
Abstract
Target leaf spot (TLS) of sorghum, a foliar disease caused by the necrotrophic fungus Bipolaris cookei (also known as Bipolaris sorghicola), can affect grain yield in sorghum by causing premature drying of leaves and defoliation. Two sorghum recombinant inbred line (RIL) populations, BTx623/BTx642 and BTx623/SC155-14E, were assessed for TLS resistance in replicated trials. Using least square mean trait data, four TLS resistance QTL were identified, two in each population. Of these, three were previously unidentified while a major QTL on chromosome 5 in the BTx623/BTx642 RIL population corresponded to the previously identified TLS resistance gene ds1. A set of sorghum lines were assessed for production of reactive oxygen species induced by treatment with the microbe-associated molecular pattern (MAMP) flg22 (a derivative of flagellin). Flg22-induced ROS production varied between lines in a consistent fashion. One QTL associated with variation in the flg22 response was detected in the RIL populations. No evidence was found to link variation in the MAMP response to variation in TLS resistance
Similar content being viewed by others
Introduction
Target leaf spot (TLS) of sorghum, a foliar disease caused by the necrotrophic fungus Bipolaris cookei (also known as Bipolaris sorghicola), can affect grain yield in sorghum by causing premature drying of leaves and defoliation. TLS is a significant disease of sorghum in the southeastern US and the lower Mississippi river valley1 and has been reported worldwide including India2, Japan3, and the United States4.
Few studies have been published on the genetics of TLS resistance in sorghum. A major recessive TLS resistance gene, ds1, on chromosome 5 appears to be caused by a loss-of-function or loss-of-expression allele of a gene encoding a putative plasma membrane receptor with leucine-rich repeat and serine-threonine kinase domains3. Mohan et al.5 identified quantitative trait loci (QTL) associated with resistance to TLS on chromosomes 3 and 6. The chromosome 6 QTL was associated with resistance to two other diseases, zonate leaf spot and drechslera leaf blight, in addition to TLS6. To our knowledge, these are the only three loci that to have been associated with TLS resistance.
Plants recognize pathogens using a suite of receptors located on the plasma membrane and in the cytoplasm. Plasma membrane-bound receptors, known as pattern recognition receptors (PRRs) generally recognize microbe-associated molecular patterns (MAMPs, also known as pathogen-associated molecular patterns or PAMPs), highly conserved molecules generally found in large classes of microbes7. The archetypal MAMP is flg22, a 22-amino acid epitope derived from bacterial flagellin8,9. Other well-studied MAMPs include epitopes of chitin, lipopolysaccharides (LPSs), peptidoglycans (PGNs), and translation elongation factor Tu (EF-Tu)10. MAMP recognition by PRRs leads to a relatively low-level defense response termed the MAMP response or the MAMP-triggered immunity (MTI) response. In cases in which pathogens are specifically adapted to the host plant, pathogen derived proteins known as effectors are often introduced into the cytoplasm or apoplast. Some effectors can suppress the MTI response while others play various roles in enhancing pathogenesis11. Cytoplasmic resistance proteins (R-proteins) recognize effectors either indirectly or via direct binding, leading to a relatively strong defense response termed the effector-triggered immunity (ETI) response. The ETI and MTI responses are qualitatively similar though ETI is stronger. Both can include phenomena such as cell wall reinforcement by callose deposition, changes in ion flux across the plasma membrane, changes in phytohormone concentrations, induction or repression of plant defense-related genes, and production of reactive oxygen species (ROS) and nitric oxide (NO)12. ETI often (though not always) includes a so-called hypersensitive response (HR) – a rapid, localized cell death at the point of pathogen penetration13.
In some necrotrophic systems, necrotrophic effectors, also known as host-specific toxins, cause the tissue necrosis specifically in plants carrying dominant susceptibility (S-) genes14. Most of the S-genes that have been identified encode proteins similar to R-genes or, in some cases, PRRs15,16,17,18. It seems that in these cases the pathogen may induce the host to trigger HR which will result in dead cells on which the necrotrophic pathogen can grow. It is not clear how many necrotrophic pathogens use this strategy beyond the four or five systems that have been characterized already19 but new cases are being discovered regularly. There are several reasons to suspect that the TLS resistance gene ds1 may be an example of an S-gene that is triggered by a host-specific toxin; susceptibility is dominant, the structure of ds1 resembles a PRR and the causal pathogen, B. cookei, is a necrotroph which is closely related to several pathogens that produce host-specific toxins20. The host-specific toxin, if it exists, has not been identified in this case.
Quantitative trait loci (QTL) controlling the MTI response have been documented in a number of plant species including Arabidopsis thaliana21,22, maize23, soybean24, and tomato25. At the same time, it has become increasingly apparent that quantitation of the MTI response is complex. Correlations in populations between responses to different MAMPs has been reported to be low or non-existent in some cases21,25,26 though Zhang et al.23 reported a strong correlation between responses to flg22 and chitin in a maize mapping population. Moreover, the MTI response can be quantified in various ways using various assays including measuring ROS or NO production; MAP kinase phosphorylation, mRNA levels of specific MAMP-induced genes, lignin and cell wall-bound phenols, seedling growth inhibition and MAMP-induced resistance to pathogens26,27,28. Relative rankings of lines can vary substantially depending on the assay used23,27. In other words, quantification of the MTI response is not straightforward and conclusions may vary significantly depending on how it is elicited and how it is measured.
The fact that several genes resembling PRRs confer quantitative resistance in various plant species29 and that the strength of flg22 perception is negatively correlated with susceptibility to Pseudomonas syringae22 suggest some connection between variation in the MTI response and quantitative disease resistance. However, the relationship between these traits is not well understood, especially in crop plants as opposed to model systems such as Arabidopsis. The objectives of this study were to identify QTL for TLS resistance and the MAMP response and then to investigate the relationship between the MAMP response in sorghum and target leaf spot disease resistance.
Results and Discussion
Linkage map construction
A linkage map was generated for the BTx623/BTx642 RIL population using 1020 SNP markers. The map spanned a length of 4,689 cM or 632,560,609 Mb with 12 linkage groups representing the 10 chromosomes of sorghum and covering approximately 92% of the genome (File S1). Chromosome arms for 3 and 10 could not be joined into one linkage group resulting in linkage groups 3A, 3B, 10A, and 10B. The map contained an average marker coverage of one marker per 5.2 cM or 654,684 Mb (Table S2). A previously-available linkage map for the BTx623/SC155-14E RIL population was used (Table S2, File S1)30.
Field evaluation of TLS resistance
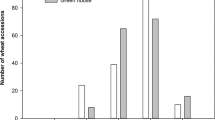
Two RIL populations, BTx623/SC155-14E and BTx623/BTx642, were selected for study based on variation of the parental lines for both TLS and MAMP response (see below). Both populations were assessed in 2016 and 2017 in randomized complete blocks with 2 full replications per year. For both populations, significant differences were identified for genotype, year of collection, replication for each experiment within each year, scoring date within each year as well as the genotype by replication and genotype by year interactions (Table 1). For the BTx623/SC155-14E RIL population, The TLS ratings of the parents, BTx623 and SC155-14E, were similar (Average of 6.07 and 5.77 respectively, Table 2 and Fig. 1). Nevertheless significant transgressive segregation was observed, RIL average values ranging from 4.1 to 6.8 (Table 2, Fig. 1) and the correlation between the average line scores for 2016 and 2017 was 0.38 (P < 0.0001, Table 3). For the BTx623/BTx642 RIL population, more divergent TLS scores were observed for the parents, with average scores 5.84 and 7.17 for BTx623 and BTx642 respectively (Table 2, Fig. 1). The range of scores for the RILs was likewise broader than for the BTx623/SC155-14E RIL population ranging on average from 4.38 to 7.75, again showing significant transgressive segregation. The correlation between the average line scores for 2016 and 2017 was 0.74 (P < 0.0001, Table 3).

Distribution of TLS resistance LS mean scores in the (A) BTx623/SC155–14E and (B) BTx623/BTx642 populations. Parental values are indicated.
TLS QTL mapping
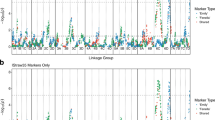
QTL for TLS severity were detected in both populations. QTL analysis was performed with phenotypic data from individual environments and also with line averages across environments (Table 4). Permutation testing estimated a genome-wide significance threshold of 2.5 for TLS scores.
In the BTx623/SC155-14E RIL population a TLS resistance QTL was detected on chromosome (chr.) 3 at around 10.6 cM in both environments and across environments. An additional TLS QTL on chr. 4 at 103.4 cM was detected in the 2017 and across environments (Table 4). The SC155-14E and BTx623 contributed the resistance alleles on chromosomes 3 and 4 respectively, which does provide explanation of the transgressive variation observed in this population.
In the BTx623/BTx642 RIL population, a major TLS QTL on chr. 5 was detected in both 2016 and 2017 using average values across environments, explaining 71.1% of the variation across environments (Table 4). Additional QTL were identified; one on chr. 9 was identified in 2017 across environments and one on chr. 6 was detected in 2016. For the QTL detected across environments, BTx642 contributed the resistance allele on chr. 5 and BTx623 contributed the resistance allele on chr.9.
Considering just the TLS QTL detected across environments, the chr. 5 QTL detected in the BTx623/BTx642 RIL population maps precisely to the position of the major TLS resistance gene ds13. Combined with its large effect the fact that it accounts for a large majority of the variation observed, this suggests that BTx642 carries the recessive ds1 resistance gene and that BTx623 does not. Since no QTL is detected at this locus in the BTx623/SC155-14E population, this also implies that SC155-14E also carries the dominant susceptibility allele at this locus. A TLS QTL was detected on chr. 3 in a previous study5 but this was not close to the QTL mapped here.
MAMP response evaluation
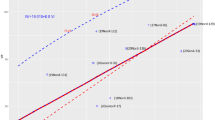
To initially assess variation within available germplasm, ROS production of sixteen sorghum parental lines (Table S1) was assessed in response to two MAMP treatments, flg22 and chitin. Variable responses between lines were identified for both flg22 and chitin treatments however, chitin responses were inconsistent between replications, so only results of the flg22 response are reported here (Fig. 2). It is not clear why the chitin response data were inconsistent between replications since we had achieved relatively consistent results using a very similar assay in maize23. While every effort was made to standardize conditions between replications, it is possible that there were sources of variation that were not accounted for. Lines BTx378, BTx642, and SC155-14E, and SC372 were the highest responders to flg22 and lines Az9594, BTx3197, BTx623, DL1366, Rio, and SC748-5 were among the lowest responders (Fig. 2).

flg22-induced ROS production in various sorghum lines.
We assessed both sorghum RIL populations for flg22-induced ROS production response. Substantial variation in levels of ROS production was observed for both RIL populations (Fig. 3). Significant effects were identified for genotype, run, and the interaction between genotype and run in both populations (Table 1). The correlations between runs were 0.52 and 0.31 for the BTx623/SC155-14E and BTx623/BTx642 populations respectively (both P < 0.0001). Other parameters for the flg22 response in the two population are shown in Table 1. For the average flg22 response values, one QTL on chr. 6 was identified In the BTx623/SC155-14E RIL population and none in the BTx623/BTx642 population (Table 4). Permutation testing estimated a genome-wide significance threshold of 2.3 for flg22 response scores.

Distribution of total ROS production (RLU) in response to flg22 in (A) BTx623/SC155–14E RIL population and (B) BTx623/BTx642 RIL population.
Comparison of TLS and MAMP response data
There was no significant correlation between TLS resistance scores and flg22-induced ROS response scores in either population (Table 3), nor was there any correspondence between QTL identified for the two traits. It seems therefore that the loci and genes controlling variation in these two traits are entirely distinct. Arguably this is not surprising as flg22 is a bacterial MAMP while TLS is caused by a fungus. It was unfortunate in this respect that we were not able to detect a consistent response to chitin. As discussed in the introduction, Zhang et al.23 found a strong correlation between responses to flg22 and chitin in a maize mapping population. Nevertheless, no correspondence to response to fungal disease was detected in this previous study either.
Also as discussed above, the MAMP response can be measured in a number of different ways and these different methods give substantially different results23,27. It is therefore premature to say that variation in the strength of the MAMP response did not affect levels of disease resistance in our experiments. However we were not able to generate any evidence to support this hypothesis.
In conclusion, this study detected several previously unidentified QTL for TLS resistance. It also reports variation between sorghum lines in the flg22-induced ROS response and identifies one QTL associated with variation in this response. No evidence was found to link variation in the MAMP response to variation in TLS resistance
Materials and Methods
Plant materials
All sorghum lines were provided by Dr. William Rooney (Texas A&M University,) and Dr. Stephen Kresovich (Clemson University). These consisted of 17 diverse lines that were parents of several mapping populations and two recombinant Inbred Line (RIL) populations (Table S1). The two RIL populations were both developed at Texas A&M and were at the F5:6 stage. BTx623 – the standard sorghum line used for genetics31- was a parent of both populations, the other parents being BTx642 and SC155-14E. BTx623, is a TLS susceptible elite line3, SC155-14E, is a line developed for anthracnose resistance30 and BTx642, is a derivative of the stay-green conversion line SC35 developed by Rosenow et al.32. The BTx623/SC155-14E and BTx623/BTx642 populations consisted of 103 and 149 lines respectively.
MAMP Response Testing
Two MAMPs, flg22 and chitin, were used in this study. Flg22 is a peptide corresponding to a conserved domain of bacterial flagellin and was ordered from Genscript (catalog# RP19986). The 20 X chitin mixture is made by adding 20 mg of chitin powder (Sigma-Aldrich, catalog# C3641) to 10 ml ddH2O, vortexing for 30 s, letting settle for 10 min, vortexing again for 30 sec and letting settle for 10 min Any undissolved chitin settled to the bottom of the tube and the clear supernatant above was used for the experiment.
ROS assays were carried out according to Zhang et al.23 with modification. One leaf disc (diameter 5.5 mm) from four 15 day-old sorghum plants of each genotype were excised from and floated on 50 µl ddH20 in a white 96-well plate (Fisher Scientific, catalog# 353296) overnight in dark. Immediately before testing, 50 µl of 2X reaction solution carrying 1 µl of 5mM L-012 (a chemiluminescence probe that reacts with superoxide anions and produces luminescence at long wavelengths; ordered from Wako Pure Chemical Industries, Ltd. catalog# 120-04891) in ddH2O, 1 µl of 2 mg/ml horseradish peroxidase (Sigma-Aldrich catalog# P6782), and 1 µl of 100 µM flg22 or 5 µl of the chitin mixture, was added to the 96-well plate. Immediately after treatment, the chemiluminescent signal from each well was recorded for 30 min using a Photek CCD camera 216 (Photek ltd., East Sussex, U.K).
To assess the variation of the MAMP response within individual genotypes, each genotype was assessed in sixteen wells of each 96-well plate. Eight wells consisted of a mock treatment (without MAMPs) and eight wells consisted of treatment (with MAMPs). The parents of the populations, BTx623, SC155-14E, and BTx642, were always included as repeated checks on each 96-well plate while we test the ROS product of the RILs populations. Eight biological replicates of responses to flg22 and chitin mixture were measured in every case with genotypes re-randomized in each of the two runs.
Inoculum preparation and inoculation procedure
Inoculum was prepared essentially as described previously33. Sorghum grain was soaked from 3 to 4 days in water, placed in 1 L flasks, and autoclaved for one hour (120 PSI and 121 °C). Autoclaved grain was inoculated with Bipolaris cookei isolate LSLP18 received from Dr. Burt Bluhm (University of Arkansas). The fungus grew at room temperature (23–25 °C) for approximately 14 days until the sorghum was colonized with the fungus. The sorghum was air-dried and stored at 4 °C. The dried infested sorghum was used to inoculate the entire trial. Sorghum plants were inoculated 30–40 days after planting by adding 6 to 10 infested sorghum kernels placed into the whorl of each plant.
Field evaluation of target leaf Spot resistance
RIL populations along with parental checks were planted at the Central Crops Research Station in Clayton, NC on May 15 2016 and June 14–15 2017 in a randomized complete block design with two experimental replications. Experiments were planted in 1.8 m single rows with a 0.9 m row width using 10 seeds per plot. Two border rows were planted around the periphery of each experiment to prevent edge effects. Rows were inoculated on July 1 2016 and July 15 2017 of each plant at sorghum growth stage 3. Visual disease ratings were taken two times at ~14-day intervals starting three weeks after inoculation. Ratings were taken visually on a one to nine percentage scale with 1 = a dead plant; 2 = > 90% of leaf area showing lesions; 3 = > 75%; 4 = > 60%; 5 = > 45%; 6 = > 30%; 7 = > 15%; 8 = > 5%; and 9 = no visible symptoms on leaves.
Phenotypic data analysis
Data was analyzed with SAS software v9.434. An analysis of variance (ANOVA) and least square (LS) means were generated using the GLM procedure for target leaf spot severity and ROS response to flg22 treatment. Mock treatment values for each line were subtracted from the mean of each line response before analysis as a normalization technique. Standard F-tests in all analyses were used to determine significance of main effects and interactions. In the model, replications and environments were considered random, while the main effects were considered fixed. Individual hypothesis testing of effects and interactions was designated using the appropriate error term in the GLM procedure. Pearson correlation coefficients were calculated for TLS severity and ROS response to flg22 and chitin treatments using the CORR procedure in SAS.
Genotypic analysis and linkage mapping
Genotypic data and a genetic linkage map for the BTx623/SC155-14E RIL population was provided by Dr. Patricia Klein in the Department of Horticultural Sciences and Institute for Plant Genomics and Biotechnology at Texas A&M University, College Station, TX and has been previously published30. Genotypic data for the BTx623/BTx642 RIL population were generated using Illumina sequencing of reduced-representation of genomic libraries as described previously35. Briefly, DNA was extracted using a modified CTAB protocol, quantified using PicoGreen, double-digested with PstI-HF and HinP1I, and ligated to barcoded adapters. Individual libraries were then pooled, bead-cleaned, PCR amplified, and bead-cleaned again. Average size and concentration of pooled libraries were estimated using a DNA7500 chip on an Agilent Bioanalyzer, and libraries were sequenced on an Illumina HiSeq. 2000 instrument. SNP calling was performed using the TASSEL5 GBS pipeline36 and missing data were imputed using Beagle4 (Browning and Browning 2007). To create the linkage map for the BTx623/BTx642 population a χ² test was performed for each marker to test for segregation distortion. Markers showing the appropriate Mendelian segregation ratios for individual markers were used to construct a genetic linkage map using Joinmap® 4.0 software37. Linkage groups were assigned using the LOD score of 5.0. Map distances were calculated using the Kosambi mapping function38. Summary statistics for each map are shown in Table S2 and the actual map positions of each marker are provided in File S1.
QTL Mapping
The composite interval mapping (CIM) option in Windows QTL cartographer 2.539 was used to identify QTL, estimate LOD scores, and measure the proportion of phenotypic variation explained by individual QTL for individual environments as well as across environments. Default settings for CIM were used with the standard model, a control marker number set to 5, a window-size of 10, and a walk speed (cM) of 2. A permutation test with 500 iterations was conducted to establish a P < 0.05 threshold for the detection of putative QTL (Churchill and Doerge 1994).
References
Zaccaron, A. Z. & Bluhm, B. H. The genome sequence of Bipolaris cookei reveals mechanisms of pathogenesis underlying target leaf spot of sorghum. Sci. Rep. 7, 17217 (2017).
Kandan, A. et al. Genetic diversity analysis of fungal pathogen Bipolaris sorghicola infecting Sorghum bicolor in India. J. Environ. Biol. 37, 1323 (2016).
Kawahigashi, H. et al. Positional cloning of ds1, the target leaf spot resistance gene against Bipolaris sorghicola in sorghum. Theor. Appl. Genet. 123, 131–142 (2011).
Dalmacio, S. C. Target leaf spot. in Compendium of sorghum diseases, 2nd edn. 16–17 (The American Phytopathological Society/APS Press, 2000).
Murali Mohan, S. et al. Identification of quantitative trait loci associated with resistance to foliar diseases in sorghum [Sorghum bicolor (L.) Moench]. Euphytica 176, 199–211 (2010).
Mohan, S. M. et al. Co‐localization of quantitative trait loci for foliar disease resistance in sorghum. Plant Breed. 128, 532–535 (2009).
Segonzac, C. & Zipfel, C. Activation of plant pattern-recognition receptors by bacteria. Curr. Opin. Microbiol. 14, 54–61 (2011).
Oakeley, E. J. et al. Bacterial disease resistance in Arabidopsis through flagellin perception. Nature 428, 764–767 (2004).
Sun, Y. et al. Structural Basis for flg22-Induced Activation of the Arabidopsis FLS2-BAK1 Immune Complex. Science (80-.). 342, 624–628 (2013).
Newman, M.-A., Sundelin, T., Nielsen, J. T. & Erbs, G. MAMP (microbe-associated molecular pattern) triggered immunity in plants. Front. Plant Sci. 4, 139 (2013).
Hogenhout, S. A., V der Hoorn, R. A. L., Terauchi, R. & Kamoun, S. Emerging concepts in effector biology of plant-associated organisms. Mol. Plant-Microbe Interact. 22, 115–122 (2009).
Thomma, B. P. H. J., Nurrnberger, T. & Joosten, M. H. A. J. Of PAMPs and Effectors: The Blurred PTI-ETI Dichotomy. Plant Cell Online 23, 4–15 (2011).
Mur, L. A., Kenton, P., Lloyd, A. J., Ougham, H. & Prats, E. The hypersensitive response; the centenary is upon us but how much do we know? J Exp Bot 59, 501–520 (2008).
Oliver, R. P., Friesen, T. L., Faris, J. D. & Solomon, P. S. Stagonospora nodorum: From Pathology to Genomics and Host Resistance. Annu. Rev. Phytopathol. 50, 23–43 (2012).
Lorang, J. M., Sweat, T. A. & Wolpert, T. J. & Anonymous. Plant disease susceptibility conferred by a ‘resistance’ gene. Proc. Natl. Acad. Sci. 104, 14861–14866 (2007).
Nagy, E. D. & Bennetzen, J. L. Pathogen corruption and site-directed recombination at a plant disease resistance gene cluster. Genome Res. 18, 1918–1923 (2008).
Faris, J. D. et al. A unique wheat disease resistance-like gene governs effector-triggered susceptibility to necrotrophic pathogens. Proc. Natl. Acad. Sci. 107, 13544–13549 (2010).
Shi, G. et al. The hijacking of a receptor kinase–driven pathway by a wheat fungal pathogen leads to disease. Sci. Adv. 2, 10 (2016).
Lorang, J. Necrotrophic Exploitation and Subversion of Plant Defense: A Lifestyle or Just a Phase, and Implications in Breeding Resistance. Phytopathology 109, 332–346 (2018).
Goodwin, S. B., Dunkle, L. D. & Zismann, V. L. Phylogenetic analysis of Cercospora and Mycosphaerella based on the internal transcribed spacer region of ribosomal DNA. Phytopathology 91, 648–658 (2001).
Vetter, M., Karasov, T. L. & Bergelson, J. Differentiation between MAMP Triggered Defenses in Arabidopsis thaliana. PLoS Genet. 12, e1006068 (2016).
Vetter, M. M. et al. Flagellin perception varies quantitatively in arabidopsis thaliana and its relatives. Mol. Biol. Evol. 29, 1655–1667 (2012).
Zhang, X., Valdés-López, O., Arellano, C., Stacey, G. & Balint-Kurti, P. Genetic dissection of the maize (Zea mays L.) MAMP response. Theor. Appl. Genet. 130, 1155–1168 (2017).
Valdes-Lopez, O. et al. Identification of Quantitative Trait Loci Controlling Gene Expression during the Innate Immunity Response of Soybean. Plant Physiol. 157, 1975–1986 (2011).
Hind, S. R., Martin, G. B., Panthee, D. R., Dunham, D. M. & Veluchamy, S. Natural Variation for Responsiveness to flg22, flgII-28, and csp22 and Pseudomonas syringae pv. tomato in Heirloom Tomatoes. PLoS One 9, e106119 (2014).
Lloyd, S. R., Ridout, C. J. & Schoonbeek, H. Methods to Quantify PAMP-Triggered Oxidative Burst, MAP Kinase Phosphorylation, Gene Expression, and Lignification in Brassicas. Plant Pattern Recognit. Recept. Methods Protoc. 325–335 (2017).
Lloyd, S. R., Schoonbeek, H., Trick, M., Zipfel, C. & Ridout, C. J. Methods to Study PAMP-Triggered Immunity in Brassica Species. Mol. Plant-Microbe Interact. 27, 286–295 (2014).
Zeidler, D. et al. Innate immunity in Arabidopsis thaliana: Lipopolysaccharides activate nitric oxide synthase (NOS) and induce defense genes. Proc. Natl. Acad. Sci. USA 101, 15811–15816 (2004).
Nelson, R., Wiesner-Hanks, T., Wisser, R. & Balint-Kurti, P. Navigating complexity to breed disease-resistant crops. Nature Reviews Genetics 19, 21–33 (2018).
Patil, N. Y. et al. Quantitative Trait Loci Associated with Anthracnose Resistance in Sorghum. Crop Sci. 57, 877–890 (2017).
Chapman, J. et al. The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556 (2009).
Rosenow, D. T. et al. Release of four sorghum parental lines ATx642 through ATx645. Int Sorghum Millets Newsl 43, 24–30 (2002).
Sermons, S. M. & Balint-Kurti, P. J. Large Scale Field Inoculation and Scoring of Maize Southern Leaf Blight and Other Maize Foliar Fungal Diseases. Bio-protocol 8, e2745 (2018).
SAS, S. A. S. v9. 4 User’s Guide. SAS Inst. Inc., Cary, NC (2013).
Burks, P. S., Kaiser, C. M., Hawkins, E. M. & Brown, P. J. Genomewide association for sugar yield in sweet sorghum. Crop Sci. 55, 2138–2148 (2015).
Glaubitz, J. C. et al. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS One 9 (2014).
Van Ooijen, J. W. JoinMap® 4, Software for the calculation of genetic linkage maps in experimental populations. Kyazma BV, Wageningen 33 (2006).
Kosambi, D. D. The estimation of map distances from recombination values. in DD Kosambi 125–130 (Springer, 2016).
Wang, S., Basten, C. J. & Zeng, Z. B. Windows QTL Cartographer 2.5. (Department of Statistics, North Carolina State University, North Carolina, USA, 2007).
Acknowledgements
We thank Drs. Steve Kresovich for provision of sorghum lines and advice on seed handling and agronomic practice, Dr. Patricia Klein for genotypic data of the Btx623/SC155-14E RIL populations and Dr Burt Bluhm for providing isolates of B. cookei and for general advice. We thank Cathy Herring and the staff at Central Crops Research Station for their work facilitating the field trials. Dr. Shannon Sermons and Greg Marshall assisted with several aspects of the field research. This work was funded by the DOE Plant Feedstock Genomics for Bioenergy program grant #DE-SC0014116.
Author information
Authors and Affiliations
Contributions
J.K. planned and conducted research, analyzed data, wrote manuscript; Y.C. conducted research, co-wrote manuscript; D.C. conducted research; P.B. developed genotypic data, co-wrote manuscript; W.R. developed mapping population, edited manuscript; G.S. planed research, edited manuscript; P.J.B.-K. planned research, wrote manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kimball, J., Cui, Y., Chen, D. et al. Identification of QTL for Target Leaf Spot resistance in Sorghum bicolor and investigation of relationships between disease resistance and variation in the MAMP response. Sci Rep 9, 18285 (2019). https://doi.org/10.1038/s41598-019-54802-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-54802-x
This article is cited by
-
Methylomes as key features for predicting recombination in some plant species
Plant Molecular Biology (2024)
-
The diverse functions of Pseudomonas syringae syringae van Hall effectors in regulating the plant immune response
Phytopathology Research (2023)
-
NB-LRR-encoding genes conferring susceptibility to organophosphate pesticides in sorghum
Scientific Reports (2021)
-
CRISPR/Cas-mediated genome editing in sorghum — recent progress, challenges and prospects
In Vitro Cellular & Developmental Biology - Plant (2021)
-
Genome-wide association analysis of the strength of the MAMP-elicited defense response and resistance to target leaf spot in sorghum
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
