and dioxane limited significantly the side product formation.

Reagents and conditions: (i) HCl, EtOH, reflux, o.n., 79–90%; (ii) NEt3, THF, -10 °C to r.t., 3 h, 75–99%; (iii) 1 M NaOH/EtOH, EtOH, r.t., 30 min., 33–55%; (iv) Pyridine, r.t., o.n., 68–87%; (v) K3PO4, Pd(dppf)Cl2, H2O/DMF, MW, 130 °C, 75 min., r.t., o.n. OR Na2CO3, Pd(OAc)2, PPh3, H2O/DME, MW, 85 °C,10 min., 34–51%; (vi) AcOH, 120 °C, o.n., OR EtOH, reflux, o.n., OR MeOH, reflux, 2 h, 48–65%; (vii) LiOH, THF/MeOH/H2O, r.t., o.n., OR 2 M NaOH, 1,4-Dioxane, o.n., 49–55%; (viii) HCl, NaNO2, SnCl2, 0 °C, 3 h, 99%.
Synthesis of increased-flexibility analogue 7
Compound 7 was prepared in order to initially investigate the influence of the new central N,2-diphenylacetamide linker in the RdRp inhibitory activity, in order to preliminarily test the Core Hopping approach. The synthetic pathway reported in Fig. 4 was followed.

Reagents and conditions: (i) Pyr, 0 °C, 4 h, 91%; (ii) 5 M NaOH, MeOH, 70 °C, 3 h, 89%; (iii) Na2CO3, Pd(PPh3)4, PhMe/EtOH/H2O, 100 °C, 24 h, 75%; (iv) LiOH, MeOH/H2O, r.t., 2 h, 93%; (v) TBTU, DiPEA, DMF, r.t., o.n., 63%.
Sulfonamide 42 was prepared in almost quantitative yield by reacting aniline 40 and commercially available N-acetylsulfanilyl chloride 41 in pyridine at 0 °C. The ester group in 42 was then hydrolysed using NaOH in methanol to afford the free carboxylic acid 43. A Suzuki coupling reaction, using sodium carbonate (Na2CO3) as base and tetrakis (Pd[Ph]3]4) as catalyst, between phenylboronic acid 45 and methyl 2-(3-bromophenyl)acetate 44, in a solvent mix of toluene/water/ethanol, gave compound 46 in good yield (75%). Hydrolysis of 46 with LiOH in water/methanol gave the free acid 47, which was then reacted with 43 in dimethylformamide (DMF), N,N-diisopropylethylamine (DiPEA) as base and TBTU as coupling agent, to afford the desired final product 7 in moderate yield after flash column chromatography purification.
Biological Evaluation and Molecular Modelling studies
Human norovirus RdRp activity inhibition
The newly synthesised compounds, including the carboxylate ester derivatives 34–35 and 7 from the Core Hopping approach, were evaluated for their enzymatic inhibition of human norovirus Sydney 2012 (HuNoV (GII.4)) RdRp activity in vitro, using a quantitative fluorescent assay29. Compounds were tested at six different concentrations (concentration range examined: 0.1–100 µM) and compared to the relative activity of mock treated samples containing the vehicle only (0.5% DMSO [vol/vol]). PPNDS [Pyridoxal-5′-phosphate-6-(2′-naphthylazo-6′-nitro-4′,8′-disulfonate) tetrasodium salt], a previously reported potent inhibitor of NoV RdRp activity27, and 1 were used as positive controls. Dose-dependent inhibitory response curves were used to establish IC50 values for the eight test compounds, as reported in Table 1 (see also Supplementary Fig. S1). While IC50 values could be attained for six of the eight compounds tested, 7 only reached 20% inhibition at the maximum concentration tested, while 4 reached 15% inhibition at the maximum concentration tested (Supplementary Fig. S2).
Compounds bearing modifications at different positions of the terminal hydrophobic ring 1 (2–3, 34–35) seem to retain the RdRp inhibitory activity with an IC50 in the range of 13–22 µM, slightly higher in comparison with 1 (5.6 µM), but in the same order of magnitude. These results further prove the RdRp inhibitory activity associated with the general scaffold of 1 and suggest that introduction of hydrophilic groups into terminal hydrophobic ring 1 could improve aqueous solubility without disrupting RdRp inhibitory activity. A similar effect is obtained when replacing the terminal hydrophobic ring 2 with an oxazole (5) or benzoimidazole (6), with an IC50 value of 12.8 and 26.7 µM, respectively. On the other hand, the insertion of a pyrimidine ring at this position (4) causes loss of activity, with only partial RdRp inhibition seen at 100 µM.
As mentioned above, 7 did not reach 50% inhibition at the test concentrations, showing only 20% RdRp inhibition at 100 µM. This result suggests that compound 1 planarity and rigidity might have an important role for its biochemical activity, as the flexibility conferred by the N,2-diphenylacetamide central core almost completely abolishes RdRp inhibition. This effect could be linked with a better overall occupation of the RdRp active site by the active inhibitors, as indicated by molecular docking results shown below.
To further confirm RdRp inhibition and exclude any potential false positives from the first fluorescence RdRp assay, the eight molecules were then assessed using a gel-shift enzyme activity assay at a fixed concentration of 100 μM, following a procedure previously adopted by our research group29. PPNDS was used as positive control and exhibited complete inhibition of transcription at 100 µM (no extension of 32 nucleotide RNA template (PE44-NoV) to 44 nucleotides in the presence of an active RdRp). Seven of the eight compounds exhibited complete or almost complete inhibition of norovirus RdRp activity at 100 μM, comparable to the effect observed for 1, further confirming their activity as pure polymerase inhibitors (Fig. 5a,b). On the contrary, 7 did not demonstrate inhibition of primed elongation activity in this assay (second higher band on gel shift almost identical to the 0.5% DMSO control, Fig. 5c), confirming that this linker modification abolishes RdRp inhibitory activity.

A counter-screen gel-shift assay was used to confirm HuNoV (GII.4) RdRp inhibitory activity of compounds 2, 3, 34 and 35 (A), 4, 5, 6 (B) and 7 (C). The eight compounds were examined for inhibition of primed elongation activity. PE44-NoV RNA templates (32 nucleotides) were extended (44 nucleotides) by the RdRp in the absence of any test compounds (0.5% DMSO [vol/vol] negative control) or with test compounds at a fixed concentration of 100 μM. PPNDS and compound 1 and were used as positive controls (100 μM) to demonstrate complete inhibition, and no RdRp was used as a negative control. Full-length gels are presented in Supplementary Fig. S3.
Molecular docking studies
In our previously published study29, both competitive binding studies with PPNDS and molecular docking simulations indicated that 1 likely occupies the central core of site-B and the initial part of site-A of the human norovirus RdRp (Fig. 6). Molecular docking evaluations of the new derivatives on the binding site of 1 in the structure of human norovirus RdRp were performed using Glide SP33. Compounds 4–6 are predicted to occupy the main nucleic acid binding site of HuNoV RdRp in a similar orientation compared to 1, with the N-phenyl/heteroaromatic-benzenesulfonamide portion pointing out from site-B towards the RdRp portion in which site-B and site-A overlap (area defined by Arg392), and the differently substituted phenyl-pyrazolidine portion occupying the area where PPNDS places its naphthylazo part (Fig. 7a–c), within the core area of site-B. The added carboxylic group appears to confer an extra interaction with Lys166, further stabilising the ligand-protein complex (Fig. 8a, compound 2). This interaction appears to be maintained also in the case of the ester analogues 34–35 (Fig. 8b, compound 34). An interesting observation can be made examining the predicted binding mode of 4. The presence of the pyrimidine ring seems to increase the attraction towards Arg182, pushing the compound away from the predicted binding site, potentially explaining its reduced enzymatic activity (Fig. 7a). This effect, which is also present at minor extent for 5, could be a consequence of a different electron/charge delocalisation on the terminal part of the molecule due to the different heteroaromatic rings. Interestingly, 7, due to the presence of the more flexible N,2-diphenylacetamide central core, assumes a series of conformations that do not allow an optimal occupation of the binding area, supporting the observed reduced inhibitory effect of RdRp activity (Fig. 9).

Proposed binding mode for 1 in the human norovirus RdRp. 1 (carbon atoms in lilac) occupies site-B (turquoise surface) and part of site-A (yellow surface). 1 binds in a different orientation compared to PPNDS (carbon atoms in grey), with its phenyl- benzenesulfonamide portion pointing out from the site-B interacting through its sulphonamide group with Glu407 and Arg413 and through an arene-cation interaction between Arg392 and the benzene ring. Arg392 is in the RdRp portion in which site-B and site-A overlap. The phenyl-pyrazolidine portion of 1 occupies the area where PPNDS places its naphthylazo portion, the core area of site-B, making interactions with the surrounding amino acids (Asp507, Glu510). The binding site area is represented as molecular surface. Human norovirus RdRp is represented as turquoise ribbon.

Proposed binding mode for 4 (A), 5 (B) and 6 (C) in the human norovirus RdRp (PDB ID 4LQ3). All the three compounds are predicted to occupy the main nucleic acid binding site of HuNoV RdRp in a similar orientation compared to 1. The presence of the pyrimidine ring seems to increase the attraction towards Arg182, pushing the compound away from the predicted binding site. The binding site area is represented as molecular surface. Compound 1 binding is reported as comparison (carbon atoms in lilac).

Proposed binding mode for 2 (A) and 34 (B) in the human norovirus RdRp (PDB ID 4LQ3). The added carboxylic acid group appears to confer an extra interaction with Lys166, further stabilising the compound 2-protein complex. This interaction appears to be maintained also in the case of the ester analogue 34. Moreover, the carboxylic acid group is placed at an optimal distance to interact with Arg419. The binding site area is represented as molecular surface. Compound 1 binding is reported as comparison (carbon atoms in lilac).

Multiple potential binding modes were obtained for compound 7 in the human norovirus RdRp (PDB ID 4LQ3). The N,2-diphenylacetamide central core confers flexibility to the molecule, which assumes a series of conformations that do not allow for an optimal occupation of the binding area. The binding site area is represented as molecular surface. Compound 1 binding is reported as comparison (carbon atoms in lilac).
Cell-based antiviral effect evaluation
The newly prepared compounds 2–7 and 34–35 were evaluated for their antiviral effect against the genogroup V mouse norovirus (MNV) using the mouse macrophage cell line RAW264.7. This assay evaluates the ability of the compounds to protect infected cells from the virus-induced cytopathic effect, thus being suitable to identify inhibition of virus replication at every step of the virus life cycle, and representing a robust cellular assay available for the rapid evaluation of potential norovirus inhibitors. Compounds identified using MNV have shown effects against HuNoV in vitro and in vivo, as in the case of nucleoside RdRp inhibitor 2′-C-methylcytidine (2CMC)34,35,36. Unfortunately, despite not presenting the aqueous solubility issues we had previously encountered for 129, no significant antiviral effect could be observed in this assay for the new compounds up to the maximum concentration tested. The new structural modifications, despite retaining inhibition of the RdRp in an isolated enzyme activity assay and showing no cytotoxic effects at the test concentrations in the MNV antiviral assay (Supplementary Table S1), are correlated with a lack of antiviral activity. As these new structures present the same limitation of most non-nucleoside HuNoV polymerase inhibitors reported so far22, we were prompted to apply a different computer-aided strategy to further modify the scaffold of 1, with the aim to achieve an antiviral effect in cellular systems. prompting us to attempt further modifications on their structure.
Computer-aided flexible alignment approach: new structural modifications on the structure of 1
Since both attempts at modifying the structure of 1 did not give the desired result of identifying antiviral hits against norovirus replication in cell-based systems, an alternative strategy to identify potential linker replacements for 1 was followed. A conformational search, using MOE conformational search tool37, was performed on 1 to identify its lowest energy conformation. This conformation was then kept rigid and used to run a flexible alignment analysis with the Flexible Alignment tool in MOE37, in order to search for potential three-dimensional and functional similarities with an in-house small-molecule database of previously reported non-nucleoside inhibitors of viral polymerases, including hepatitis C, Zika and Dengue virus RdRps38,39,40. In particular, the chosen database contains small-molecule compounds reported to have inhibitory activity against viral polymerases, and also to show antiviral effects in cell-based assays. From this study, TPB [3-chloro-N-[({4-[4-(2-thienylcarbonyl)-1-piperazinyl]phenyl}amino)carbonothioyl]-1-benzothiophene-2-carboxamide] (48), a recently identified inhibitor of Zika virus RdRp39, resulted as the best match for its structural overlapping with 1, as highlighted in Fig. 10.

Chemical structure of TPB (48, carbon atoms and molecular surface in green) and Flexible Alignment results showing its significant degree of structural and molecular surface overlapping with 1 (carbon atoms and molecular surface in lilac).
Evaluation of inhibition of norovirus RdRp activity using an in vitro fluorescent, de novo RdRp activity assay at a fixed concentration of 20 μM revealed that 48, which we promptly synthesised in our laboratory, has a mild inhibition of human norovirus RdRp activity (20% inhibition at 20 μM), as reported in the biological evaluation studies section below. This preliminary encouraging result, together with the reported antiviral activity demonstrated for 48 in Vero cells against ZIKV replication39, prompted us to explore this scaffold in the search of new norovirus RdRp inhibitors, and most importantly new agents to inhibit HuNoV replication. The structure of 48 was therefore combined with some structural features from 1 and from some of our previously published norovirus RdRp inhibitors29, leading to the design of a series of new compounds with a non-planar central linker. These new molecules could present improved solubility and better cell-based antiviral profiles in comparison with 1 and its analogues, as shown in Fig. 11. In addition, in order to verify the activity associated with the scaffold of 48, a few small structural modifications on its scaffold were also planned and carried out.

New modifications designed from the structural combination of 1 and 48.
Chemistry
Synthesis of the newly designed “hybrid” molecules
The 12 newly designed analogues, along with the Flexible Alignment hit 48, were synthesised according to a four-step synthetic pathway, as shown in Fig. 12.

Reagents and conditions: (i) TBTU, DiPEA, DMF, r.t., 5 h, OR NEt3, DCM, 0 °C to r.t., o.n., 81–99%; (ii) H2, 10% Pd/C, EtOH, r.t., 24 h, 78–99%; (iii) CNSH*NH3, Me2CO, reflux, 1 h, quant.; (iv) Me2CO, reflux, 2 h, 41–78%.
Briefly, commercial 1-(4-nitrophenyl)piperazine 61 was treated with the appropriately substituted aryl-carboxylic acids or aryl sulfonyl chlorides, to give intermediate nitro-compounds 62a–67a. In particular, amide-compounds 62a–65a were obtained through a TBTU-assisted coupling reaction in the presence of DiPEA, in DMF at r.t., while sulfonamide intermediates 66a–67a were obtained in DCM at 0 °C to r.t., in the presence of NEt3. Nitro-intermediates 62a–67a were then converted into the corresponding substituted aromatic amines 62b–67b following a catalytic hydrogenation in EtOH, in the presence of wet activated palladium on carbon. Isothiocyanates intermediates 68b–72b were obtained by treating the corresponding aryl-carbonyl chlorides 68a–72a with ammonium isothiocyanate, in acetone under reflux conditions. These intermediates were not isolated, but treated in situ with the differently substituted aromatic amines 62b–67b, to give the desired final products 48–60. In most cases, formation of unwanted amide-byproducts was observed for this reaction, thus explaining the moderate yields obtained in the final step for most of these products. By-products 73–76 were formed in a particularly high amount in the course of the reaction, therefore they were isolated and fully characterised.
Biological Evaluation and Molecular Modelling Studies
Evaluation of the inhibitory activity of HuNoV RdRp for analogues 48–60
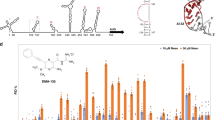
The newly prepared compounds 48–60 were initially evaluated against norovirus Sydney 2012 RdRp activity in vitro using a quantitative fluorescent assay. Compounds were tested at a fixed concentration of 20 µM and compared to the relative activity of mock treated samples. PPNDS and 1 were used as positive controls. Results of this assay are reported in Fig. 13. Removal of the chlorine atom from position 2 of the benzothiophene ring (49) or its replacement with a bigger trifluoromethyl group (50) appears not to affect much the RdRp inhibitory activity, with an observed inhibition which is similar to the effect of 48 (15–20% inhibition at 20 µM). A similar effect is obtained for 51 and 52 (~15% inhibition at 20 µM), in which the benzothiophene ring has been replaced with a phenyl ring and a 4-methylphenyl ring respectively. These two rings are structural features present in 1 and in one of its derivatives we have previously reported25. Replacement of the thiophene by a furan (53) or by an unsubstituted phenyl ring (54) increased the degree of HuNoV RdRp inhibition up to 60% at 20 µM, whereas introduction of a thiazole ring (56) does not affect enzymatic inhibition activity. From these observations, the thiophene ring appears not to influence inhibitory activity. Derivatives in which the central core of 48 has been used to link the two terminal hydrophobic rings of 1 or of its derivatives we have previously reported25 (55, 57, 58) appear to retain the RdRp inhibitory activity found for 48 (~20% at 20 µM). A similar effect is obtained when a sulfonyl group, as in our hit 1, is used to link ring 2, regardless if it is a thiazole (59) or an unsubstituted phenyl ring (60), to the rest of the molecule. Interestingly, 73, one of the amide side products formed during the last synthetic step for this series of compounds, reduced the polymerase activity of ~40% at 20 µM. 53, the most active compound found in this series, also exhibits a significant inhibition of HuNoV RdRp activity at 100 µM in the gel-shift assay, confirming the polymerase inhibitory function of the new scaffold. Overall, even if showing a reduced biochemical activity in comparison with 1, some of the new derivatives can be considered as modest HuNoV RdRp inhibitors at 20 µM. This reduced activity in comparison with 1 could to some extent have been expected, since, as seen for 7, the reduction in central core planarity/rigidity in the new derivatives would likely reduce their interactions and optimal fitting of the HuNoV RdRp active site. This hypothesis is in line with molecular docking results obtained for 51 and 53, as shown in Fig. 14.

Screening of new derivatives for inhibition of HuNoV (GII.4) RdRp activity. (A) The inhibitory effects of 8 compounds were examined against norovirus RdRp activity using a fluorescent activity assay at 20 μM. Compounds 53 and 54 exhibited almost 60% inhibition. PPNDS (green bar) and compound 1 (black bar) were used as positive controls. Percentage of inhibition was normalised to control DMSO (red bar). The mean values of triplicate datasets with standard error of the mean are shown. (B) A counter-screen-gel- shift assay was used to confirm norovirus RdRp inhibitory activity of 53, the most active compound identified in panel (A). PE44-NoV RNA templates (32 nucleotides) were extended (44 nucleotides) by the RdRp in the absence of any test compounds (0.5% DMSO [vol/vol] negative control) or with test compounds at a fixed concentration of 100 μM. PPNDS and compound 1 and were used as positive controls (100 μM) to demonstrate complete inhibition, and no RdRp was used as a negative control. Full-length gels are presented in Supplementary Fig. S4.

Proposed binding mode for 51 (A) and 53 (B) to the human norovirus RdRp (PDB ID 4LQ3). Although the sulfur atom of the thiourea forms different H-bonds with the surrounding amino acids, the reduction in central core planarity/rigidity of the new derivatives does not allow an optimal occupation of the HuNoV RdRp active site, potentially reducing the inhibitory activity. The binding site area is represented as molecular surface. Compound 1 binding is reported as comparison (carbon atoms in lilac). Human norovirus RdRp is represented as turquoise ribbon.
Cell-based antiviral effect evaluation
Since the main scope of this work was to find new norovirus inhibitors which show antiviral effects against both the HuNoV RdRp and in norovirus-infected cells, priority was given to the evaluation of most of the new molecules for their antiviral activity against MNV in infected RAW cells. The nucleoside 2′-C-methylcytidine was used as positive control. The test compounds were evaluated at eight different concentrations (range 0.6–100 µM) and their ability to reduce the virus-induced cytopathic effect was assessed.
Five out of 10 compounds tested were found to have an interesting antiviral EC50 in the 20–100 µM range in this assay, as reported in Table 2. These new derivatives showed a slightly better EC50 if compared with what we had previously found for 129, but more interestingly they were not cytotoxic at the test concentrations, whereas 1 exhibited a mean half maximal cytotoxic concentration (CC50) of 62.8 μM in a similar MNV cell-based assay29.
As a further step, the five hit compounds which showed activity in the MNV assay were tested against HuNoV using a replicon system, i.e. a human gastric tumor-1 (HGT-1) cell line which stably expresses the genome of a HuNoV GI.1 virus41. Given that in this system the gene encoding for the major capsid protein is replaced by a neomycin resistance gene, no new virus particles will be produced but the non-structural proteins are expressed and the replication of the genomic RNA can be studied42.
While 48 and 56 do not display any significant antiviral activity in this second assay, 52, 59 and 60 inhibit the viral replication of HuNoV GI with EC50 values in the low micromolar range, as shown in Table 3. The lack of activity encountered for 48 and 56 in this assay could be a consequence of several factors, such as differences on the envelope and capsid proteins between MNV and HuNoV, which could affect the ability of the two compounds to reach the viral RdRp, or different readouts and sensitivity of the two assays in detecting the inhibitory activity of the compounds.
Even if showing an interesting EC50 of 8 µM, 60 was found to form crystals starting from a 25 µM concentration, thus indicating that its solubility should still be improved. 52 and 59 display instead the most interesting antiviral activity profile found so far, with EC50 values against HuNoV GI replication of 6 µM and 10 µM, respectively, providing a new antiviral scaffold for norovirus which shows promise for additional investigations. Overall, due to their low-micromolar inhibitory activity against norovirus replication in vitro, 52 and 59 represent one of the few successful examples of non-nucleoside HuNoV polymerase inhibitors which also show antiviral activity against the viral replication in a cell-based system. These compounds represent a promising starting point for further structural optimisation and SAR evaluations. In particular, current research efforts are now ongoing to further improve the drug-like properties of these compounds, and to increase their potency in both biochemical and cellular assays.
Conclusions and Future Work
Starting from a rational approach to modify the structure of one HuNoV RdRp inhibitor, 1, which was recently identified in our research group, we have explored different modifications on its scaffold, in order to increase its drug-like potential as an antiviral agent. Despite the fact that most of the newly modified compounds retain inhibitory activity on the viral polymerase, none of them resulted as an antiviral hit when tested in a MNV CPE reduction assay. In an attempt to further modify the scaffold of 1 and achieve an antiviral effect in cellular systems, we have performed a computer-aided approach in order to identify potential similarities between 1 and previously reported non-nucleoside inhibitors of viral polymerases, which also display good antiviral activities against the replication of different viruses in cell-based assays. Following this rationale, we have recognised significant structural similarities between our biochemical hit 1 and a recently reported inhibitor of Zika virus polymerase, 48. Rationally combining together different structural features of 1 and 48, we have designed and synthesised a new family of 12 modified analogues. Among them, different displayed at least partial inhibition of HuNoV RdRp activity in a quantitative fluorescent assay. Most interestingly, five of these compounds were found to reduce MNV-induced cytopathic effect with EC50 values in the micromolar range, without being associated with significant cytotoxicity. These five hits were then evaluated in a human norovirus replicon assay harbouring a genogroup GI, and two of them, 52 and 59, were found to inhibit HuNoV replication with EC50 values in the low micromolar range, revealing a new antiviral scaffold for human norovirus. These compounds represent one of the few examples of non-nucleoside polymerase inhibitors also showing significant antiviral effect against human norovirus replication in a cell-based system. Based on these findings, additional investigations on these new structures are currently ongoing to further explore their potential as antiviral agents. In particular, further structural optimisation is the main focus of present research efforts, which aim to identify suitable preclinical antiviral candidates for the treatment of norovirus infections.
Experimental
Chemistry
All solvents and reagents were used as obtained from commercial sources unless otherwise indicated. All solvents used for chromatography were HPLC grade from Fisher Scientific (UK). All reactions were performed under a nitrogen atmosphere29. 1H and 13C-NMR spectra were recorded with a Bruker Avance III HD spectrometer operating at 500 MHz for 1H and 125 MHz for 13C, with Me4Si as internal standard. Deuterated chloroform was used as the solvent for NMR experiments, unless otherwise stated. 1H chemical shifts values (δ) are referenced to the residual non-deuterated components of the NMR solvents (δ = 7.26 ppm for CHCl3, etc.)29. The 13C chemical shifts (δ) are referenced to CDCl3 (central peak, δ = 77.0 ppm). TLC was performed on silica gel 60 F254 plastic sheets. Flash column chromatography was performed using silica an Isolera Biotage system. UPLC-MS analysis was conducted on a Waters UPLC system with both Diode Array detection and Electrospray (+′ve and −′ve ion) MS detection. The stationary phase was a Waters Acquity UPLC BEH C18 1.7 um 2.1 × 50 mm column. The mobile phase was LC-MS grade H2O containing 0.1% formic acid (A) and LC-MS grade MeCN containing 0.1% formic acid (B). Column temperature: 40 °C. Sample diluent: MeCN. Sample concentration 1 µg/mL. Injection volume 2 µL29. Three alternative methods were used:
Linear gradient standard method (A): 90% A (0.1 min), 90–0% A (2.5 min), 0% A (0.3 min), 90% A (0.1 min); flow rate 0.5 mL/min.
Linear gradient standard method (B): 90% A (0.1 min), 90–0% A (2.1 min), 0% A (0.8 min), 90% A (0.1 min); flow rate 0.5 mL/min.
Linear gradient standard method (C): 90% A (0.1 min), 90–0% A (1.5 min), 0% A (1.4 min), 90% A (0.1 min); flow rate 0.5 mL/min.
All compounds tested in biological assays were>95% pure. Purity of intermediates was>90%, unless otherwise stated. All intermediates were generally prepared according to literature procedures, which are described in detail along with compound characterisation in the Supporting Information. Details for the preparation and full characterisation of the new target final compounds are given below.
General method for the preparation of N-aryl-4-(5-((3,5-dioxo-1-phenylpyrazolidin-4-ylidene)methyl)furan-2-yl)benzenesulfonamides 5, 34–35
The appropriately substituted 1-phenylpyrazolidine-3,5-dione 17–19 (1 mmol) and the differently substituted N-(aryl)-(5-formylfuran-2-yl)benzenesulfonamides 30, 32 (1 mmol) were suspended in AcOH (15 ml/mmol) and the mixture was stirred at 120 °C for 3 hours. The mixture was then cooled to room temperature, quenched with water and the precipitate obtained was filtered, washed with water and dried under vacuum. The crude product was purified by trituration to afford the title compound.
(E)-4-(5-((3,5-dioxo-1-phenylpyrazolidin-4-ylidene)methyl)furan-2-yl)-N-(oxazol-2-yl)benzenesulfonamide (5)
Purified by trituration from DCM. Obtained in 65% yield as a light brown solid. 1H-NMR (DMSO-d6), δ: 12.25 (bs, 1 H), 11.19 (bs, 1 H), 8.54–8.50 (m, 1 H), 8.14–8.11 (m, 2 H), 7.99–7.96 (m, 2 H), 7.77–7.70 (m, 3 H), 7.64–7.63 (m, 1 H), 7.59–7.58 (m, 1 H), 7.47–7.43 (m, 2 H), 7.32–7.30 (m, 1 H), 7.24–7.18 (m, 1 H). 13C-NMR (DMSO-d6), δ: 158.5, 158.4, 156.7, 150.7, 150.6, 144.1, 131.7, 129.4, 129.3, 129.2, 127.4, 127.3, 125.8, 118.7, 116.3, 115.1, 113.7, 113.4, 113.3. UPLC-MS (Method C): Rt 1.84 min, MS [ESI, m/z]: 477.1 [M + H].
Anal. Calcd for C23H16N4O6S: C, 57.98; H, 3.38; N, 11.76. Found: C, 57.69; H, 3.12; N, 11.95.
Ethyl(E)-4-(3,5-dioxo-4-((5-(4-(N-phenylsulfamoyl)phenyl)furan-2-yl)methylene)pyrazolidin-1-yl)benzoate (34)
Purified by trituration from DCM. Obtained in 48% yield as a light brown solid. 1H-NMR (DMSO-d6), δ: 11.53 (bs, 1 H), 10.36 (bs, 1 H), 8.56–8.50 (m, 1 H), 8.11 (d, J = 8.4 Hz, 2 H), 8.03 (d, J = 8.6 Hz, 2 H), 7.96–7.90 (m, 2 H), 7.87–7.84 (m, 2 H), 7.70–7.65 (m, 1 H), 7.58–7.56 (m, 1 H), 7.26–7.23 (m, 2 H), 7.11 (d, J = 8.6 Hz, 2 H), 7.06–7.03 (m, 1 H), 4.31 (q, J = 7.1 Hz, 2 H), 1.33 (t, J = 7.1 Hz, 3 H). 13C-NMR (DMSO-d6), δ: 175.9, 165.7, 165.6, 158.1, 158.0, 150.8, 140.3, 138.8, 132.5, 130.7, 130.6, 129.7, 128.1, 128.0, 126.1, 124.9, 120.9, 120.8, 115.1, 114.9, 113.9, 61.1, 14.7. UPLC-MS (Method C): Rt 2.30 min, MS [ESI, m/z]: 558.1 [M + H]. Anal. Calcd for C29H23N3O7S: C, 62.47; H, 4.16; N, 7.54. Found: C, 62.61; H, 3.97; N, 7.39.
Ethyl(E)-3-(3,5-dioxo-4-((5-(4-(N-phenylsulfamoyl)phenyl)furan-2-yl)methylene)pyrazolidin-1-yl)benzoate (35)
Purified by trituration from DCM. Obtained in 58% yield as a light brown solid. 1H-NMR (DMSO-d6), δ: 11.28 (bs, 1 H), 10.35 (bs, 1 H), 8.51–8.39 (m, 2 H), 8.10 (d, J = 8.5 Hz, 2 H), 8.01–7.97 (m, 1 H), 7.85 (d, J = 8.5 Hz, 2 H), 7.78–7.76 (m, 1 H), 7.71–7.68 (m, 1 H), 7.61–7.55 (m, 2 H), 7.26–7.23 (m, 2 H), 7.12–7.10 (m, 2 H), 7.06–7.03 (m, 1 H), 4.35 (q, J = 7.1 Hz, 2 H), 1.34 (t, J = 7.1 Hz, 3 H). 13C-NMR (DMSO-d6), δ: 176.6, 165.9, 165.8, 150.8, 140.3, 140.2, 137.8, 132.5, 131.1, 131.0, 129.9, 129.8, 129.7, 128.2, 128.1, 126.1, 124.8, 120.9, 120.8, 115.1, 113.9, 113.8, 61.5, 14.6. UPLC-MS (Method C): Rt 2.32 min, MS [ESI, m/z]: 558.1 [M + H].
Anal. Calcd for C29H23N3O7S: C, 62.47; H, 4.16; N, 7.54. Found: C, 62.32; H, 4.24; N, 7.39.
Preparation of (E)-4-(5-((3,5-dioxo-1-phenylpyrazolidin-4-ylidene)methyl)furan-2-yl)-N-(pyrimidin-2-yl)benzenesulfonamide (4)
To a suspension of 1-phenylpyrazolidine-3,5-dione 19 (0.02 g, 0.13 mmol) in EtOH (10 ml/mmol) were added 4-(5-formylfuran-2-yl)-N-(pyrimidin-2-yl)benzenesulfonamide 31 (0.05, 0.15 mmol) and one drop of pyridine. The reaction was stirred under reflux overnight, then cooled to room temperature and the precipitate formed was filtered off and washed with cold EtOH and dried under vacuum. The crude product was purified by automated flash column chromatography (Biotage Isolera One, SNAP KP Sil 10 g) eluting with DCN:MeOH:Net3 100:0:0 v/v increasing to 80:15:5 v/v in 15 CV. Obtained in 51% yield as a light brown solid. 1H-NMR (DMSO-d6), δ: 11.90 (bs, 2 H), 8.54–8.47 (m, 3 H), 8.16–8.13 (m, 2 H), 8.10–8.08 (m, 2 H), 7.79–7.76 (m, 3 H), 7.59–7.58 (m, 1 H), 7.48–7.44 (m, 2 H), 7.23–7.19 (m, 1 H), 7.08–7.04 (m, 1 H). 13C-NMR (DMSO-d6), δ: 158.1, 157.3, 157.2, 150.8, 150.7, 132.5, 129.4, 129.3, 128.9, 128.3, 125.7, 125.2, 119.1, 118.8, 117.2, 115.5, 115.3, 113.8, 113.7, 113.0. UPLC-MS (Method C): Rt 1.94 min, MS [ESI, m/z]: 488.1 [M + H].
Anal. Calcd for C24H17N5O5S: C, 59.13; H, 3.52; N, 14.37. Found: C, 59.28; H, 3.40; N, 14.51.
Preparation of (E)-N-(1H-benzo[d]imidazol-2-yl)-4-(5-((3,5-dioxo-1-phenylpyrazolidin-4-ylidene) methyl)furan-2-yl)benzenesulfonamide (6)
To a solution of 1-phenylpyrazolidine-3,5-dione 19 (0.19 mmol) in MeOH (10 ml/mmol) was added N-(1H-benzo[d]imidazol-2-yl)-4-(5-formylfuran-2-yl)benzenesulfonamide 33 (0.19 mmol). The reaction was stirred at reflux for 2 hours. The precipitate formed was filtered off, washed with cold MeOH and dried under vacuum. The crude product was purified by automated flash column chromatography (Biotage Isolera One, SNAP KP Sil 10 g) eluting with DCN:MeOH 100:0:0 v/v increasing to 90:10 v/v in 10 CV. Obtained in 55% yield as a light brown solid. 1H-NMR (DMSO-d6), δ: 11.38 (bs, 1 H), 8.43–8.35 (m, 1 H), 8.19–8.15 (m, 4 H), 7.79–7.73 (m, 2 H), 7.72–7.70 (m, 1 H), 7.68–7.63 (m, 1 H), 7.62–7.60 (m, 1 H), 7.46–7.43 (m, 2 H), 7.21–7.18 (m, 3 H), 7.17–7.14 (m, 2 H), 7.06–7.03 (m, 1 H). 13C-NMR (DMSO-d6), δ: 157.1, 152.5, 151.2, 151.1, 143.3, 136.8, 136.7, 134.6, 130.4, 129.4, 129.3, 128.3, 128.2, 126.5, 126.4, 125.3, 121.1, 116.5, 116.1, 114.7, 114.6, 114.1, 112.7. UPLC-MS (Method C): Rt 1.93 min, MS [ESI, m/z]: 526.2 [M + H]. Anal. Calcd for C27H19N5O5S: C, 61.71; H, 3.64; N, 13.33. Found: C, 61.54; H, 3.52; N, 13.55.
Preparation of (E)-4-(3,5-dioxo-4-((5-(4-(N-phenylsulfamoyl)phenyl)furan-2-yl)methylene) pyrazolidin-1-yl)benzoic acid (2)
To a solution of ethyl (E)-4-(3,5-dioxo-4-((5-(4-(N-phenylsulfamoyl)phenyl)furan-2-yl)methylene)pyrazolidin-1-yl) benzoate 34 (0.18 mmol) in 18 ml of THF:MeOH:H2O (4:1:1) was added LiOH (0.53 mmol). The reaction was stirred at room temperature overnight. The mixture was acidified with 1 N HCl aqueous solution. The solvent was reduced in volume to remove MeOH and THF, and the aqueous layer was extracted with EtOAc (3 × 20 ml). The organic layer was dried over MgSO4 and concentrated under vacuum. The crude product was purified by trituration from DCM. Obtained in 58% yield as a light brown solid. 1H-NMR (DMSO-d6), δ: 13.05 (bs, 1 H), 11.88 (bs, 1 H), 10.27 (s, 1 H), 8.07 (d, J = 8.7 Hz, 2 H), 7.90–7.88 (m, 4 H), 7.81 (d, J = 8.7 Hz, 2 H), 7.30 (d, J = 3.6 Hz, 1 H), 7.26–7.22 (m, 2 H), 7.16 (d, J = 3.6 Hz, 1 H), 7.12–7.09 (m, 2 H), 7.05–7.02 (m, 1 H). 13C-NMR (DMSO-d6), δ: 167.1, 155.6, 151.8, 150.9, 146.2, 144.9, 138.3, 138.0, 134.0, 130.1, 129.8, 129.6, 128.0, 125.3, 124.7, 124.2, 120.9, 120.8, 117.1, 110.9, 103.6. UPLC-MS (Method C): Rt 2.04 min, MS [ESI, m/z]: 528.1 [M-H]. Anal. Calcd for C27H19N3O7S: C, 61.24; H, 3.62; N, 7.94. Found: C, 61.50; H, 3.51; N, 8.03.
Preparation of (E)-3-(3,5-dioxo-4-((5-(4-(N-phenylsulfamoyl)phenyl)furan-2-yl)methylene) pyrazolidin-1-yl)benzoic acid (3)
Ethyl (E)-3-(3,5-dioxo-4-((5-(4-(N-phenylsulfamoyl)phenyl)furan-2-yl)methylene)pyrazolidin-1-yl)benzoate 35 (0.07 mmol) was added to a mixture of 2 M NaOH aqueous solution (1 ml) and dioxane (1 ml). The reaction was stirred vigorously at room temperature overnight. The solvent was reduced in volume under vacuum and acidified by addition of 1 M HCl solution. The water layer was then extracted with EtOAc (3 × 20 ml). The organic portions were combined, dried over MgSO4 and concentrated under vacuum. The crude product was purified by automated flash column chromatography (Biotage Isolera One, SNAP KP Sil 10 g) eluting with DCM:MeOH 100:0:0 v/v increasing to 80:20 v/v in 15 CV. Obtained in 49% yield as a light brown solid. 1H-NMR (DMSO-d6), δ: 13.04 (bs, 2 H), 10.35 (bs, 1 H), 8.54–8.50 (m, 1 H), 8.43–8.39 (m, 1 H), 8.12–8.09 (m, 2 H), 8.05–7.99 (m, 1 H), 7.86–7.84 (m, 2 H), 7.75–7.73 (m, 1 H), 7.67–7.63 (m, 1 H), 7.58–7.55 (m, 2 H), 7.26–7.22 (m, 2 H), 7.12–7.10 (m, 2 H), 7.06–7.03 (m, 1 H). 13C-NMR (DMSO-d6), δ: 167.4, 156.2, 151.6, 150.8, 146.3, 145.5, 140.2, 137.8, 132.6, 129.6, 129.5, 128.0, 125.9, 124.8, 124.3, 120.9, 120.3, 117.4, 109.9, 103.4. UPLC-MS (Method C): Rt 2.04 min, MS [ESI, m/z]: 528.1 [M-H]. Anal. calcd for C27H19N3O7S: C, 61.24; H, 3.62; N, 7.94. Found: C, 61.01; H, 3.43; N, 8.06.
Preparation of 2-([1,1′-biphenyl]-3-yl)-N-(4-(N-phenylsulfamoyl)phenyl)acetamide (7)
DiPEA (1.1 mmol) was added to a solution of 2-([1,1′-biphenyl]-3-yl)acetic acid 47 (0.44 mmol), 4-amino-N-phenylbenzenesulfonamide 43 (0.48 mmol) and TBTU (0.48 mmol) in 3 mL of anhydrous DMF under N2 atmosphere at room temperature. The reaction was stirred at r.t. over night, then diluted with EtOAc (20 mL) and washed with saturated aqueous NH4Cl solution (15 mL), saturated aqueous NaHCO3 solution (15 mL) and brine (10 mL). The organic phase was dried over MgSO4 and concentrated under vacuum.
The crude product was purified by automated flash column chromatography (Biotage Isolera One, SNAP KP Sil 10 g) eluting with n-hexane:EtOAc 100:0 v/v increasing to 0:100 v/v in 12 CV. Obtained in 63% yield as an off-white solid. 1H-NMR (DMSO-d6), δ: 3.75 (s, 2 H), 6.98–7.02 (m, 1 H), 7.06 (d, J = 8.2 Hz, 2 H), 7.19–7.22 (m, 2 H), 7.30–7.32 (m, 1 H), 7.35–7.43 (m, 2 H), 7.46–7.49 (m, 2 H), 7.54 (d, J = 7.1 Hz, 1 H), 7.62–7.65 (m, 3 H), 7.68 (d, J = 8.8 Hz, 2 H), 7.73 (d, J = 8.8 Hz, 2 H), 10.14 (bs, 1 H), 10.55 (bs, 1 H). 13C-NMR (DMSO-d6), δ: 170.2, 143.4, 140.7, 140.5, 138.5, 136.6, 133.9, 129.5, 129.4, 128.6, 128.3, 128.1, 127.9, 127.1, 125.5, 124.3, 120.5, 119.8, 119.2, 43.7. UPLC-MS (Method C): Rt 1.96 min, MS [ESI, m/z]: 443.2 [M + H]. Anal. calcd for C26H22N2O3S: C, 70.57; H, 5.01; N, 6.33. Found: C, 70.40; H, 5.27; N, 6.17.
General method for the preparation of N-((4-(4-arylpiperazin-1-yl)phenyl)carbamothioyl) arylamides 48–58 and N-((4-(4-(arylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)arylamides 59–60
The appropriate aryl chloride 68a-72a (1 mmol) and ammonium thiocyanate (1 mmol) were dissolved in acetone (4 mL/mmol) at 0 °C. Stirring was continued for 1 h at r.t., then the formed precipitate (NH4Cl) was filtered off. To the freshly filtered solution, the appropriate aromatic amine 62b–67b (1 mmol) was added. The mixture was then stirred under reflux for 1 h. Upon completion of reaction, the resulting precipitate was collected by filtration. The crude product was purified by trituration, re-crystallisation or flash column chromatography to afford the title compound.
3-Chloro-N-((4-(4-(thiophene-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)benzo[b]thiophene-2-carboxamide (48)
Purified by trituration from acetone. Obtained in 58% yield as a yellow solid. 1H-NMR (DMSO-d6), δ: 11.95 (bs, 1 H), 11.29 (bs, 1 H), 8.19 (dd, J1=7.9 Hz, J2 = 2.0 Hz, 1 H), 7,98 (dd, J1 = 7.9 Hz, J2 = 2.3 Hz, 1 H) 7.79 (dd, J1 = 5.0 Hz, J2 = 1.2 Hz, 1 H), 7.70–7.63 (m, 2 H), 7.57 (d, J = 9.0 Hz, 2 H), 7.48 (dd, J1 = 3.7 Hz, J2 = 1.1 Hz, 1 H), 7.16 (dd, J1 = 5.5 Hz, J2 = 3.6 Hz, 1 H), 7.02 (d, J = 9.0 Hz, 2 H), 3.81–3.79 (m, 4 H), 3.28–3.26 (m, 4 H). 13C-NMR (DMSO-d6), δ: 177.4, 162.8, 161.2, 149.4, 138.0, 137.5, 136.2, 130.2, 130.1, 129.9, 129.7, 128.8, 127.6, 126.8, 125.6, 124.1, 123.5, 122.5, 115.8, 48.7. UPLC-MS (Method C): Rt 2.18 min, MS [ESI, m/z]: 541.0, 543.0 [M + H]. Anal. Calcd for C25H21ClN4O2S3: C, 55.49; H, 3.91; N, 10.35. Found: C, 55.70; H, 4.08; N, 10.2239.
N-((4-(4-(Thiophene-2-carbonyl)piperazin-1- yl)phenyl)carbamothioyl)benzo[b]thiophene-2-carboxamide (49)
Re-crystallised from MeOH/H2O. Obtained in 45% yield as a yellow solid. 1H-NMR (DMSO-d6), δ: 12.25 (bs, 1 H), 11.76 (bs, 1 H), 8.76 (s, 1 H), 8.10 (dd, J1=8.7 Hz, J2=0.8 Hz, 1 H), 8.02 (d, J = 7.8 Hz, 1 H), 7.79 (dd, J1 = 5.0 Hz, J2 = 1.1 Hz, 1 H), 7.56–7.48 (m, 3 H), 7.52–7.50 (m, 1 H), 7.49–7.48 (m, 1 H), 7.16 (dd, J1 = 5.2 Hz, J2 = 3.6 Hz, 1 H), 7.00 (d, J = 9.1 Hz, 2 H), 3.81–3.79 (m, 4 H), 3.27–3.25 (m, 4 H). 13C-NMR (DMSO-d6), δ: 178.4, 176.6, 163.0, 162.8, 149.4, 141.9, 139.4, 137.5, 137.1, 130.3, 130.1, 129.7, 127.9, 127.6, 126.7, 125.8, 125.7, 123.4, 115.9, 48.8. UPLC-MS (Method C): Rt 2.02 min, MS [ESI, m/z]: 507.1[M + H]. Anal. Calcd for C25H22N4O2S3: C, 59.27; H, 4.38; N, 11.06. Found: C, 59.34; H, 4.23; N, 10.92.
N-((4-(4-(Thiophene-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)-3- (trifluoromethyl)benzo[b] thiophene-2-carboxamide (50)
The crude product was purified by automated flash column chromatography (Biotage Isolera One, SNAP KP Sil 10 g) eluting with n-hexane:DCM:MeOH 100:0:0 v/v increasing to 0:90:10 v/v in 20 CV. Obtained in 41% yield as a yellow solid. 1H-NMR (DMSO-d6), δ: 12.40 (bs, 1 H), 11.94 (bs, 1 H), 8.27–8.24 (m, 1 H), 7.99–7.96 (m, 1 H), 7.79 (dd, J1 = 6.1 Hz, J2=1.1 Hz, 1 H), 7.73–7.70 (m, 2 H), 7.66–7.63 (m, 2 H), 7.56 (d, J=9 Hz, 1 H), 7.16 (dd, J1 = 7.1 Hz, J2=3.6 Hz, 1 H), 7.01 (d, J = 9.1 Hz, 2 H), 3.82–3.80 (m, 4 H), 3.28–3.26 (m, 4 H). 13C-NMR (DMSO-d6), δ: 177.6, 176.6, 172.6, 163.1, 162.8, 162.6, 149.4, 138.8, 137.5, 133.9, 130.1, 129.8, 129.7, 127.6, 126.9, 125.7, 124.0, 123.3, 115.8, 100.0, 55.4, 48.7. 19F-NMR (DMSO-d6), δ ppm: −56.59 (s, 3 F). UPLC-MS (Method C): Rt 2.05 min, MS [ESI, m/z]: 575.1 [M + H]. Anal. Calcd for C26H21F3N4O2S3: C, 54.34; H, 3.68; N, 9.75. Found: C, 54.11; H, 3.53; N, 9.98.
N-((4-(4-(Thiophene-2-carbonyl)piperazin-1- yl)phenyl)carbamothioyl)benzamide (51)
The desired product did not precipitate after cooling the reaction at r.t., as the precipitated solid was in this case by-product 73, which was collected by filtration and triturated from acetone. The filtrate was dried under vacuum and the crude product was purified by automated flash column chromatography (Biotage Isolera One, SNAP KP Sil 10 g) eluting with n-hexane:DCM:MeOH 100:0:0 v/v increasing to 0:95:5 v/v in 20 CV. Obtained in 42% yield as a yellow solid. 1H-NMR (CDCl3), δ: 12.47 (bs, 1 H), 9.09 (bs, 1 H), 7.93–7.91(m, 2 H), 7.70–7.67 (m, 1 H), 7.64–7.62 (m, 2 H), 7.60–7.56 (m, 2 H), 7.50 (dd, J1 = 5.0 Hz, J2 = 1.1, 1 H),7.38 (dd, J1=4.8 Hz, J2 = 1.1, 1 H), 7.10 (dd, J1 = 5.0 Hz, J2 = 3.6, 1 H), 7.00–6.97 (m, 2 H), 3.95 (t, 4 H), 3.30 (t, 4 H). 13C-NMR (CDCl3), δ: 178.2, 176.6, 166.9, 163.7, 149.5, 133.7, 131.7, 130.2, 129.2, 129.0, 128.8, 127.5, 126.8, 125.3, 116.3, 49.4. UPLC-MS (Method B): Rt 2.18 min, MS [ESI, m/z]: 451.1 [M + H]. Anal. Calcd for C23H22N2O4S2: C, 61.31; H, 4.92; N, 12.43. Found: C, 61.20; H, 5.13; N, 12.29.
Isolated by-product N-(4-(4-(thiophene-2-carbonyl)piperazin-1-yl)phenyl)benzamide (73)
Obtained in 37% yield as a yellow solid. 1H-NMR (CDCl3), δ: 10.35 (bs, 1 H), 8.03–8.02 (m, 2 H), 7.88–7.86 (m, 3 H), 7.66–7.57 (m, 5 H), 7.57 (bs, 1 H), 7.22 (dd, J1=1.2 Hz, J2=3.7 Hz, 1 H), 4.06 (m, 4 H), 3.49 (m, 4 H).13C-NMR (CDCl3), δ: 176.6, 165.8, 162.9, 137.1, 135.2, 132.1, 130.4, 129.9, 128.8, 128.1, 127.6, 121.8, 37.7. UPLC-MS (Method B): Rt 1.92 min, MS [ESI, m/z]: 392.3 [M + H].
4-Methyl-N-((4-(4-(thiophene-2-carbonyl)piperazin-1- yl)phenyl)carbamothioyl)benzamide (52)
The crude product was purified by trituration from acetone. Obtained in 78% yield as a yellow solid. 1H-NMR (DMSO-d6), δ: 12.54 (bs, 1 H),11.37 (bs, 1 H), 7.90 (d, J=8.2 Hz, 2 H), 7.79 (dd, J1 = 6.4 Hz, J2 = 1.1 Hz, 1 H), 7.55 (d, J = 9.1 Hz, 2 H), 7.48 (dd, J1 = 4.2 Hz, J2 = 1.1 Hz, 1 H), 7.34 (d, J = 8.2 Hz, 2 H, H- aromatic), 7.16 (dd, J1 = 6.4 Hz, J2 = 4.2 Hz, 1 H, H-aromatic), 7.00 (d, J = 9.1 Hz, 2 H, H- aromatic), 3.81–3.80 (m, 4 H), 3.27–3.25 (m, 4 H), 2.40 (s, 3 H). 13C-NMR (DMSO-d6), δ: 179.1, 176.6, 168.5, 162.8, 149.3, 144.0, 139.7, 137.5, 130.1, 129.7, 129.5, 129.2, 127.6, 125.6, 115.9, 48.8, 21.6. UPLC-MS (Method B): Rt 1.97 min, MS [ESI, m/z]: 465.2 [M+H]. Anal. Calcd for C24H24N4O2S2: C, 62.05; H, 5.21; N, 12.06. Found: C, 62.27; H, 4.97; N, 11.99.
3-Chloro-N-((4-(4-(furan-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)benzo[b]thiophene-2-carboxamide (53)
The crude product was purified by trituration from acetone. Obtained in 74% yield as a light orange solid. 1H-NMR (CDCl3), δ: 12.11 (bs, 1 H), 10.08 (bs, 1 H), 7.92–7.90 (m, 1 H), 7.83–7.82 (m, 1 H), 7.55–7.48 (m, 4 H), 7.45–7.44 (m, 1 H), 6.99 (dd, J1=4.4 Hz, J2 = 1.4 Hz, 1 H), 6.91–6.88 (m, 2 H), 6.44 (dd, J1=5.2 Hz, J2 = 1.4 Hz,), 3.91 (m, 4 H), 3.23–3.21 (m, 4 H). 13C-NMR (CDCl3), δ: 177.4, 176.6, 160.1, 159.1, 149.5, 147.9, 143.8, 138.9, 136.8, 129.9, 128.8, 126.1, 125.2, 123.9, 122.9, 122.4, 116.8, 116.3, 111.4, 49.3. UPLC-MS (Method C): Rt 2.14 min, MS [ESI, m/z]: 525.1 [M + H]. Anal. Calcd for C25H21ClN4O3S2: C, 57.19; H, 4.03; N, 10.67. Found: C, 56.93; H, 3.88; N, 10.91.
N-((4-(4-Benzoylpiperazin-1-yl)phenyl)carbamothioyl)-3-chlorobenzo[b]thiophene-2-carboxamide (54)
The crude product was purified by trituration from acetone. Obtained in 53% yield as a yellow solid. 1H-NMR (CDCl3), δ: 12.20 (bs, 1 H), 10.17 (bs, 1 H), 8.02–8.00 (m, 1 H), 7.93–7.91 (m, 1 H), 7.64–7.57 (m, 4 H), 7.48–7.46 (m, 5 H), 7.00–6.96 (m, 2 H), 3.97 (m, 2 H), 3.64 (m, 2 H), 3.32–3.20 (m, 4 H). 13C-NMR (CDCl3), δ: 177.2, 170.4, 160.2, 149.6, 138.9, 136.8, 135.5, 130.1, 129.9, 129.6, 128.9, 128.6, 127.1, 126.1, 125.2, 124.0, 122.9, 122.5, 116.5, 49.1. UPLC-MS (Method C): Rt 2.18 min, MS [ESI, m/z]: 535.1 [M + H]. Anal. Calcd for C27H23ClN4O2S2: C, 60.61; H, 4.33; N, 10.47. Found: C, 60.38; H, 4.51; N, 10.29.
N-((4-(4-Benzoylpiperazin-1-yl)phenyl)carbamothioyl)benzamide (55)
The crude product was purified by trituration from acetone. Obtained in 62% yield as a white solid. 1H-NMR (DMSO-d6), δ: 10.28 (bs, 1 H), 7.97–7.95 (m, 2 H), 7.77 (d, J = 8.6 Hz, 2 H), 7.61 (m, 1 H), 7.55–7.52 (m, 2 H), 7.49–7.47 (m, 6 H), 7.39 (bs, 2 H), 3.91 (m, 2 H), 3.65 (m, 2 H), 3.36 (m, 4 H). 13C-NMR (DMSO-d6), δ: 178.5, 176.6, 169.5, 165.8, 135.9, 135.3, 132.0, 130.2, 128.9, 128.8, 128.1, 127.5, 121.8, 36.4. UPLC-MS (Method C): Rt 1.89 min, MS [ESI, m/z]: 445.2 [M + H]. Anal. Calcd for C25H24N4O2S: C, 67.55; H, 5.44; N, 12.60. Found: C, 67.61; H, 5.17; N, 12.47.
3-Chloro-N-((4-(4-(thiazole-2-carbonyl)piperazin-1-yl)phenyl)carbamothioyl)benzo[b]thiophene-2-carboxamide (56)
The crude product was purified by trituration from acetone. Obtained in 43% yield as an off-white solid. 1H-NMR (CDCl3), δ: 12.20 (bs, 1 H) 10.17 (bs, 1 H), 8.01–8.01 (m, 1 H),7.93 (d, J = 3.2 Hz, 1 H), 7.93–7.91 (m, 1 H), 7.65–7.57 (m, 5 H), 7.02–6.99 (m, 2 H), 4.64 (m, 2 H), 4.00 (m, 2 H), 3.36 (m, 4 H). 13C-NMR (CDCl3), δ: 177.2, 176.6, 165.0, 160.1, 154.2, 149.6, 143.2, 138.9, 136.8, 129.9, 129.6, 128.9, 126.1, 125.2, 124.2, 123.9, 122.9, 116.3, 49.6, 49.1, 46.1, 43.3. UPLC-MS (Method C): Rt 2.20 min, MS [ESI, m/z]: 543.0 [M + H]. Anal. Calcd for C24H20ClN5O2S3: C, 53.18; H, 3.72; N, 12.92. Found: C, 52.96; H, 3.79; N, 13.03.
N-((4-(4-Benzoylpiperazin-1-yl)phenyl)carbamothioyl)-4-methylbenzamide (57)
The desired product did not precipitate after cooling the reaction at r.t., as the precipitated solid was in this case by-product 74, which was collected by filtration and triturated from acetone. The filtrate was dried under vacuum and the crude product was purified by automated flash column chromatography (Biotage Isolera One, SNAP KP Sil 10 g) eluting with n-hexane:DCM:MeOH 100:0:0 v/v increasing to 0:95:5 v/v in 25 CV. Obtained in 41% yield as a yellow solid. 1H-NMR (CDCl3), δ: 12.50 (bs 1 H), 9.05 (bs 1 H), 7.80 (d, J = 8.3 Hz, 2 H), 7.61 (d, J = 8.9 Hz, 2 H), 7.35 (d, J = 7.9 Hz, 2 H), 7.48–7.46 (m, 5 H), 6.99–6.95 (m, 2 H), 3.97 (m, 2 H), 3.63 (m, 2 H), 3.31–3.19 (m, 4 H), 2.38 (s, 3 H). 13C-NMR (CDCl3), δ: 178.3, 170.4, 166.8, 149.5, 144.7, 135.6, 130.4, 129.9, 129.8, 128.9, 128.5, 127.5, 127.1, 125.2, 116.5, 49.5, 21.6. UPLC-MS (Method C): Rt 2.29 min, MS [ESI, m/z]: 459.2 [M + H]. Anal. Calcd for C26H26N4O2S: C, 68.10; H, 5.72; N, 12.22. Found: C, 68.25; H, 5.53; N, 11.97.
Isolated by-product N-(4-(4-benzoylpiperazin-1-yl)phenyl)-4-methylbenzamide (74)
Obtained in 35% yield as a yellow solid. 1H-NMR (CDCl3), δ: 10.20 (bs, 1 H), 7.88 (d, J = 8.2 Hz, 2 H), 7.78 (d, J = 8.5 Hz, 2 H), 7.50–7.47 (m, 6 H), 7.37–7.33 (m, 3 H), 3.92 (m, 2 H), 3.63 (m, 2 H), 3.36 (m, 4 H), 2.39 (s, 3 H). 13C-NMR (CDCl3), δ: 195.6, 176.6, 169.5, 165.6, 142.0, 135.9, 132.4, 130.2, 129.4, 129.3, 128.1, 127.6, 121.8, 38.1, 21.5. (Method C): Rt 1.75 min, MS [ESI, m/z]: 400.3 [M + H].
N-((4-(4-(Thiazole-2-carbonyl)piperazin-1- yl)phenyl)carbamothioyl)benzamide (58)
The desired product did not precipitate after cooling the reaction at r.t., as the precipitated solid was in this case by-product 75, which was collected by filtration and triturated from acetone. The filtrate was dried under vacuum and the crude product was purified by automated flash column chromatography (Biotage Isolera One, SNAP KP Sil 10 g) eluting with n-hexane:DCM:MeOH 100:0:0 v/v increasing to 0:90:10 v/v in 20 CV. Obtained in 47% yield as a yellow solid. 1H-NMR (DMSO-d6), δ: 12.46 (bs, 1 H), 9.07 (bs, 1 H), 7.94–7.91 (m, 3 H), 7.70–7.67 (m, 1 H), 7.65–7.62 (m, 2 H), 7.59–7.56 (m, 3 H), 7.02–7.09 (m, 2 H), 4.64 (m, 2 H), 4.01 (m, 2 H), 3.55 (m, 4 H). 13C-NMR (DMSO-d6), δ: 178.1, 166.9, 165.1, 159.2, 149.5, 149.6, 143.2, 133.7, 131.8, 129.2, 127.5, 125.3, 124.1, 116.3, 49.7. UPLC-MS (Method C): Rt 1.89 min, MS [ESI, m/z]: 452.2 [M + H]. Anal. Calcd for C22H21N5O2S2: C, 58.52; H, 4.69; N, 15.51. Found: C, 58.48; H, 4.87; N, 15.34.
Isolated by-product N-(4-(4-(thiazole-2-carbonyl)piperazin-1-yl)phenyl)benzamide (75)
Obtained in 29% yield as a yellow solid. 1H-NMR (DMSO-d6), δ: 10.29 (bs, 1 H), 8.07 (m, 2 H), 7.98–7.96 (m, 2 H), 7.77 (d, J = 8.9 Hz, 2 H), 7.61–7.58 (m, 1 H), 7.55–7.51 (m, 2 H), 7.37–7.34 (m, 2 H), 4.71–4.59 (m, 4 H), 3.99 (m, 2 H), 3.43 (m, 2 H). 13C-NMR (DMSO-d6), δ: 176.7, 172.4, 165.7, 164.8, 158.9, 144.0, 135.4, 131.9, 128.8, 128.1, 126.1, 121.9, 31.1. (Method B): Rt 1.89 min, MS [ESI, m/z]: 393.2 [M + H].
N-((4-(4-(Thiophen-2-ylsulfonyl)piperazin-1- yl)phenyl)carbamothioyl)benzamide (59)
The desired product did not precipitate after cooling the reaction at r.t., therefore the reaction mixture was dried under vacuum and the crude product was purified by automated flash column chromatography (Biotage Isolera One, SNAP KP Sil 10 g) eluting with n-hexane:DCM:MeOH 100:0:0 v/v increasing to 0:95:15 v/v in 20 CV. Obtained in 69% yield as a yellow solid. 1H-NMR (DMSO-d6), δ: 12.47 (bs, 1 H), 11.47 (bs, 1 H), 8.09 (dd, J1 = 6.3 Hz, J2 = 1.3, 1 H), 7.98–7.96 (m, 2 H), 7.00 (dd, J1 = 3.8 Hz, J2 = 1.3, 1 H), 7.68–7.64 (m, 1 H), 7.56–7.51 (m, 4 H), 7.32 (dd, J1=6.3 Hz, J2 = 3.8, 1 H), 6.96 (d, J = 9.1 Hz, 2 H), 3.30–3.28 (m, 4 H), 3.09–3.07 (m, 4 H). 13C-NMR (DMSO-d6), δ: 179.1, 176.6, 168.7, 148.9, 134.8, 134.6, 133.8, 133.5, 132.7, 130.5, 129.1, 128.9, 125.6, 116.3, 48.2, 46.3. UPLC-MS (Method C): Rt 2.00 min, MS [ESI, m/z]: 487.1 [M + H]. Anal. Calcd for C22H22N4O3S3: C, 54.30; H, 4.56; N, 11.51. Found: C, 54.45; H, 4.36; N, 11.44.
N-((4-(4-(Phenylsulfonyl)piperazin-1-yl)phenyl)carbamothioyl)benzamide (60)
The desired product did not precipitate after cooling the reaction at r.t., as the precipitated solid was in this case by-product 76, which was collected by filtration and triturated from acetone. The filtrate was dried under vacuum and the crude product was purified by automated flash column chromatography (Biotage Isolera One, SNAP KP Sil 25 g) eluting with n-hexane:DCM:MeOH 100:0:0 v/v increasing to 0:95:5 v/v in 20 CV. Obtained in 77% yield as a yellow solid. 1H-NMR (DMSO-d6), δ: 12.45 (bs, 1 H), 11.46 (bs, 1 H), 7.98–7.96 (m, 2 H), 7.81–7.75 (m, 3 H), 7.71–7.64 (m, 3 H), 7.55–7.50 (m, 4 H), 6.94 (d, J=9.1 Hz, 2 H), 3.26–3.24 (m, 4 H), 3.04–3.02 (m, 4 H). 13C-NMR (DMSO-d6), δ: 207.4, 199.5, 180.8, 179.0, 176.6, 138.1, 135.1, 133.9, 130.0, 129.1, 128.9, 128.1, 125.6, 116.3, 48.3, 46.2. UPLC-MS (Method B): Rt 2.37 min, MS [ESI, m/z]: 481.2 [M + H]. Anal. Calcd for C24H24N4O3S2: C, 59.98; H, 5.03; N, 11.66. Found: C, 59.76; H, 4.91; N, 11.90.
Isolated by-product N-(4-(4-benzoylpiperazin-1-yl)phenyl)benzamide (76)
Obtained in 14% yield as a yellow solid. 1H-NMR (DMSO-d6), δ: 10.19 (bs, 1 H), 7.95–7.93 (m, 2 H), 7.81–7.76 (m, 3 H), 7.71–7.64 (m, 4 H), 7.59–7.56 (m, 1 H), 7.53–7.50 (m, 2 H), 7.22–7.00 (m, 2 H), 3.39–3.24 (m, 4 H), 3.17–3.07 (m, 4 H).13C-NMR (DMSO-d6), δ: 176.6, 165.6, 162.6, 135.4, 135.0, 134.0, 131.9, 130.0, 128.8, 128.1, 128.0, 121.8, 118.0, 49.8, 46.0. (Method B): Rt 2.15 min, MS [ESI, m/z]: 422.2 [M + H].
Biology
RdRp enzyme inhibition assays
Quantitative RdRp activity and gel-based assays
Fluorescent RdRp activity assays were carried out as previously described12,29. Briefly, RdRp activity was quantified by monitoring the formation of double-stranded RNA (dsRNA) from a single stranded homopolymeric template, poly(C) (Sigma Aldrich), using the fluorescent dye PicoGreen (Life Technologies, Carlsbad, CA, USA). RdRp assays were performed in 384-well plates, and each reaction mixture contained 400 ng enzyme, 45 μM GTP, 10 ng/μL poly(C) RNA, 2.5 mM MnCl2, 5 mM dithiothreitol, (DTT), 0.01% bovine serum albumin (BSA), and 0.005% Tween 20 in 20 mM Tris-HCl, pH 7.5, with a final reaction volume of 25 μL. The PPNDS and compound 1 were used as a positive controls. RdRps were incubated for 10 mins at 30 °C in the presence of the test compounds or the compound vehicle DMSO (0.5% vol/vol) before addition into the reaction mixture, which was then allowed to run 15 min at 30 °C then terminated with 10 mM EDTA, followed by PicoGreen staining and dsRNA quantitation. GraphPad Prism V6.05 (La Jolla, CA, USA) was used to plot the IC50 values. A secondary gel-based polymerase activity assay was used as a counter-screen as described below, to exclude the possibility of RdRp activity enhancement. Primed elongation activity was examined in a gel-based assay as previously described12,29 using the RNA template (PE44-NoV). Gels were imaged using BioRad (Hercules, CA, U.SA) Geldoc Universal Hood II, running BioRad Image Lab software, V4.1, build 16.
MNV and HuNoV cell-based assays
Norovirus
Cells and virus: MNV (virus strain MNV-1.CW1) was propagated in RAW 264.7 cells grown in DMEM (Life Technologies, Gent, Belgium) supplemented with 10% or 2% FBS, 2 mM L-glutamine, 20 mM HEPES, 0.075 g/L sodium bicarbonate, 1 mM sodium pyruvate, 100 U penicillin/mL and 100 lg/mL streptomycin at 37 °C in a humidified atmosphere of 5% CO2. The human norovirus GI.1 replicon harboring gastric tumor-1 cell line (HGT-NV) (kindly provided by Dr. Ian Goodfellow, University of Cambridge) was maintained in DMEM supplemented with 10% FBS, 2mM L-glutamine, 0.075 g/L sodium bicarbonate and 1.0 mg/ml of Geneticin (G418; Life Technologies), at 37 °C in a humidified atmosphere of 5% CO234.
Antiviral assay with MNV: The antiviral activity of the Compounds was determined using an MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium]-based cytopathic effect (CPE) reduction assay. RAW 264.7 cells (1 × 104 cells/well) were seeded in a 96-well plate and infected with MNV (MOI of 0.001) in the presence (or absence) of a dilution series of Compounds Cells were incubated for 3 days, i.e. until complete CPE was observed in infected untreated cells. Then, a MTS-phenazinemethosulfate (MTS/PMS) stock solution [(2 mg/mL MTS (Promega, Leiden, The Netherlands) and 46 g/mL PMS (Sigma–Aldrich, Bornem, Belgium) in PBS at pH 6–6.5)] was diluted 1/20 in MEM (Life Technologies, Gent, Belgium) and 75 µL were added to each well. After 2 h, the optical density (OD) was read at 498 nm. The %CPE reduction was calculated as [(ODtreated)MNV − ODVC]/[ODCC − ODVC] × 100, where ODCC represents the OD of the uninfected untreated cells, whereas ODVC and (ODtreated)MNV represent the OD of infected untreated cells and virus-infected cells treated with a compound concentration, respectively. The 50% effective concentration (EC50) was defined as the compound concentration that protected 50% of the cells from virus-induced CPE34.
Cytotoxicity: The cytotoxicity of the Compounds was evaluated by the MTS method, by exposing uninfected cells to the same concentrations of compounds for 3 days. The %cell viability was calculated as (ODtreated/ODCC) × 100, where ODCC is the OD of uninfected untreated cells and ODtreated are uninfected cells treated with compound. The CC50 was defined as the compound concentration that reduces the number of viable cells by 50%34.
Antiviral assay with the HGT-NV replicon: The inhibitory effect of Compounds on human norovirus replication was assessed by quantification of the levels of HuNoV GI replicon RNA and of the reference (housekeeping) gene β-actin mRNA [by quantitative reverse transcription polymerase chain reaction (qRT-PCR)]. To this end, HGT cells (2000 cells/well) were seeded in 96-well plates, in complete DMEM without the selection marker G418. Following an incubation period of 24 h, a serial dilution of rupintrivir was added to the cultures. After 72 h of incubation, cell monolayers were washed with phosphate-buffered saline (PBS) and collected for quantification of RNA load by qRT-PCR. Intracellular RNA was extracted from cells using the cell-to-cDNA lysis buffer (Ambion, Life Technologies). For detection of HuNoV GI replicon RNA, forward (5′-CCG GCT ACC TGC CCA TTC-3′), reverse (5′-CCA GAT CAT CCT GAT CGA CAA G-3′) primers and probe (5′-FAM-ACA TCG CAT CGA GCG AGC ACG TAC-TAMRA-3′) for the neomycin gene were used. For detection of β-actin mRNA, forward (5′-GGC ATC CAC GAA ACT ACC TT-3′), reverse (5′-AGC ACT GTG TTG GCG TAC AG-3′) primers and probe (5′-HEX-ATC ATG AAG TGT GAC GTG GAC ATC CG-BHQ1–3′) were used. One-step qRT-PCR was performed in a 20 µL reaction mixture containing 10 µl 2X iTaq Universal SYBR® Green one step reaction mix (Bio-rad, California, USA), 5.17 µl RNAse free water, 0.5 µl of iScript reverse transcriptase (Bio-rad, California, USA), 4 µl of the template RNA and either 300 nM of HuNoV GI replicon primers and probe, 300 nM of β-actin primers and 200 nM of probe. Cycling conditions were: reverse transcription at 50 °C for 10 min, initial denaturation at 95 °C for 3 min, followed by 40 cycles of denaturation at 95 °C for 15 s, annealing and extension at 60 °C for 30 seconds (Roche Lightcycler®96, Roche Diagnostics, Belgium). To determine the relative expression levels of HuNoV GI replicon RNA, β-actin was used as a normalizer and ratios were calculated using the Pfaffl method. The Expression Ratio (HuNoV GI Replicon/β-actin) was calculated as: Expression Ratio = (E HuNoV)ΔCT, HuNoV (CC – TC)/(Eβ-actin)ΔCT, β-actin (CC – TC), where E HuNoV and Eβ-actin represent the amplification efficiency (E = 10–1/slope) for the HuNoV GI replicon and β-actin qRT-PCR reactions, respectively. ΔCT, HuNoV (CC – TC) is the Ct of untreated control cells (CC) minus the Ct of cells treated with a compound concentration (TC) obtained with HuNoV GI replicon primers and probe. ΔCT, β-actin (CC – TC) is the Ct of untreated control cells (CC) minus the Ct of cells treated with a compound concentration (TC) obtained with β-actin primers and probe. Efficiency values (E HuNoV and Eβ-actin) were determined for each qRT-PCR reaction. The 50% effective concentration (EC50) was defined as the compound concentration that resulted in a 50% reduction of the relative HuNoV replicon RNA levels34.
Molecular modelling
All molecular docking studies were performed on a Viglen Genie Intel®CoreTM i7–3770 vPro CPU@ 3.40 GHz × 8 running Ubuntu 16.04. Molecular Operating Environment (MOE) 2018.1033 and Maestro (Schrödinger Release 2017–1)33 were used as molecular modelling software. The RdRp structure n complex with PPNDS was downloaded from the PDB data bank (http://www.rcsb.org/; PDB code 4LQ3). The protein was pre-processed using the Schrödinger Protein Preparation Wizard by assigning bond orders, adding hydrogens and performing a restrained energy minimization of the added hydrogens using the OPLS_2005 force field. Ligand structures were built with MOE and then prepared using the Maestro LigPrep tool by energy minimising the structures (OPLS_2005 force filed), generating possible ionization states at pH 7 ± 2, generating tautomers and low-energy ring conformers. An 11 Å docking grid (inner-box 10 Å and outer-box 21 Å) was prepared using as centroid the co-crystallised PPNDS. Molecular docking studies were performed using Glide SP precision keeping the default parameters and setting 5 as number of output poses per input ligand to include in the solution. The output poses were saved as mol2 file. The docking results were visually inspected for their ability to bind the active site.
The Flexible Alignment was performed using MOE2018.10. The MOE flexible alignment tool generates different possible conformations for each molecule present in the input database (in-house small-molecule database of reported non-nucleoside inhibitors of viral polymerases, previously prepared in MOE) that could overlap the conformation of the assigned template, which is kept rigid (compound 1). The quality of the alignment is evaluated by a score which is a sum of the internal strain of the obtained conformation (the smaller, the better) and the overlap of molecular features (aromatic regions, donors/acceptors). MOE, for each alignment performed, evaluates the average internal energy of the ligands U, the similarity score F (the lower value is, the better the two structures overlap) and the value S (sum of U and F values obtained for each alignment). A good alignment should present a dU value (the average strain energy of the molecules in the alignment in kcal/mol) lower than 1 kcal/mol meaning that the obtained conformations are not energetically disadvantaged. The obtained data from the Flexible Alignment with dU of 0.0 (no energy penalty) were kept and ranked according to the lowest S value. TPB (48) resulted the best match for its structural overlapping.
The conformation of 1 required for the Flexible Alignment was obtained using MOE conformational search tool, using the default parameters, selecting Stochastic as search method and setting RMS Gradient to 0.001 and the RMSD Limit to 0.15. 159 conformations were generated and among them the best in terms of dE (the strain of the conformation relative to the lowest energy conformation with the same stereochemistry configuration. Value of 0 indicate the best result) and E (value of the potential energy of the conformation) was chosen as template for the Flexible Alignment.
The Glide-Based Core Hopping tool of Maestro32 was used to perform a scaffold replacement. The structure of compound 1 docked into the RdRp binding site was used as template ligand keeping as fixed the two terminal hydrophobic rings and let the program replacing the original scaffold with new optimal linkers. The new ligands are then automatically docked on the receptor and ranked and scored according to the docking results. The default parameters were used, using as docking grid the one previously generated for the SP docking studies (see above).
References
Lopman, B. A., Steele, D., Kirkwood, C. D. & Parashar, U. D. The vast and varied global burden of norovirus: prospects for prevention and control. PloS Med 13, e:1001999 (2016).
Petrignani, M., Verhoef, L., de Graaf, M., Hendrik Richardus, J. & Koopmans, M. Chronic sequelae and severe complications of norovirus infection: A systematic review of literature. J. Clin. Virol. 105, 1–10 (2018).
Shah, M. P. & Hall, A. J. Norovirus illnesses in children and adolescents. Infect Dis Clin North Am. 32, 103–118 (2018).
Lucero, Y., Vidal, R. & O’Ryan, G. M. Norovirus vaccines under development. Vaccine 36, 5435–5441 (2018).
Ettayebi, K. et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 353, 1387–1393 (2016).
Jones, M. K. et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 346, 755–759 (2014).
Rossignol, J.-F. & El-Gohary, Y. M. Nitazoxanide in the treatment of viral gastroenteritis: A randomized double-blind placebo controlled clinical trial. Aliment. Pharmacol. Ther. 24, 1423–1430 (2006).
Siddiq, D. M., Koo, H. L., Adachi, J. A. & Viola, G. M. Norovirus gastroenteritis successfully treated with nitazoxanide. J. Infect. 63, 394–397 (2011).
Lanier, R.S. D. et al. CMX521: A nucleoside with pan-genotypic activity against norovirus. In Proceedings of 31st International Conference on Antiviral Research, Porto, Portugal (2018).
Eltahla, A. A. et al. Non nucleoside inhibitors of norovirus RNA polymerase: scaffolds for rational drug design. Antimicrob. Agents Chemother. 58, 3115–3123 (2014).
Jin, Z. et al. Biochemical evaluation of the inhibition properties of favipiravir and 2′-c-methyl-cytidine triphosphates against human and mouse norovirus RNA polymerases. Antimicrob. Agents Chemother. 59, 7504–7516 (2015).
Netzler, N. E. et al. Broad-spectrum non-nucleoside inhibitors for caliciviruses. Antiviral. Res. 146, 65–75 (2017).
Tarantino, D. et al. Naphthalene-sulfonate inhibitors of human norovirus RNA-dependent RNA-polymerase. Antiviral. Res. 102, 23–28 (2014).
Hussey, R. J. et al. A structural study of norovirus 3C protease specificity: binding of a designed active site-directed peptide inhibitor. Biochemistry 50, 240–249 (2011).
Prior, A. M. et al. Design, synthesis, and bioevaluation of viral 3C and 3C-like protease inhibitors. Bioorg. Med. Chem. Lett. 23, 6317–6320 (2013).
Tiew, K. C. et al. Design, synthesis, and evaluation of inhibitors of Norwalk virus 3C protease. Bioorg. Med. Chem. Lett. 21, 5315–5319 (2011).
Kaufman, S. S., Green, K. Y. & Korba, B. E. Treatment of norovirus infections: moving antivirals from the bench to the bedside. Antiviral. Res. 105, 80–91 (2014).
Hansman, G. S. et al. Intergenogroup recombination in sapoviruses. Emerg. Infect. Dis. 11, 1916–1920 (2005).
Alam, I. et al. Crystal structure of murine norovirus-1 RNA-dependent RNA polymerase in complex with 2-thiouridine or ribavirin. Virology 426, 143–151 (2012).
Rohayem, J., Robel, I., Jager, K., Scheffler, U. & Rudolph, W. Protein-primed and de novo initiation of RNA synthesis by norovirus 3Dpol. J Virol 80, 7060–7069 (2006).
Yates, M. K. & Seley-Radtke, K. L. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antivir. Res. 162, 5–21 (2019).
Bassetto, M., Van Dycke, J., Neyts, J., Brancale, A. & Rocha-Pereira, J. Targeting the viral polymerase of diarrhea-causing viruses as a strategy to develop a single broad-spectrum antiviral therapy. Viruses 11, 173 (2019).
Zamyatkin, D. F. et al. Structural insights into mechanisms of catalysis and inhibition in Norwalk virus polymerase. J. Biol. Chem. 283, 7705–7712 (2008).
Zamyatkin, D. F., Parra, F., Machin, A., Grochulski, P. & Ng, K. K. Binding of 2′amino-2′-deoxycytidine-5′-triphosphate to norovirus polymerase induces rearrangements in the active site. J. Mol. Biol. 390, 10–16 (2009).
Zamyatkin, D. et al. Structure of a backtracked state reveals conformation changes similar to the state following nucleotide incorporation in human norovirus polymerase. Acta Crystallogr. Sect. D: Biol. Crystallogr. 70, 3099–3109 (2014).
Croci, R. et al. Structural bases of norovirus RNA dependent RNA polymerase inhibition by novel suramin-related compounds. PLoS One 9, e91765 (2014).
Croci, R. et al. PPNDS inhibits murine norovirus RNA-dependent RNA-polymerase mimicking two RNA stacking bases. FEBS Lett. 588, 1720–1725 (2014).
Mastrangelo, E. et al. Structure-based inhibition of norovirus RNA-dependent RNA polymerases. J. Mol. Biol. 419, 198–210 (2012).
Ferla, S. et al. In silico screening for human norovirus antivirals reveals a novel non-nucleoside inhibitor of the viral polymerase. Sci. Rep. 8, 4129 (2018).
Ishikawa, M. & Hashimoto, Y. Improvement in aqueous solubility in small molecule drug discovery programs by disruption of molecular planarity and symmetry. J. Med. Chem. 54, 1539–1554 (2011).
Bergström, C. A. S., Wassvik, C. M., Johansson, K. & Hubatsch, I. Poorly soluble marketed drugs display solvation limited solubility. J. Med. Chem. 50, 5858–5862 (2007).
Schrödinger Release 2017-21 Core Hopping, Schrödinger, LLC, New York, NY (2017).
ULC, C. C. G. Molecular Operating Environment (MOE), 2018.10, 1010 Sherbooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7 (2018).
Rocha-Pereira, J. et al. The viral polymerase Inhibitor 2′-C-Methylcytidine inhibits norwalk virus replication and protects against norovirus-induced diarrhea and mortality in a mouse model. J. Virol. 87, 11798–11805 (2013).
Kolawole, A. O., Rocha-Pereira, J., Elftman, M. D., Neyts, J. & Wobus, C. E. Inhibition of human norovirus by a viral polymerase inhibitor in the B cell culture system and in the mouse model. Antivir. Res. 132, 46–49 (2016).
Van Dycke, J. et al. A robust human norovirus replication model in zebrafish larvae. BioRxiv, https://doi.org/10.1101/528364v1 (2019).
Schrödinger Release 2017-1: Maestro, Schrödinger, LLC, New York, NY (2017).
Hirashima, S. et al. Benzimidazole derivatives bearing substituted biphenyls as hepatitis C virus NS5B RNA-dependent RNA polymerase inhibitors: structure-activity relationship studies and identification of a potent and highly selective inhibitor JTK-109. J. Med. Chem. 49, 4721–4736 (2006).
Pattnaik, A. et al. Discovery of a non-nucleoside RNA polymerase inhibitor for blocking Zika virus replication through in silico screening. Antivir. Res. 151, 78–86 (2018).
Niyomrattanakit, P. et al. Inhibition of dengue virus polymerase by blocking of the RNA Tunnel. J. Virol. 84, 5678–5686 (2010).
Kitano, M., Hosmillo, M., Emmott, E., Lu, J. & Goodfellow, I. Selection and characterization of rupintrivir-resistant norwalk virus replicon cells in vitro. Antimicrob. Agents Chemother. 62, e00201–18 (2018).
Rocha-Pereira, J. et al. The enterovirus protease inhibitor rupintrivir exerts cross-genotypic anti-norovirus activity and clears cells from the norovirus replicon. Antimicrob. Agents Chemother. 58, 4675–4681 (2014).
Acknowledgements
M.B. and S.F. were supported by the Sêr Cymru II programme which is part-funded by Cardiff University and the European Regional Development Fund through the Welsh Government. G.G. was supported by the Wellcome Trust through an ISSF3 Translational Kickstart Award. The authors would like to thank Prof. Peter White at the UNSW, Sydney, Australia, for kindly providing the purified HuNoV RdRp used in this work and for sharing useful insights on enzymatic assay setup. We thank Jasper Rymenants and Lindsey Bervoets for excellent technical assistance.
Author information
Authors and Affiliations
Contributions
M.B. and S.F. conceived the study and supervised the project; G.G. and J.R.-P. performed and interpreted the biological experiments; S.F. and M.B. worked on the molecular modelling; G.G. and I.R. worked on the synthesis and compounds characterization under the supervision of M.B.; J.N. and A.B. co-supervised the biological and molecular modelling work. G.P. and M.T.Y. contributed to biochemical assay set-up and data interpretation. S.F. and M.B. wrote the manuscript with the help of G.G. and J.R.-P.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Giancotti, G., Rigo, I., Pasqualetto, G. et al. A new antiviral scaffold for human norovirus identified with computer-aided approaches on the viral polymerase. Sci Rep 9, 18413 (2019). https://doi.org/10.1038/s41598-019-54903-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-54903-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
