
Abstract
DNA double-strand breaks (DSBs), selectively visualized as γ-H2AX+ foci, occur during the development of the central nervous system, including the retina, although their origin and biological significance are poorly understood. Mutant mice with DSB repair mechanism defects exhibit increased numbers of γ-H2AX+ foci, increased cell death during neural development, and alterations in axonogenesis in the embryonic retina. The aim of this study was to identify putative sources of DSBs. One of the identified DSBs sources is LINE-1 retrotransposition. While we did not detect changes in LINE-1 DNA content during the early period of cell death associated with retinal neurogenesis, retinal development was altered in mice lacking RAG-2, a component of the RAG-1,2-complex responsible for initiating somatic recombination in lymphocytes. Although γ-H2AX+ foci were less abundant in the rag2−/− mouse retina, retinal ganglion cell death was increased and axonal growth and navigation were impaired in the RAG-2 deficient mice, a phenotype shared with mutant mice with defective DNA repair mechanisms. These findings demonstrate that RAG-2 is necessary for proper retinal development, and suggest that both DSB generation and repair are genuine processes intrinsic to neural development.
Similar content being viewed by others


Introduction
The assembly of a functional central nervous system (CNS) is a complex process that requires coordination between proliferation, differentiation, and programmed cell death during early stages of development1,2,3,4,5. In different areas of the CNS, including the hippocampus, frontal cortex, and retina, the transition from neuroepithelial cell to mature neuron coincides with massive cell death and an increase in the number of phosphorylated histone H2AX (γ-H2AX) foci, a selective marker of DNA double-strand breaks (DSBs)1,6,7. The generation of DSBs is immediately followed by the recruitment of γ-H2AX to induce active DNA repair, particularly that mediated by the nonhomologous end-joining (NHEJ) pathway. DNA repair is critical for proper neural development. Indeed, DNA repair defects usually lead to severe neural, as well as immune, alterations, in some cases resulting in embryonic lethality or microcephaly, while other tissues remain unaffected3,8,9.
A collection of genomic variations associated with somatic mosaicism in neurons, including deletions, insertions, copy number variations, and retrotransposition, have been described in the human and mouse CNS10,11. Most if not all of these DNA alterations depend, at some point, on DSB generation and repair pathways12. DSB repair is thus increasingly recognized as a fundamental element of CNS cellular dynamics during development and in adulthood. While the deleterious impact of unresolved DSBs in the CNS is well described9,13,14,15,16, how these DSBs are generated and their physiological role during neuronal differentiation remains poorly understood.
The mouse retina is a part of CNS that serves as a convenient model for the analysis of cellular processes involved in early neural development16. Particularly, the different phases of cell death that occur during retinal development are well characterized1,17. We previously described in the embryonic mouse retina an early phase of neural cell death during which γ-H2AX+ foci number increases, a peculiarity absent in later phases of retinal cell death18. One consequence of deficient DSB repair is programmed cell death; defects in NHEJ-mediated DSB repair result in increases in cell death that selectively affect recently born neurons, as described in the retinas of DNA-polymerase-µ- and Ku86-deficient mice, and SCID (DNA-PK) mutant mice18,19,20, as well as in the cerebral cortex of NHEJ-1-deficient mice7.
While proliferative and metabolic stressors have been proposed to give rise to DNA damage and DSBs21, other potential sources in the nervous system include active mechanisms related to the generation of neuronal somatic mosaicism4,12. To date, LINE-1 retrotransposition is the only DSB-generating mechanism identified in mouse neural tissues during development22,23. LINE-1 is an autonomous DNA retrotransposon that can replicate and intercalate itself into various locations within the genome, thereby generating DSBs and disrupting gene expression (reviewed in23). In human tissues, relative copy numbers of LINE-1 retrotransposon are higher in the brain than in non-neural tissue, although the role of LINE-1 retrotransposition in neural development remains controversial24,25. In addition to LINE-1, recent studies have characterized several endonucleases responsible for the generation of sequence-directed DNA breaks in adult neurons26 and other cell types27. In the immune system, the endonuclease RAG-1,2-complex is responsible for generating directed DSBs at sequence-specific targets and regulates DNA repair, primarily via the NHEJ pathway28,29. The RAG-1,2 functional complex consists of a catalytic subunit, RAG-1, and a regulatory subunit, RAG-2. RAG-1 is widely distributed throughout the nervous system30, and mutant mice defective for this subunit exhibit defects in memory formation31 and alterations in the olfactory system32. By contrast, RAG-2 is expressed at low levels in the zebrafish nervous system, except for the olfactory bulb33, and rag2 mRNA expression has been reported in the mammalian brain and retina34,35.
Here, we demonstrate that RAG-2 protein is present in the embryonic mouse retina and is involved in early retinal development. The absence of RAG-2 in the embryonic retina increases cell death at E13.5 and leads to abnormal axonal growth, supporting that RAG-2 is required for proper retinal development in mice.
Results
Possible sources of DSBs in the developing mouse retina
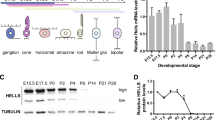
The origins of DSBs in the developing retina remain unclear18,19,20,36. Characterization of potential sources of DSBs could provide important clues as to their physiological relevance. To investigate the role in retinal development of LINE-1, a putative source of DSBs, we measured the relative levels of its orf2 sequence by genomic quantitative PCR (Fig. 1a).

Possible sources of DSBs in the developing retina. (a) Relative levels of LINE-1 DNA detected by genomic qPCR in WT mouse liver and retinal extracts collected at different developmental stages and in adulthood. Dotted red line indicates the mean LINE-1 DNA content in the adult liver. Each datapoint represents a pool of littermates in the case of embryonic tissue samples, and a single animal in the case of adult tissue samples (only 2 animals in P2). Histograms depict the mean ± SEM. *P < 0.05 vs. LINE-1 adult liver content. (b) Western blot analysis of RAG-2 protein levels in retinal extracts from E13.5 WT and rag2−/− mice. WT adult thymus was used as positive control; WT adult muscle was used as negative control. E, embryonic day; P, postnatal day.
We observed no significant changes in relative amounts of LINE-1 between E12.5 and E14.5, the period during which γ-H2AX+ foci incidence peaks and retinal ganglion cells (RGCs) are generated and selectively targeted by programmed cell death18. Genomic levels of LINE-1 in the developing embryonic retina were comparable to those found in both the embryonic and adult liver (Fig. 1a). Significantly higher levels of LINE-1 were detected in the adult retina, an interesting observation that is beyond the scope of this study.
Based on these findings, which suggest that LINE-1 is not involved in the generation of DSBs during this stage of retinal development, we investigated the RAG-1,2-complex, which exerts an essential endonuclease activity in the immune system37, as a possible source of DSBs during early retinal development. The detection of RAG-2 protein in the E13.5 WT retina (Fig. 1b), together with previous reports of RAG-1 expression in the retina30,31,32,38, establishes the expression of the two subunits known to be required for stable DNA binding and cleavage activity in the immune system and suggests a physiological function involving RAG-1,2-complex endonuclease activity in DSB generation in the developing retina.
RAG-2 is active in the E13.5 mouse retina
To confirm that RAG-2 has a function in retinal development, we compared rag2−/− and WT retinas (Fig. 2). Immunostaining for γ-H2AX in retinal cells dissociated at E13.5 revealed the presence of the characteristic γ-H2AX+ foci associated with the presence of DSBs39 (Fig. 2a,b). γH2AX immunostaining was reduced in rag2−/− versus WT retinas (Fig. 2), an observation that correlates with its expected role as endonuclease. In dissociated retinal cells from RAG-2-deficient mice the number of γ-H2AX+ foci per cell and the number of cells containing γ-H2AX+ foci were reduced as compared with WT controls (Fig. 2c,d). In line with this observation, the density of γ-H2AX+ cells in whole-mount retinas was lower in rag2−/− than WT retinas (Fig. 2e). This decrease was evident in the two cell populations found in the E13.5 retina, i.e., PCNA+ proliferative neuroepithelial cells and recently differentiated TUJ-1+ neurons (Fig. 2f). These results support the involvement of RAG-2 in retinal development. Although our experimental approach allowed only indirect assessment, our findings are compatible with the interpretation that RAG-2 is at least partially responsible for the generation of naturally occurring DSBs in the developing retina18,19,20,36.

DSB number is reduced in the retinas of E13.5 rag2−/− mice. (a,b) Presence of DSBs in WT and rag2−/− mice as determined by γH2AX immunostaining (green). Nuclei were counterstained with DAPI (cyan). Quantification of nuclear γH2AX positive foci per cell (c) and percentage of γH2AX+ cells (d) in dissociated retinal cells from WT and rag2−/− mice (n > 100). (e) Density of γH2AX+ cells in whole-mount retinas. (f) Immunostaining for γH2AX, PCNA, and TUJ-1 in dissociated retinal cells from WT and rag2−/− animals. The percentage of γH2AX+ proliferative cells (PCNA+) and γH2AX+ neurons (TUJ-1+) are shown. Histograms depict the mean ± SEM values. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001 vs. corresponding controls. Individual values are depicted as circles (WT) and squares (rag2−/−). Scale bar, 5 µm in a and b.
RGC death is increased in the E13.5 mouse retina in the absence of RAG-2
We next examined the effect of RAG-2 deficiency on retinal neurogenesis, specifically during the early neural cell death phase, which affects recently born RGCs1,17. Compared with WT retinas, the number of apoptotic TUNEL+ cells was significantly higher in both whole-mount retinas (Fig. 3a–c) and dissociated cells (Fig. 3d) from rag2−/− mice, even though RAG-2 deficiency was associated with fewer γ-H2AX+ foci (Fig. 2). Double TUNEL and PCNA immunostaining of dissociated retinal cells revealed no significant differences in numbers of dying neuroepithelial cells between WT and rag2−/− mice (Fig. 3e,g). Conversely, TUNEL combined with specific Islet-1/2 immunostaining of RGCs at this embryonic stage revealed increased cell death in this population of young neurons in rag2−/− versus WT mice (Fig. 3f,h).

Neuronal cell death is increased in E13.5 rag2−/− retinas. (a,b,e,f) Apoptotic cells as visualized by TUNEL (green) in WT and rag2−/− mouse retinas. Density of TUNEL+ nuclei in whole-mount retinas (c) and percentage of TUNEL+ cells in dissociated retinal cells (d) are shown. (e,g) Dissociated retinal cells were immunostained for PCNA (red) and processed for TUNEL (green), and labelled cells were scored. (f,h) Apoptotic neurons were scored in whole-mount retinas immunostained for Islet 1/2 (red) and processed for TUNEL. Histograms show the mean ± SEM values. *P < 0.05, **P < 0.01 vs. corresponding controls. Individual values are depicted as circles (WT) and squares (rag2−/−). Scale bar, 50 µm in a and b, 12 µm in e and f.
Contrary to expectations, the neuronal cell death phenotype observed in RAG-2-deficient mice, in which the number of γ-H2AX+ foci is reduced versus WT (Fig. 2), was in line with that described in DNA-PK (SCID) and DNA polymerase μ (polμ−/−) DSB repair mutants, which accumulate γ-H2AX+ foci18,19. Based on this observation, we next sought to characterize other phenotypic consequences of RAG-2 deficiency.
RGC axonal growth is altered in RAG-2-deficient mice
RGC axonal growth is impaired in mice that lack repair DNA polymerase μ (Pol μ)19. We therefore investigated whether RAG-2 deficiency gives rise to a similar phenotype. Intraretinal axonal trajectories were visualized in E13.5 WT and rag2−/− mice by TUJ-1 immunostaining of retinal whole-mounts (Fig. 4a–d,f). WT retinas typically exhibited regular fasciculation, with axons converging at the optic nerve head (Fig. 4a,c). By contrast, rag2−/− mouse retinas showed alterations in axonal growth, including markedly disorganized fasciculation, reduced axonal density, and tangential axonal trajectories (Fig. 4b,d,f). According to the images of TUJ-1-immunostained retinas, fasciculation was classified as normal when ≥90% of the image area showed regular fasciculation; as mildly defective, when axonal density was reduced below 90%; or as severely defective, when besides a reduction in axonal density, tangential axonal trajectories were observed (quantified in e). Moreover, we also observe that midline crossing of optic fibres in E13.5 rag2−/− mice was delayed relative to WT counterparts (Supp. Fig. 1), but the distribution of cell adhesion molecules involved in axonal guidance, namely L1-CAM and Bravo, that were altered in the polμ−/− mutant mice19, were not affected (data not shown).

In vivo axonal navigation is altered in E13.5 rag2−/− mice. RGC axonal trajectories were visualized by TUJ-1 immunostaining in whole-mount retinas from E13.5 WT (a,c) and rag2−/− mice (b,d,f). The optic nerve head is indicated with an asterisk. According to the axonal trajectory observed, fasciculation was classified as normal (a-b), mildly defective (c,d), or severely defective (f). The histogram (e) depicts the proportion (%) and absolute number (indicated within each bar) of retinas for which each phenotype was observed. Scale bar, 50 µm in a-d and f.
To further characterize the axonal disturbances observed in the rag2−/− mouse retina, we performed 2D electrophoresis and protein identification in E15.5 retinal extracts from WT and rag2−/− mice. Significant alterations (p < 0.05; Supp. Fig. 2 and Table 1) in the protein levels of tubulin β3 and tubulin α1C, constituents of the axonal cytoskeleyon, fascin and platelet-activating factor acetylhydrolase 1, also implicated in axonal function, were observed in the mutant retinas supporting the view that RAG-2 deficiency affects axonal structure and function (Table 1).
Neurite outgrowth is dysregulated in dissociated retinal cell cultures from RAG-2-, Pol μ-deficient, and DNA-PK-mutant mice
Axonal growth and navigation is a complex process mediated by interactions between environmental cues and cell autonomous determinants40. Based on our previous observation of abnormal neurite outgrowth in dissociated polμ−/− retinal cells19, we examined neurite phenotype in rag2−/− retinal cells (Fig. 5). To minimize the influence of neighbouring cells on axonal growth, dissociated retinal cells from E13.5 WT and rag2−/− mice were cultured at low density in homogeneous, defined media. After culture for 18 hours the proportions of neurite-bearing cells among isolated TUJ-1+ cells were comparable in WT and rag2−/− mice (62.9 ± 6.1% and 58.1 ± 5.9% of total TUJ-1+ cells, respectively; n = 6 litters). However, neurite morphology differed between the groups; while over half of WT TUJ-1+ cells emitted straight unbranched neurites (53.7 ± 6.2% of labelled cells), the proportion of these cells was lower in rag2−/− TUJ-1+ cells (41.8 ± 10.3%). RAG-2-deficient neurons more frequently had morphologically aberrant phenotypes, including self-contacting neurites (Fig. 5a,c), as well as abrupt changes in neurite trajectory (Fig. 5b,d). Interestingly, the presence of aberrant and shorter neurites was common to cultured rag2−/−, polμ−/−, and SCID retinal neurons (Fig. 5e–g), although in the case of SCID retinal cells a significant difference relative to WT controls was observed only in neurite length (Fig. 5f). These data altogether define a cell-autonomous phenotype related to axonal growth and navigation that is common to rag2−/−, polμ−/−, and SCID mouse retinas, despite the opposite role of these proteins on DSB generation (RAG-2) and repair (Pol μ and DNA-PK).

Altered neurite growth in E13.5 rag2−/−, SCID, and polμ−/− cultured retinal cells. E13.5 dissociated retinal cells from the indicated mouse genotypes were cultured on surfaces pretreated with polyornithine and laminin. Neurons and their neurites were visualized by TUJ-1 immunostaining and classified based on trajectory as straight (e), self-contacting (a), or with abrupt changes in their directionality (b), and plotted by typology (c,d). Neurite length (e) was measured in retinal cells from rag2−/−, polμ−/−, and SCID mutant mice after culture for 18 hours and compared with that of WT counterparts (WT1-C57BL/6; WT2-Balb/C; WT3-C57BL/10) (n > 100 cells) (f). The total percentage of neurites exhibiting alterations in directionality was determined in retinal cultures from the 3 mutant mice and compared with that observed in corresponding WT controls (n > 100 cells) (g). Percentages in c, d and g refer to the number of TUJ-1+ cells emitting neurites. Histograms show the mean ± SEM. *P < 0.05, **P < 0.01, **P < 0.001 vs. corresponding controls. Individual values are depicted as circles (WT) and squares (mutant mice). Scale bar, 10 µm in a, b and e.
Together, our results support the involvement of the endonuclease RAG-2 in early retinal development, and suggest direct or indirect roles in DSB generation, the early phase of neural cell death, which affects RGC generation, and cell autonomous control of RGC axonal growth.
Discussion
Our previous studies have demonstrated that DSBs, as visualized by γ-H2AX immunostaining, are generated during the early stages of retinal development, and have characterized the impact of deficient DNA repair mechanisms on RGC survival and axonal growth in SCID and polμ−/− mutant mice18,19. In this study, we sought to characterize sources of DSBs and determine their physiological relevance.
Evidence suggests that LINE-1, retrotransposition of which gives rise to DSBs, may be involved in the generation of somatic mosaicism in the nervous system22,23,41,42. We found that relative amounts of LINE-1 DNA were modestly but significantly increased in the adult retina, but not during retinal neurogenesis, ruling out LINE-1 as a relevant source of the observed γ-H2AX+ foci18 in retina at E13.5. Widespread expression of the RAG-1 subunit in the nervous system, including the retina, had been previously described30,38, while nervous system expression of RAG-2 was poorly characterized34,35. Our analyses revealed the presence of RAG-2 protein in retina at E13.5, an observation suggestive of a physiological role in DSB generation. Our finding is compatible with the formation of RAG-1,2-complexes in the developing retina, a situation that has not been described in non-immune cell types30,33,43,44. In line with the demonstrated role of the RAG-1,2-complex in the immune system, we observed fewer γ-H2AX+ foci in RAG-2-deficient versus WT retinas. Our observations in the rag2−/− mouse retina provide for the first time clear evidence of a role of RAG-2 in retinal development.
Our previous studies confirmed that one direct consequence of deficient DSB repair is increased cell death, which in the developing retina particularly affects RGCs, the first neuronal cell type generated during retinal neurogenesis18,19. However, this causal relationship is called into question by our findings in RAG-2-deficient mice. In the E13.5 rag2−/− mouse retina we observed a decrease in γ-H2AX+ foci number that were compatible with reduced endonuclease activity, but, unexpectedly, increased numbers of dying RGCs. Furthermore, altered RGC axonal growth and navigation, the characteristic phenotype of DSB repair mutants, were also observed in RAG-2-deficient retinas, specifically alterations in axonal growth and intra- and extra-retinal axonal navigation, as well as cell-autonomous defects in neurite outgrowth. The selective RGC death in the rag2−/− mice, in contrast to the non-significantly affected population of proliferative neuroepithelial cells, might point to the existence of a still unknown selective mechanism active on RGCs, essential for their correct development and the generation of neuronal diversity, as has been previously proposed12.
There are several possible direct and indirect mechanisms that could link RAG-2 deficiency with defects in RGC development. Increased RGC death could affect axonal pathfinding, since some axonal guidance cues are exposed on the axonal surface of pioneer or surrounding neurons40,45. This hypothesis, which can also be applied to SCID and polμ−/− mutant mice, does not explain the cell-autonomous neurite phenotype observed in dissociated retinal-cell cultures from rag2−/− mice (Fig. 5). Although beyond the scope of this study, another potential explanation is that neural development of these mutants is impaired as a consequence of maternal immunodeficiency46,47 caused by the absence of RAG-2, DNA-PK, or Pol µ48,49,50.
The coupling of DNA endonuclease activity and DNA repair mechanisms to generate cellular diversity may not be exclusive to the immune system, and may be a feature of other biological processes requiring programmed DNA cleavage. It is tempting to speculate that RAG-2-mediated DSBs promote the acquisition of somatic alterations in recently born RGCs. This phenomenon, mediated by the RAG-1,2-complex and NHEJ repair mechanisms, is well established during lymphocyte generation and diversification37,51. The RAG-1,2-complex binds DNA in specific sequences, at canonical sites within immunoglobulin genes or lymphocyte receptor gene rearrangement sites (RSSs). However, cryptic, non-canonical RSSs are spread throughout the genome and, at least in lymphocytes, usually correspond to potential RAG-1,2 binding and cutting sites52, although off-target endonuclease activity in immune cells is generally scarce52,53. It should be noted that, according to the RSS-site database54, the 4 axonal proteins for which we observed altered expression in rag2−/− mice (Table 1) contain putative RSSs that can be recognized by the RAG-1,2-complex endonuclease, supporting a possible direct role of RAG-2 in axonogenesis and, maybe, axonal guidance. These observations, taken together with the alterations in retinal development observed in RAG-2-deficient mice and the somatic mosaicism described in the brain10,12, make it tempting to propose a role for RAG-2-mediated DNA breaks in the generation of neuronal mosaicism in the retina, and a role of mosaicism in proper neural connectivity. In line with this view, a recent study has proposed that DSBs may play important roles in brain physiology and disease, and research attention is beginning to focus on the mechanisms responsible for DSB generation and repair55.
In summary, our findings in the rag2−/− mouse underscore the importance of this component of the DNA cleavage mechanism in RGC viability and axonogenesis. These data, together with previous findings in DNA-repair-deficient mice, describe a possible scenario in which the RAG-1,2-complex mediates DSB generation and induces NHEJ repair during retinal development. Further studies employing single-neuron whole-genome sequencing will be required to establish a direct link between those processes and neuronal diversification, as already demonstrated in the immune system.
Materials and Methods
Mice
All experimental procedures in this paper were performed in accordance with European Union regulations for the use and treatment of animals in research (RD53/2013, BOE, Spain), and were approved by the CSIC bioethics committee for animal experimentation and the Dirección General de Medio Ambiente, Comunidad de Madrid. Male and female mouse embryos and adult tissues were used in all experiments. C57BL/10SnJrag2−/− mice (rag2−/−; Taconic, Hudson, NY, USA) were provided by Dr. J.A. García-Sanz (CIB) and Balb/C-JHAhsd-prkdc SCID (SCID) mice were obtained from Harlan (Gannat, France). DNA-polymerase-mu-deficient mice (polμ−/−) were kindly provided by Dr. Luis Blanco and backcrossed to a C57BL/6 J background as described in19. Three different wild type strains (WT) were used, according to the three different mutants respective genetic background: C57BL/10JCrl mice were obtained from Charles River (Ecully, France), while Balb/C JHA and C57BL6/J were both obtained from Harlan. WT and mutant mice were bred in local facilities and euthanized at the indicated ages.
LINE-1 levels in tissue
LINE-1 levels were determined by quantitative PCR of genomic DNA from liver and retina. SYBR-green qPCR assays were performed using 0.1 ng of genomic DNA and primers against 5 S DNA (normalizer gene) and LINE-1 orf2 (target gene). PCR was performed using the following conditions: 95 °C for 10 min followed by 40 cycles of 95 °C for 30 s, 60 °C for 30 s, and 72 °C for 1 min. The following primers were used: 5 S.2, 3′ GCCTACAGCACCCGG; 5 S.3, 5′ GTCTACGCCATACCACC; L1.1, 5′ TAAAGAACTCAAGAAGGTGG; L1.2, 3′ AGGGTTGTTTTGATTTGC. Data were analyzed using the ΔCt method relative to the normalizer gene56. LINE-1 levels in the different tissues were compared with those found in the adult liver.
Western blot
Individual adult tissues and embryonic retinas were processed as previously described19. Briefly, tissues or retinas were lysed, and 50 μg of total protein was resolved by PAGE and transferred to PVDF membranes. Membranes were blocked and incubated with antibodies against RAG-2 (1/1000, Abcam #ab133609; Cambridge, UK), and subsequently with a polyclonal goat anti-rabbit HRP secondary antibody (Dako Cytomation, Glostrup, Germany).
Immunostaining of dissociated cells
Freshly dissected retinas from E13.5 embryos were dissociated and the dissociated cells were fixed onto glass slides by cytospin as previously described18. Cytospin slides were incubated overnight at 4 °C with primary antibodies against γH2AX (1/1000, Ser 139, Abcam #ab22551); human β-III tubulin clone TUJ-1 (1/1000, PRB-435P; Covance, Paris, France); and PCNA [1/250, PCNA(C19) antibody, DB051; Delta Biolabs, Gilroy, CA, USA] and then for 1 h at room temperature (RT) with Alexa Fluor-conjugated secondary antibodies (1/250–1/500, depending on the primary antibody; Molecular Probes/Thermo Scientific, Rockford, IL, USA). Cell death was detected using the TUNEL technique (TdT-mediated dUTP nick end-labelling of fragmented DNA; Promega, Madison, WI, USA) according to manufacturer instructions, and combined with immunohistochemistry when necessary. Preparations were counterstained with DAPI and mounted with Fluoromount-G mounting medium (Southern Biotech, Birmingham, AL, USA). Scoring of 100–500 cells from non-adjacent fields was performed using a fluorescence microscope (Zeiss Axioplan, Oberkochen, Germany) coupled to a CCD camera (Leica, Wetzlar, Germany) or a confocal microscope (Leica TCS-SP5-A0BS) with a 40X objective.
Immunostaining and TUNEL analysis of whole-mount retinas
Freshly dissected E13.5 retinas were flat-mounted onto nitrocellulose membranes. Tissue fixation, permeation and antigen blocking were performed as previously described18. Retinas were immunostained with primary antibodies against γH2AX (1/1000), β-III tubulin (1/1000) or Islet-1/2 (1/200; Developmental Studies Hybridoma Bank #39.4D5, Iowa city, IA, USA), and then incubated for 1 h at RT with Alexa Fluor-conjugated secondary antibodies. The retina preparations were mounted with Fluoromount-G mounting medium and analyzed by confocal microscopy with a 40X objective. Cell death was detected using the TUNEL technique, combined with immunohistochemistry when necessary. Samples were counterstained with DAPI, and the density of TUNEL-positive cells was determined under a fluorescence microscope using a 100X objective. In all other cases, cell density was determined using FIJI software by calculating the number of cells present in 4 confocal images (40X) per retina, and normalizing according to the size of the imaged area.
Primary dissociated neuroretina cell culture
Neuroretinas were dissected from E13.5 embryos, pooled per litter, and dissociated as previously described18,19. Dissociated cells from 3 different litters were plated in DMEM/F12 medium (Gibco, Life Technologies, Rockford, IL, USA) with N2 supplement at a density of 85,000 cells/cm2 in 2-cm2 Permanox chamber slides (NUNC, Thermo Fisher Scientific, Waltham, MA, USA) pre-treated with 0.5 mg/mL polyornithine (Sigma-Aldrich, San Luis, MI, USA) and 1 μg/mL laminin (Sigma-Aldrich), as previously described19. After incubation for 18–24 h at 37 °C and 5% CO2, cells were fixed for 20 min with 4% (w/v) paraformaldehyde (Sigma-Aldrich) in 0.1 M phosphate buffer (PB) at pH 7.4 and RT, and were processed for immunostaining as described above for dissociated cells.
Anterograde DiI staining
E13.5 embryos were euthanized, the heads dissected out, and the lens removed from the eye cups, prior to fixation as previously described20. The optic nerve head was covered with Neurotrace DiI lipophilic marker (Molecular Probes) and, to ensure diffusion of the dye along the entire optic nerve, the heads were maintained in PB at 37 °C for 2 weeks. Optic nerve length and decussation at the optic chiasma were evaluated as previously described19.
Two-dimensional gel electrophoresis and protein identification
For two-dimensional gel electrophoresis analysis, 25 μg of total protein from E13.5 mouse retina was loaded on ReadyStrip IPG strips for isoelectric focusing (linear 7-cm strips, pH 3–10; Bio-Rad, Hercules, CA, USA), following the manufacturer´s instructions. The strips were then equilibrated, applied to 12% SDS gels, and stained with SYPRO Ruby (Bio-Rad) following the manufacturer´s instructions. An EXQuest Spot Cutter was used for image acquisition and spot picking. The images were analyzed with PDQuestTM 2D Analysis 7.4 Software (Bio-Rad) as previously described57.
MALDI peptide mass fingerprinting, MS/MS analysis and database searching
Spots of interest were manually excised and processed automatically in a Proteineer DP (Bruker Daltonics, Bremen, Germany). The sample was digested and prepared as previously described58, with minor modifications59. For MALDI-TOF/TOF analysis, samples were automatically acquired in an ABi 4800 MALDI TOF/TOF mass spectrometer (Applied Biosystems, Framingham, MA, USA) in positive ion reflector mode (ion acceleration voltage, 25 kV for MS acquisition and 1 kV for MS/MS). PMF and MS/MS fragment ion spectra were smoothed and corrected to zero baseline using routines embedded in ABi 4000 Series Explorer Software v3.6. Each PMF spectrum was internally calibrated with the mass signals of trypsin autolysis ions to reach a typical mass measurement accuracy of <25 ppm. GPS Explorer v4.9 was used to submit the combined PMF and MS/MS data to MASCOT software v.2.1 (Matrix Science, London, UK), searching in the non-redundant NCBI protein database. The mascot total score is −10*Log(P), where P is the probability that the observed match is a random event. The percentage of sequence coverage was calculated as the ratio of amino acids (number identified in peptides / number of theoretical peptides from sequence data).
Statistical analysis
Animals from at least 3 independent litters were used for each experiment, unless indicated. All experiments were conducted at least twice. Data size was estimated in accordance with previous literature to fulfil the 3 R principles for animal experimentation. Bars in graphs represent the mean and the standard error of the mean, and data points represent individual mice or pools from a given litter, as indicated. In all cases, data were checked for normality using D’Agostino-Pearson omnibus and Shapiro-Wilk normality tests sequentially, and considered to fit a normal distribution only if they passed both tests. For normal data, homoscedasticity was determined using Fisher’s test. Normal data were compared using an unpaired Student’s T-test, applying Welch’s correction in cases of non-homoscedasticity. Non-normal samples were compared using the Mann Whitney nonparametric U-test. All analyses were performed at a fixed 95% confidence interval and outliers were excluded from further analysis according to Grubbs’ outlier test. Data analyses were performed using GraphPad Prism version 5.01 for Windows (GraphPad Software, San Diego, CA, USA). Statistically significant differences are indicated as follows: *p < 0.05; **p < 0.01; ***p < 0.001.
References
Chavarria, T., Baleriola, J., Mayordomo, R., de Pablo, F. & de la Rosa, E. J. Early neural cell death is an extensive, dynamic process in the embryonic chick and mouse retina. ScientificWorldJournal 2013, 627240 (2013).
Yeo, W. & Gautier, J. Early neural cell death: dying to become neurons. Dev Biol 274, 233–244 (2004).
Boya, P. & de la Rosa, E. J. Cell death in early neural life. Birth Defects Res C Embryo Today 75, 281–293 (2005).
de la Rosa, E. J. & de Pablo, F. Cell death in early neural development: beyond the neurotrophic theory. Trends Neurosci 23, 454–458 (2000).
Dekkers, M. P., Nikoletopoulou, V. & Barde, Y. A. Cell biology in neuroscience: Death of developing neurons: new insights and implications for connectivity. J Cell Biol 203, 385–393 (2013).
Gatz, S. A. et al. Requirement for DNA ligase IV during embryonic neuronal development. J Neurosci 31, 10088–10100 (2011).
El Waly, B., Buhler, E., Haddad, M. R. & Villard, L. Nhej1 Deficiency Causes Abnormal Development of the Cerebral Cortex. Mol Neurobiol (2014).
McKinnon, P. J. & Caldecott, K. W. DNA strand break repair and human genetic disease. Annu Rev Genomics Hum Genet 8, 37–55 (2007).
McKinnon, P. J. Maintaining genome stability in the nervous system. Nat Neurosci 16, 1523–1529 (2013).
Paquola, A. C. M., Erwin, J. A. & Gage, F. H. Insights into the role of somatic mosaicism in the brain. Curr Opin Syst Biol 1, 90–94 (2017).
Evrony, G. D., Lee, E., Park, P. J. & Walsh, C. A. Resolving rates of mutation in the brain using single-neuron genomics. Elife, 5 (2016).
Weissman, I. L. & Gage, F. H. A Mechanism for Somatic Brain Mosaicism. Cell 164, 593–595 (2016).
Barzilai, A. & McKinnon, P. J. Genome maintenance in the nervous system; insight into the role of the DNA damage response in brain development and disease. DNA Repair (Amst) 12, 541–542 (2013).
Frappart, P. O. & McKinnon, P. J. Mouse models of DNA double-strand break repair and neurological disease. DNA Repair (Amst) 7, 1051–1060 (2008).
Jeppesen, D. K., Bohr, V. A. & Stevnsner, T. DNA repair deficiency in neurodegeneration. Prog Neurobiol 94, 166–200 (2011).
Mason, C. The development of developmental neuroscience. J Neurosci 29, 12735–12747 (2009).
Valenciano, A. I., Boya, P. & de la Rosa, E. J. Early neural cell death: numbers and cues from the developing neuroretina. Int J Dev Biol 53, 1515–1528 (2009).
Baleriola, J., Suarez, T. & de la Rosa, E. J. DNA-PK promotes the survival of young neurons in the embryonic mouse retina. Cell Death Differ 17, 1697–1706 (2010).
Baleriola, J. et al. Increased neuronal death and disturbed axonal growth in the Polmu-deficient mouse embryonic retina. Sci Rep 6, 25928 (2016).
Karanjawala, Z. E., Hinton, D. R., Oh, E., Hsieh, C. L. & Lieber, M. R. Developmental retinal apoptosis in Ku86−/− mice. DNA Repair (Amst) 2, 1429–1434 (2003).
Hoeijmakers, J. H. DNA damage, aging, and cancer. N Engl J Med 361, 1475–1485 (2009).
Coufal, N. G. et al. L1 retrotransposition in human neural progenitor cells. Nature 460, 1127–1131 (2009).
Thomas, C. A., Paquola, A. C. & Muotri, A. R. LINE-1 retrotransposition in the nervous system. Annu Rev Cell Dev Biol 28, 555–573 (2012).
Evrony, G. D. et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell 151, 483–496 (2012).
Evrony, G. D. et al. Cell lineage analysis in human brain using endogenous retroelements. Neuron 85, 49–59 (2015).
Madabhushi, R. et al. Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell 161, 1592–1605 (2015).
Arya, R. & Bassing, C. H. V(D)J Recombination Exploits DNA Damage Responses to Promote Immunity. Trends Genet 33, 479–489 (2017).
Helmink, B. A. & Sleckman, B. P. The response to and repair of RAG-mediated DNA double-strand breaks. Annu Rev Immunol 30, 175–202 (2012).
Gigi, V. et al. RAG2 mutants alter DSB repair pathway choice in vivo and illuminate the nature of ‘alternative NHEJ’. Nucleic Acids Res 42, 6352–6364 (2014).
Chun, J. J., Schatz, D. G., Oettinger, M. A., Jaenisch, R. & Baltimore, D. The recombination activating gene-1 (RAG-1) transcript is present in the murine central nervous system. Cell 64, 189–200 (1991).
McGowan, P. O., Hope, T. A., Meck, W. H., Kelsoe, G. & Williams, C. L. Impaired social recognition memory in recombination activating gene 1-deficient mice. Brain Res 1383, 187–195 (2011).
Rattazzi, L., Cariboni, A., Poojara, R., Shoenfeld, Y. & D’Acquisto, F. Impaired sense of smell and altered olfactory system in RAG-1(–) immunodeficient mice. Front Neurosci 9, 318 (2015).
Jessen, J. R., Jessen, T. N., Vogel, S. S. & Lin, S. Concurrent expression of recombination activating genes 1 and 2 in zebrafish olfactory sensory neurons. Genesis 29, 156–162 (2001).
Laszkiewicz, A. et al. Bidirectional activity of the NWC promoter is responsible for RAG-2 transcription in non-lymphoid cells. PLoS One 7, e44807 (2012).
Niu, N. et al. Expression and distribution of immunoglobulin G and its receptors in an immune privileged site: the eye. Cell Mol Life Sci 68, 2481–2492 (2011).
Muller, B., Ellinwood, N. M., Lorenz, B. & Stieger, K. Detection of DNA Double Strand Breaks by gammaH2AX Does Not Result in 53bp1 Recruitment in Mouse Retinal Tissues. Front Neurosci 12, 286 (2018).
Nishana, M. & Raghavan, S. C. Role of recombination activating genes in the generation of antigen receptor diversity and beyond. Immunology 137, 271–281 (2012).
Hirano, T., Murata, T. & Hayashi, T. Physiological significance of recombination-activating gene 1 in neuronal death, especially optic neuropathy. FEBS J 282, 129–141 (2015).
Sharma, A., Singh, K. & Almasan, A. Histone H2AX phosphorylation: a marker for DNA damage. Methods Mol Biol 920, 613–626 (2012).
Erskine, L. & Herrera, E. The retinal ganglion cell axon’s journey: insights into molecular mechanisms of axon guidanc. e. Dev Biol 308, 1–14 (2007).
Kano, H. et al. L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev 23, 1303–1312 (2009).
Muotri, A. R. et al. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435, 903–910 (2005).
Eggan, K. et al. Mice cloned from olfactory sensory neurons. Nature 428, 44–49 (2004).
Li, J., Ishii, T., Feinstein, P. & Mombaerts, P. Odorant receptor gene choice is reset by nuclear transfer from mouse olfactory sensory neurons. Nature 428, 393–399 (2004).
Pittman, A. J., Law, M. Y. & Chien, C. B. Pathfinding in a large vertebrate axon tract: isotypic interactions guide retinotectal axons at multiple choice points. Development 135, 2865–2871 (2008).
Weiner, J. A. & Chun, J. Maternally derived immunoglobulin light chain is present in the fetal mammalian CNS. J Neurosci 17, 3148–3156 (1997).
Singer, H. S. et al. Prenatal exposure to antibodies from mothers of children with autism produces neurobehavioral alterations: A pregnant dam mouse model. J Neuroimmunol 211, 39–48 (2009).
Shinkai, Y. et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell 68, 855–867 (1992).
Taccioli, G. E. et al. Targeted disruption of the catalytic subunit of the DNA-PK gene in mice confers severe combined immunodeficiency and radiosensitivity. Immunity 9, 355–366 (1998).
Bertocci, B., De Smet, A., Berek, C., Weill, J. C. & Reynaud, C. A. Immunoglobulin kappa light chain gene rearrangement is impaired in mice deficient for DNA polymerase mu. Immunity 19, 203–211 (2003).
Lieber, M. R. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 79, 181–211 (2010).
Teng, G. et al. RAG Represents a Widespread Threat to the Lymphocyte Genome. Cell 162, 751–765 (2015).
Canela, A. et al. DNA Breaks and End Resection Measured Genome-wide by End Sequencing. Mol Cell 63, 898–911 (2016).
Merelli, I. et al. RSSsite: a reference database and prediction tool for the identification of cryptic Recombination Signal Sequences in human and murine genomes. Nucleic Acids Res 38, W262–267 (2010).
Alt, F. W. & Schwer, B. DNA double-strand breaks as drivers of neural genomic change, function, and disease. DNA Repair (Amst) (2018).
Schmittgen, T. D. & Livak, K. J. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3, 1101–1108 (2008).
Salvachua, D. et al. Differential proteomic analysis of the secretome of Irpex lacteus and other white-rot fungi during wheat straw pretreatment. Biotechnol Biofuels 6, 115 (2013).
Shevchenko, A., Wilm, M., Vorm, O. & Mann, M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Anal Chem 68, 850–858 (1996).
Mohedano Mde, L. et al. A partial proteome reference map of the wine lactic acid bacterium Oenococcus oeni ATCC BAA-1163. Open Biol 4, 130154 (2014).
Adams, J. C. Roles of fascin in cell adhesion and motility. Curr Opin Cell Biol 16, 590–596 (2004).
Keays, D. A. et al. Mutations in alpha-tubulin cause abnormal neuronal migration in mice and lissencephaly in humans. Cell 128, 45–57 (2007).
Poirier, K. et al. Mutations in the neuronal ss-tubulin subunit TUBB3 result in malformation of cortical development and neuronal migration defects. Hum Mol Genet 19, 4462–4473 (2010).
Tischfield, M. A. et al. Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140, 74–87 (2010).
Sasaki, S. et al. A LIS1/NUDEL/cytoplasmic dynein heavy chain complex in the developing and adult nervous system. Neuron 28, 681–696 (2000).
Hirotsune, S. et al. Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet 19, 333–339 (1998).
Acknowledgements
This work was supported by the Ministerio de Economía y Competitividad, Spain (Grants SAF2013-41059-R and SAF2016-75681R to EJdlR). We thank Drs. Owen Howard and Carolina Isiegas for critical reading of the manuscript. We thank María Donina Hernández-Fuentes and the staff of the CIB animal house, microscopy and proteomics facilities for technical support. Islet-1/2 monoclonal antibody, developed by Thomas M. Jessell and Susan Brenner-Morton, was obtained from the Developmental Studies Hybridoma Bank, developed under the auspices of the NICHD and maintained by the Department of Biology, University of Iowa, Iowa City, IA 52242.
Author information
Authors and Affiliations
Contributions
J.B. performed the initial analysis of the rag 2−/− mutant mice and provided data to all figures. N.A.L. finished the experimentation, analyzed the result, prepared the final figures and wrote an initial version of the manuscript. V.d.l.R. performed the proteomic analysis. T.S. and E.J.d.l.R. supervised the experimentation and data analysis, and wrote the final manuscript. E.J.d.l.R. elaborated the project for grant applications.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Álvarez-Lindo, N., Baleriola, J., de los Ríos, V. et al. RAG-2 deficiency results in fewer phosphorylated histone H2AX foci, but increased retinal ganglion cell death and altered axonal growth. Sci Rep 9, 18486 (2019). https://doi.org/10.1038/s41598-019-54873-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-54873-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
