
Abstract
Chromosomal microarray (CMA) is now recommended as first tier for the evaluation in individuals with unexplained neurodevelopmental disorders (ND). However, in developing countries such as Brazil, classical cytogenetic tests are still the most used in clinical practice, as reflected by the scarcity of publications of microarray investigation in larger cohorts. This is a retrospective study which analyses the reading files of CMA and available clinical data from 420 patients from the south of Brazil, mostly children, with neurodevelopmental disorders requested by medical geneticists and neurologists for diagnostic purpose. Previous karyotyping was reported for 138 and includes 17 with abnormal results. The platforms used for CMA were CYTOSCAN 750K (75%) and CYTOSCAN HD (25%). The sex ratio of the patients was 1.625 males :1 female and the mean age was 9.5 years. A total of 96 pathogenic copy number variations (CNVs), 58 deletions and 38 duplications, were found in 18% of the patients and in all chromosomes, except chromosome 11. For 12% of the patients only variants of uncertain clinical significance were found. No clinically relevant CNV was found in 70%. The main referrals for chromosomal microarrays (CMA) were developmental delay (DD), intellectual disability (ID), facial dysmorphism and autism spectrum disorder (ASD). DD/ID were present in 80%, facial dysmorphism in 52% and ASD in 32%. Some phenotypes in this population could be predictive of a higher probability to carry a pathogenic CNV, as follows: dysmorphic facial features (p-value = < 0.0001, OR = 0.32), obesity (p-value = 0.006, OR = 0.20), short stature (p-value = 0.032, OR = 0.44), genitourinary anomalies (p-value = 0.032, OR = 0.63) and ASD (p-value = 0.039, OR = 1.94). The diagnostic rate for CMA in this study was 18%. We present the largest report of CMA data in a cohort with ND in Brazil. We characterize the rare CNVs found together with the main phenotypes presented by each patient, list phenotypes which could predict a higher diagnostic probability by CMA in patients with a neurodevelopmental disorder and show how CMA and classical karyotyping results are complementary.
Similar content being viewed by others


Introduction
Neurodevelopmental disorders (ND), which mostly involve developmental delay (DD), intellectual disability (ID) and/or autism spectrum disorders (ASD), affect around 3–4% of the world’s population1,2. Such disorders, when isolated, are termed non-syndromic; when associated with the presence of dysmorphisms or apparent congenital anomalies (CA), are termed syndromic3.
Individuals affected with ND usually present reduced adaptive skills and/or limited intellectual ability and face major challenges throughout their life, often including motor difficulties, CA and problems with social interaction. These are relevant characteristics which affect not only the patient, but also impact the daily life of family members due to their special care and dedication needs3,4.
Adequate diagnosis is necessary for the clinical follow-up of individuals with ND and to provide appropriate genetic counseling to the family, preventing the risk of recurrence. Hundreds of genes and many different chromosomal changes are associated with ND and, apart from the well-known and easy identifiable syndromes, the diagnosis of each affected individual remains a clinical challenge.
Due to their high phenotypic and genetic heterogeneity, studies and diagnostics of ND are intricate. Additionally both, genetic and environmental factors, isolated or together, play an important role in their pathogenesis5,6. Currently, molecular karyotyping by chromosomal microarrays (CMA) has been clinically recommended as the first-tier cytogenetic diagnostic test of choice in the investigation of patients with idiopathic ND, such as developmental delay, intellectual disability, autism spectrum disorder and multiple congenital anomalies7.
After the publication of the first comprehensive map of copy number variation in the human genome8, that lead the authors to suggest that CNV assessment should become standard in the design of all studies of the genetic basis of phenotypic variation, including disease susceptibility, a growing number of publications have reported the diagnostic yield of CMA in cohorts of patients with ND, with a worldwide average rate of 15% to 20% in recent years5,9,10,11,12,13,14,15,16,17,18, (Table 1).
Although the CMA test is considered the gold standard in the diagnostics of ND, in Latin America classic karyotyping is still the predominant genetic test in clinical practice, and in Brazil there are only a few publications of CMA in cohorts of ND patients. Pereira and coworkers7 analyzed 15 patients with ND attended by the Laboratory of Human Cytogenetics and Molecular Genetics of the PUC (Pontifical Catholic University) of Goiás between 2010 and 2012, with a diagnostic rate of 22% using the CYTOSCAN HD platform. In Espírito Santo, Pratte-Santos and coworkers19 investigated 39 individuals with ND and a normal karyotype, with the 4 × 180K CMA platform from Agilent, reporting a 15% rate of pathogenic CNVs. In the Northeast of Brazil, Vianna and coworkers20, using a 60K microarray platform (Agilent) in 200 patients with ND, found pathogenic CNVs in 33 of them, a diagnostic rate of 16.5%.
Our study analyzed a cohort of 420 patients from the south of Brazil, that underwent microarray testing from 2013–2016 for diagnostic purpose.
Results
Of the 420 participating patients, 260 (62%) were male and 160 (38%) female, from 0 to 49 years of age, with a mean age of 9.5 years (SD = 9.73, Mo = 4). For 139 patients previous karyotyping was reported, 122 with normal result and 17 with abnormal results for which CMA was requested to define the sequences involved.
For most patients’ previous genetic assessments are unclear.
From the 420 microarrays, a total of 2,468 CNVs which fulfilled the filtering criteria were selected; 1,462 duplications and 1,007 deletions which were interpreted and classified into benign CNVs, pathogenic CNVs and variants of uncertain clinical significance (VOUS).
In 18% patients (75/420) we identified a total of 96 rare CNVs which were interpreted as pathogenic (Table 2). Of these 75 patients, 15 had more than one pathogenic CNV, 9 of them had 2 pathogenic CNVs (#33, #47, #61, #127, #251, #332, #372 and #407) and 6 had 3 pathogenic CNVs (#151, #188, #196, #219, #270 and #392). Three cases (#81, #255 and #331), along with a pathogenic CNV, also presented VOUS. Of the 96 pathogenic CNVs 58 were deletions, leaving only a single copy of the sequence involved. The remaining 38 were duplications that usually result in a total of three copies of the sequence involved, however in two brothers (cases #24 and #25) the duplication of a relevant region of chromosome X resulted in two copies (in which the main reason of pathogenicity is the fact that none of the duplicated copies undergoes X-inactivation, as usual in females) and in three patients (cases #306, #422 and #443) the CNV found was in a four-copy state, of which case #422 had a previous abnormal karyotype result (Table 2). The pathogenic CNVs were found in all chromosomes, except in chromosome 11. Figure 1 illustrates the frequency and number of pathogenic CNVs found per chromosome.

Pathogenic CNVs per chromosome.
Variants of uncertain significance (VOUS), which also are rare CNVs, were the main findings in 12% (49/420) of the patients, summing up a total 56 CNVs, 17 deletions and 39 duplications, (Table 3). These variants were found on most chromosomes except for 21, 22 and Y, and contained from 1 to 48 genes (SD = 10:19, Mo = 4), of which from 1 to 28 (SD = 5.06 Mo = 2) are genes cited in the OMIM database (OMIM genes). Figure 2 illustrates the frequency and amount of VOUS per chromosome.

VOUS per chromosome.
Four of these VOUS (in cases #180, #223, #384 and #444) are discussed in greater detail, because they were considered potentially pathogenic, however with no compelling evidence at this point (Table 4).
All other CNVs were interpreted as benign or as common genetic polymorphisms. In 70% of the cases, they were the only findings present in the genome of a patient, and thus considered a negative result for clinically relevant CNVs.
Figure 3 Patients grouped according to the most relevant CNV found in their genomes.

Classification of cases per most relevant CNV found.
Phenotypic characterization
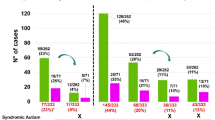
Of the 420 cases, three were not included in the phenotypic characterization because it was not possible to obtain clinical data. The features registered in our cohort are listed in Table 5. Most patients, besides the main reasons of referral (DD, ID, ASD) had additional characteristics, including dysmorphologies, psychiatric or behavioral issues, or variations in height or body weight, whose relation to the main problem often is unclear. Many have syndromic features, as can be concluded by the high presence of congenital abnormalities and atypical facial appearance. As expected, 80% of the individuals of the studied cohort had DD/ID (the main reasons for referral). DD and ID are cited here together because ID is only diagnosed above 5 years of age, however it is a known fact that most individuals with DD in early infancy will later be diagnosed with ID. Of the patients in our study 67% had DD at the time of the study or at an earlier age, with 41% considered intellectually disabled. Facial dysmorphisms (most of them minor) were reported for 53% and ASD for 32%. Other phenotypes were in lower frequencies. Univariate analysis (chi-square or Fisher’s test when more appropriate) indicated predictive phenotypes for a higher diagnostic result (a higher chance to have a pathogenic CNV) in our cohort with ND: dysmorphic facial features (p-value = <0.0001, OR = 0.32), obesity (p-value = 0.006, OR = 0.20), short stature (p-value = 0.032, OR = 0.44), genitourinary anomalies (p-value = 0.032, OR = 0.63) and ASD (p-value = 0.039, OR = 1.94) (Fig. 4). There was no significant higher diagnostic result by CMA for the other phenotypes.

Odds ratios of pathogenic CNVs in cohort study patients. Odds ratios shown in log2 scale. Odds ratios with a p-value < 0.05, two tailed were displayed in red, while others were shown in black. **p-value < 0.001. CM: Congenital malformations, ID: Intellectual disability; DD: Developmental delay and ASD: Autism spectrum disorder.
Table 5 summarizes the clinical features recorded for patients with negative and positive CMA results with the percentage (and number) of patients presenting them. Most patients have more than one relevant phenotype.
Classical karyotyping and CMA
Seventeen patients informed previous abnormal karyotyping results (Table 6), three of which are not very understandable or with a question mark (#282, #412 and #430). For 12 cases, CMA specified the sequences involved, often with unexpected findings, hinting to the mechanism of occurrence of the anomaly and explaining phenotypes that the karyotype by itself suggested otherwise. In case #196, for instance, CMA identified a deletion in the short arm of chromosome 5, whereas the chromosomal analysis of the patient (46, XX, 5p+) indicated additional DNA in chromosome 5. CMA revealed also that the additional DNA in chromosome 5 originated from a partial duplication of the long arm of chromosome 18. For another case, #263 (47, XY +mar), a large deletion was found instead of a gain. Regarding the five cases where the cytogenetic analysis was abnormal and no pathogenic CNV was identified, in one (#138) a VOUS with no apparent relation to the chromosome analysis result was found whereas the other four had a normal CMA result, including the three cases whose informed karyotype was followed by a question mark, indicating that the chromosomal analysis was not conclusive (Table 6).
Discussion
In the present study, a total of 96 pathogenic CNVs were detected in CMA results of 75 patients with ND in the state of Santa Catarina, a diagnostic yield of 18%, within the range of 15–20% diagnostic rate cited in literature for patients with ND in other cohorts5,9,11,12,13,14,15,16,17. It is important to highlight that the 75 patients with pathogenic CNVs, included 12 patients of the 17 with previous abnormal karyotype result, for whom the CMA test was requested in order to identify the DNA sequences involved. Excluding the 17 cases with known abnormal karyotype results in a diagnostic rate of 15.63%, and when considering only the 122 patients that underwent previous karyotyping and had normal results, the diagnostic rate was not different, 15.57%. However, the diagnostic yield was considered 18% because CMA was essential to uncover the sequences altered in the abnormal karyotype results, and thus was diagnostic, unveiling unexpected findings, like deletions in chromosomes whose karyotype showed additions or deletion when karyotype had suggested addition. These are exemplified by case #127 [46, XX, add(18) (q23)] CMA identified a distal trisomy of 10q with simultaneous distal 18q deletion and for #196 (46, XX, 5p+) CMA revealed a distal trisomy 18q together with a distal deletion in 5p. For case #263 (47, XY +mar), a new chromosomal analysis would be desired, because instead of additional DNA, a large pathogenic deletion in chromosome 9 was found. The CMA results of the 17 cases for whom a previous abnormal chromosomal analysis was reported, are depicted in Table 6, case by case, together with comments about the findings.
Conversely, our results also point to the usefulness of traditional karyotyping to complement the CMA results, allowing an insight into the mechanisms that gave rise to the genetic abnormality, which is relevant for genetic counselling. For instance, from the 15 cases that had more than one rare CNV (pathogenic CNV or VOUS) and no previous abnormal karyotyping, eight involved the terminal region of chromosomes, some of them quite large, combining terminal deletions with terminal duplications, suggesting that they might be derivative chromosomes that arose form a translocation. This can be seen in case #61 (Table 1) with a distal trisomy of chromosome 8q and a simultaneous deletion in the end of the long arm of chromosome 13; #151, with a terminal del18p and a terminal trisomy 7p; #188, with a terminal del21q and a terminal trisomy 19p; #251, with a terminal del20q and a terminal trisomy 19p; #270, with a terminal del18q and a terminal trisomy 3q; #332, with a terminal del7q and a terminal trisomy 3q; #372, with a terminal del8p and a terminal trisomy 4p, and case #407, with a terminal del21q and a terminal trisomy. This derivative chromosome could have been originated during meiosis, during the first mitotic divisions of the zygote or possibly were inherited from a healthy parent that carries the translocation in an equilibrated state. In latter case there is a risk of recurrence for the same or possibly the complementary derivative in another child. Three cases had 2 or 3 CNVs within the same chromosome: case #33, where the microarray result points to a circular chromosome 18, since both ends are deleted; case #331, with two deletions and one duplication, suggesting a del/dup inversion, and case #47, that had two small deletions on the tip of the p arm, surrounding the SHOX gene, indicating a possible del/del inversion including SHOX. Other cases had a combination of interstitial or terminal and interstitial CNVs in two or more chromosomes, pointing to more complex mechanisms.
In 2010, the American College of Medical Genetics recommended CMA as first-tier test for the population of individuals with DD, ID, ASD and multiple congenital anomalies. We agree with that. However, about the often-made question if CMA is a substitute for the classical chromosome analysis or even if CMA is causing karyotyping to be obsolete, we consider that a correct diagnosis requires the combination of CMA and chromosome analysis as stated by others21, which observed structural rearrangements in addition to simple deletions or duplications under the microscope in 85 (18%) of 469 cases with an abnormal CMA result. Likewise, chromosome analysis of the parents of individuals with clearly pathogenic terminal deletions/duplications or large CNVs (regardless if terminal or interstitial) should be a follow-up rule, because this knowledge is essential for genetic counselling. For instance, the karyotype of a father of two affected siblings, a girl (#149) with a large deletion in chromosome 5 [5p14.3–p15.31 (6,801,589–18,992,827)] and her brother, #445, with a duplication of the exact same region, revealed complex translocations involving at least four chromosomes, 46, XY, t (1; 2) (q44; ~p23-pter); t(5; 7) (p14.3–p15.31; p22) (Table 1). The genome of this father survived catastrophic events with no obvious clinical consequence for him which, however, left rearrangements (not detectable by MCA) whose deleterious reflexes did affect deeply the development of his two children – in two distinct (or opposite) molecular ways midst an even larger array of possibilities.
Among 17 abnormal karyotypes we had at least one equilibrated translocation, case #175 [46, XY, t(4; 7) (q31; p14)], whose CMA result showed no CNV. This is an interesting case to study because it is unlikely that this translocation has no pathogenic relevance. Possibly the translocation disrupts or interferes with the regulation of the causal gene, which could be identified by breakpoint mapping/sequencing.
The pathogenic CNVs found in this study and the reported phenotypes of the respective patients are detailed in Table 1. It is known that most pathogenic CNVs occur “de novo” because of an error during meiotic recombination, an early illegitimate mitotic recombination, or the mutagenic repair of DNA double-strand breaks during the first divisions of the embryonic cells22. They can also be consequence of a balanced chromosomal translocation in the genome of one of the parents, therefore classical karyotype test for parents of individuals with large pathogenic CNVs is advisable, since balanced translocations cannot be identified by CMA and there is a high risk of recurrence23.
We tried to draw comparisons between pathogenic CNVs detected between various studies, which is a challenge, since each study used distinct CMA platforms with probes of varying sizes, densities and characteristics. To allow a comparison, we made a circle plots with the pathogenic CNVs detected in our study together with the pathogenic CNVs detected in cohorts of North America24,25 and Europe13,14,15,26,27 using studies that made the data sufficiently available for such analysis (Fig. 5).

The circle plot compares pathogenic CNVs found in: (first, the outhermost double track) our study of a cohort of 420 individuals with neurodevelopmental disorders (ND), derived from a complex population in the south of Brazil, mostly composed by the Portuguese conquerors, German and Italian immigrants, besides descendants of slaves and of Amerindians; (second double track from the border) studies of 1.245 individuals from five affected European cohorts; (third double track) studies of 15.901 individuals from two affected North American cohorts; (fourth, innermost double track) the pathogenic CNVs detected exclusively in our study, when compared to the other studies in the plot.
Among the studies of the circle plot, the following pathogenic CNVs were detected exclusively in our sample: arr[hg19] 1p36.33p36.32(1,073,574–2,458,606)x1, arr[hg19] 2q31.1-q31.2(174,065,715–190,659,870)x1, arr[hg19] 4p16.3p16.1(68,345–9,509,606)x3, arr[hg19] 4p16.3(68,345–964,416)x1, arr[hg19] 7p22.3p21.3(43,376–9,454,786)x3, arr[hg19] 7q31.32q33(122,736,512–136,162,906)x3, arr[hg19] 8p21.1p11.21(28,393,484-41,026,001)x1, arr[hg19] 8p11.22p11.21(39,388,765–42,335,424)x3, arr[hg19] 12p13.2p13.1(10,922,516–12,937,320)x1, arr[hg19] 13q33.1q34(104,782,510–112,352,804)x1, arr[hg19] 16p13.3(85,880–2,145,951)x1, arr[hg19] 18p11.32p11.31(136,226–4,409,550)x1, arr[hg19] 19p13.3(260,911–1,434,508)x3, arr[hg19] 21q11.2q22.3(15,006,457–44,968,648)x3, arr[hg19] 21q22.12q22.2(35,834,713–39,831,660)x1, arr[hg19] 22q12.3q13.1(35,888,588–38,692,765)x4, arr[hg19] Xp22.3q28(1–247,249,719)x3, arr[hg19] Xp22.33(372,029–578,764)x1, arr[hg19] Xp22.33(679,520–950,907)x1, arr[hg19] Xq26.3q28(135,224,845–155,233,098)x2, arr[hg19] Xq27.3q28(142,412,280–155,233,098)x2, arr[hg19] Xq27.3q28(146,425,635–151,604,987)x2 and arr[hg19] Xq27.3q28(146,418,810–151,604,987)x2.
The interpretation of CNVs is not an absolute science and caution must be used in the report of the results. Palmer et al. (2013) already presented data on how the interpretation of CNVs detected by CMA had a significant change over time, with an increase in CNVs classified as pathogenic as new studies and case descriptions are reported. That is why it is important to register the CNVs interpreted as VOUS when no pathogenic CNV is found. In our study we found VOUS (as the most relevant CNV) in 12% (49/420) of the patients in the cohort (Table 2). Although we believe that most of them will have no clinical impact, some of the CNVs in this subgroup possibly will be classified as pathogenic in the future, as more data accumulates. Bellow we highlight four cases where we considered the VOUS potentially pathogenic:
Case #223 = Refers to a boy that was ten years old when he was referred for CMA. He presented short stature, intrauterine growth restriction, DD, mild ID, a narrow face, dolichocephaly, high-arched palate, microtia (small ears), nipple hypertelorism and constipation. His MCA revealed no pathogenic CNV, however three duplication VOUS (Table 4), of which two were considered potentially pathogenic: arr[hg19] 3p26.3(255,645–1,510,822)x3 and arr[hg19] 6q25.3(156,488,875–158,534,725)x3, whose inheritance is unclear. The region arr[hg19] 3p26.3(255,645–1,510,822)x3 duplicates the entire sequence of the contactin 6 gene (CNTN6), LINC01266, a long intergenic ncRNA, and the final of the CHL1 gene (cell adhesion molecule L1 like). CHL1 has been proposed as a candidate gene for intellectual disability of the 3p deletion syndrome28,29, and one partial duplication of a similar portion of the CHL1 gene as in case #223 was described, including also the complete CDS of LINC01266, and a small portion of the CNTN6 gene30. It is not clear if the partial duplication of CHL1 in was originated by some rearrangement that could have disrupted one of the complete copies of the gene. Contactin 6, encoded by CNTN6 is a neural cell adhesion molecule that has been proposed as one of the critical genes of the 3p deletion syndrome31 and deletions or duplications of CNTN6 was suggested to be associated to a wide spectrum of neurodevelopmental disorders32. The 6q25.3(156,488,875–158,534,725) genomic region contains the complete sequences of the genes ARID1B (AT-rich interaction domain 1B), TMEM242 (Transmembrane Protein 242), ZDHHC14 (Zinc Finger DHHC-Type Containing 14), SNX9 (Sorting Nexin 9), SYNJ2 (Synaptojanin 2), the beginning of the SERAC (Serine Active Site Containing 1) gene, and the microRNA genes MIR4466 and MIR3692. No complete duplication of any of these genes was found on the DGV. Of those, SYNJ2 is majoritarily expressed in the brain33 and is a member of the synaptojanin family, which are key players in the synaptic vesicle recovery at the synapse; TMEM242 is a potential multi-pass membrane protein of unknown function34, that is expressed in most tissues33), however, with highest expression in the brain; ZDHHC14 is a probable palmitoyltransferase34 whose expression is highest in the brain and utherus33; SNX9 could involved in several stages of intracellular trafficking and is espressed is most tissues, with very low brain expression33 and ARID1B is a component of the SWI/SNF chromatin remodeling complex and its haploinssuficiency is one of the most frequent causes of ID, both, syndromic (Coffin-Siris syndrome) and non-syndromic35,36,37,38. Coffin-Siris syndrome is characterized by, feeding difficulties in infancy, delayed motor skills, severe speech impairment, mild to severe ID, coarse facial features, hirsutism and its hallmark is the hypoplasia or absence of the 5th distal phalanx of the finger and/or toes. Up to now, only intragenic duplications that probably disrupt gene function were described, however no complete duplication of the gene ARID1B has been described. Duplications comprising the region of chromossome 6 that is duplicated in case #223 are much larger, with the exception of one registered in Decipher, for patient: 287902 with microcephaly and ID, that has a “de novo” duplication of about the same size as the one in our case. Other three duplications including only complete ARID1B alone or with one more gene are also in Decipher, all being the only, or the only non-inherited CNV, found.
Cases #180, #384 and #444 refer to three boys, 4, 2, and 5 years old, respectively, at the date of referral for CMA, because of DD (# 180), motor delay, chronic encephalopathy and spastic quadriparesis (# 384), and DD and ASD (# 444), all of them with a different intragenic deletion in the gene RBFOX1. The RBFOX1 gene (OMIM * 605104), also known as Ataxin-2-binding protein 1 (A2BP1) or FOX1, is one of the largest genes in the human genome and encodes a neuronal RNA binding protein that is highly conserved evolutionarily. It has a very complex transcription unit that generates transcripts from multiple promoters, and presents alternative termination sites. The inclusion of its multiple internal exons is highly regulated, yielding various nuclear and cytoplasmic protein isoforms39. In the nucleus, RBFOX1 protein isoforms act as RNA processing factors, while in the cytoplasm they act as proteins that regulate the stability and translation of RNAs involved in cortical development and autism40,41.
Changes in RBFOX1 have been related to several neurodevelopmental syndromes, including ID, epilepsy, and ASD42,43,44, with important roles in neuronal migration and synapse network formation during corticogenesis45. Specifically, intragenic deletions have been related to neuropsychiatric and neurodevelopmental disorders42,46,47.
The case #180 showed a microdeletion 593 Kbp (arr[hg19] 16p13.3(6,243,228–6,835,898)x1), eliminating exon 1 from transcript variant 6 (isoform 4 NM_001142334.1) and exons 2 and 3 from transcripts variants 4, 5 and 7 (respectively, isoform 4 NM_018723.3, isoform 5 NM_001142333.1 and isoform 6 NM_001308117.1).of the gene RBFOX1 which in the reference sequence are non-coding exons of the 5 ‘ region. Besides possibly affecting the transcription of the main isoforms, this microdeletion also affects the promoter of several isoforms of RBFOX1, whose transcription begins after exon two.
Case #384 presented one microdeletion 117 kbp in 16p13.3 (arr[hg19] (7,108,169–7,225,285)x1), involving an intronic region between exon 4 and 5 from transcripts variants 4, 5 and 7 (respectively, isoform 4 NM_018723.3, isoform 5 NM_001142333.1 and isoform 6 NM_001308117.1) and between exon 2 and 3 from from transcript variant 6 (isoform 4 NM_001142334.1) of the RBFOX1 gene, and case #444 had microdeletion of 31 kbp (arr[hg19] 16p13.3(6,644,079–6,675,606)x1) in intron 2 from transcripts variants 4, 5 and 7 (respectively, isoform 4 NM_018723.3, isoform 5 NM_001142333.1 and isoform 6 NM_001308117.1) of the RBFOX1 gene, affecting various isoforms and possibly affecting the isoform promoter region that initiates from transcript variant 6 (isoform 4 NM_001142334.1) after exon 3 of the reference sequence.
It is topic of ongoing discussion of how to communicate the CNVs findings in the reports, where the communication of VOUS is particularly challenging. In clinical practice, it is a confounding factor to have a CNV about which no one can say something for sure. The limitations of the test and, more shockingly, of the current understanding of the results are difficult for the clinician to explain and even more difficult for the patient/guardians to understand. They often cannot settle for the idea that they underwent such an expensive test and the doctors cannot say anything useful or definitive with the results. Even though adequate pre-testing explanation is provided to patients or their guardians, and they sign a consent form which also lists the limitations of the test, for many persons the real understanding of what that means only sinks in after receiving an ambiguous CMA result. It is much easier to explain a negative result that, if not answering the question of why the neurodevelopment was disturbed, at least answers that it is not caused by a genomic imbalance produced by an excess or a deletion of genetic material. A VOUS tends to represent a point of frustration for all involved. The American College of Medical Genetics allows to communicate the likelihood that a VOUS is pathogenic or benign, when well founded in the report and the uncertainty of such classification is clearly communicated. In addition, they also recommend that the report includes guidelines for the continuous monitoring of medical literature, since new knowledge can clarify the CNV’s real clinical impact.
One strategy in the interpretation of a VOUS is to investigate if it occurred “de novo” or has been inherited from one of the parents. Inherited CNVs are more likely benign, whereas “de novo” variants found in ND patients have a greater chance to be causal. However, the incomplete penetrance or variable expression of a clinical phenotype can explain the presence of a pathogenic CNV in an unaffected (or sub-clinically affected) parent. As well as a “de novo” event is indicative, but not necessarily the cause of the disorder.
In regard to their size, the pathogenic CNVs were typically very large (Fig. 6A), with a mean size of 7,770 kbp (median: 5,179 kbp), and contained multiple genes when compared with benign CNVs (mean: 483 kbp, median: 285 kbp, Fig. 6A,B) and VOUS (mean: 666 kbp, median: 382 kbp), as shown in Fig. 6A,C, in agreement to findings by others25,48,49. The variation inside each class is very large and some pathogenic CNVs are quite small whereas some benign CNVs can be very large when they are situated in gene-poor regions, like those close to centromeres. It is to expect that a VOUS is not typically very large because the more genes a CNV contains the higher chance of including known dosage-sensitive genes, regulatory regions or, in case of a deletion, to expose a recessive mutation which may be present in the remaining copy of the gene.

(A) CNV type by size variation. (B) Benign CNV size variation on a larger scale. (C) VOUS size variation on a larger scale.
Based on the clinical data, obtained from the medical records, the most frequent phenotypes reported are also the main reasons of referral: DD, ID, congenital anomalies and/or dysmorphia, and ASD (Table 4). The same phenotypes are predominant in other CMA studies for the investigation of neurodevelopmental disorders4,5,9,11,14,15,16,17,18.
For instance, congenital anomalies, along with facial dysmorphisms, were reported in more than 58% of our cohort (Table 4). This frequency similar if the findings of 50% in a cohort of 78 affected with ND in the study of Qiao et al.50 and the 55% reported by Roselló et al.5 in their study with 246 patients with DD and ID, and probably represents a selection bias by the MDs for the referral for testing. Nevertheless, there was no statistical difference of diagnostic rate for patients with neurodevelopmental disorders without an obvious congenital anomaly or dysmorphia (data not shown).
Univariate analysis showed a significant association for the presence of pathogenic CNVs with dysmorphic facial features (p-value = < 0.0001, OR = 0.32) and ASD (p-value = 0.039). Congenital anomalies only showed a higher association with pathogenic CNVs in this cohort when broken down into more specific affected systems, where genitourinary anomalies had a higher correlation with the finding of a pathogenic CNV (p-value = 0.032). Furthermore, two secondary phenotypes, obesity (p-value = 0.006) and short stature (p-value = 0.032), were shown to be phenotypes associated to higher findings of pathogenic CNVs in patients with ND. However, this are incipient results, and should be avoided to be used for testing decisions. A clinical and standardized reassessment in all cases and a larger sample would be crucial to confirm this.
As already discussed by Quintela et al.26 the interpretation of genomic variations such as CNVs is an arduous task, especially in the challenging VOUS, when the genotype is suggestive of a genomic disorder characterized by incomplete penetrance and/or variable expressivity.
Regarding the negative diagnoses of the CMA (without CNVs or with only benign CNVs) in high resolution SNP CMA platforms like the ones used in this study, the homozygous regions can also be studied. Those results with very large LCSHs (long contiguous stretches of homozygosity) indicating possible uniparental disomy (UPD) or consanguinity should be reported to the accompanying MD for follow-up investigation of eventual imprinting syndromes or autosomal recessive mutations, through methylation or exome analysis. The relevance of LCSHs, which can be identified by most modern CMA platforms, is discussed elsewhere51.
Conclusions
The diagnostic rate for CMA in this study was 18% and is within the literature (15–20%). CMA is an essential tool to decipher the sequences involved in structural karyotype abnormalities detected by classical chromosome analysis, as well as patients with abnormal CMA results should have their chromosomes analyzed - which can lead to unexpected surprises. For a correct diagnosis CMA and chromosome analysis should be used complementary. Parental chromosome analysis is essential for genetic counselling, particularly when the patient has terminal deletion/duplication or large CNVs. The main reasons for referral for CMA testing were DD/ID, dysmorphic facial features and ASD. Dysmorphic facial features and ASD (as main or secondary feature) and secondary phenotypes such as obesity, short stature, genitourinary anomalies are possible predictive phenotypes of a higher diagnostic answer by CMA.
Clinical interpretation of CNVs is still a challenge and depends in large part on information about their frequency in normal and affected populations, provided by cohort studies with significant samples.
Methods
Ethical aspects
The project was submitted and approved by the Research Ethics Committee of the Hospital Infantil Joana de Gusmão, the children´s hospital of Florianópolis-SC, Brazil, under the Nr 2,339,104, and respects the guidelines and criteria of the resolution Nr 466/12 of the Brazilian National Health Council. Patients or their parent and/or legal guardian (in cases where patient was under legal age), signed the Informed Consent Form. In cases in which it was not possible to contact the patient for any justifiable reason (loss of contact information, mainly) the data was used and a Justification of Absence of Consent approved by Research Ethics Committee and signed by the research team, ensuring the commitment to maintain confidentiality and privacy of the patients whose data and/or information was collected in the records.
Sample
The sample refers to the reading files of CMA and available clinical data from 420 patients from the south of Brazil, mostly children, with neurodevelopmental disorders. The CMAs were requested by medical geneticists and neurologists for diagnostic purposes, mainly from the Joana de Gusmão Children’s Hospital, but also from the University Hospital Professor Polydoro Ernani de São Thiago and from private clinics in Florianópolis (State of Santa Catarina), throughout the years 2013 to 2016 and performed by the Laboratório Neurogene (Florianópolis, Santa Catarina, Brazil).
Collection of clinical data
To correlate the phenotype to possible causal genes, the clinical description of the affected individuals was collected with their MDs through a questionnaire, seeking information about their clinical presentation, behavior, history of physical exams, as well as results of previous genetic and metabolic tests and prescription medication. No new appointments with the patients were made for this, and clinicians retrieved most data from their medical records.
Genomic analysis
The platforms used were CYTOSCAN 750K (75%) and CYTOSCAN HD (25%) and the resulting files were analyzed using the CHROMOSOME ANALYSIS SUITE (ChAS) AFFYMETRIX software, based on the reference genome sequence of the University of California, Santa Cruz database (https-//genome.ucsc.edu/cgi-bin/hgGateway) using the human genome version of February 2009 (GRCh37/hg19). The filter criteria for CNVs were sizes >100 Kbp for deletions and >150 Kbp for duplications, both with at least 50 markers, according to ACMG recommendations52.
CNVs interpretation and classification
To interpret CNVs, regarding their function, dosage effects (known haploinsufficiency or overexpression studies) and effects of mutations, the UCSC Genome Browser with integrated databases was widely used, mainly ClinVar (NCBI), DECIPHER (Database of Chromosomal Imbalance and Phenotype in Humans using Ensembles Resources), DGV (Database of Genomic Variants), OMIM (Online Mendelian Inheritance in Man), ISCA (International Standard Cytogenomic Array), dbGaP (Database of Genotypes and Phenotype), dbVAR (Database of Large Scale Genomic Variants), ECARUCA (European Cytogeneticists Association Register of Unbalanced Chromosome Aberrations), PUBMED (Public Medline), ClinGen (Clinical Genome Resource), MGI (Mouse Genome Informatics Database, from The Jackson Laboratory) and the private database CAGdb (Cytogenomics Array Group CNV Database).
The variants were classified into three types according to clinical interpretation as benign, variants of uncertain significance (VOUS), or pathogenic variants (causal), and the result in each case was assigned based on the CNV(s) of greatest clinical relevance detected in the genome of the patients.
Variables like location, type and size of each CNV, the CNV classification, number of CNVs detected for each patients, age, gender, clinical descriptions (phenotypes), previous genetic testing results (karyotype, fragile X, etc.), and other relevant known clinical data, were compiled (with coded identification) into simple Excel sheet for data handling with the R software (version 3.4.2, the R FOUNDATION FOR STATISTICAL COMPUTING) in order to understand the phenotypic frequency, the diagnostic rate of the study, the average age and the gender distribution in the cohort, the frequency of genomic changes in each chromosome, and the relation of the phenotype (or groups of clinical phenotypes) to the type of CNV to find if there are any indications which allow to recognize the patients with higher chance of carrying a pathogenic CNV - most suitable for submission to the CMA as a first-line test in the unfortunate setting of financial shortage.
Ethics approval and consent to participate
The project was submitted and approved by the Research Ethics Committee of the Hospital Infantil Joana de Gusmão, the children’s hospital of Florianópolis-SC, Brazil, under the Nr 2,339,104, and respects the guidelines and criteria established by the resolution 466/12 of the Brazilian National Health Council. Patients or their caregivers signed the Informed Consent Form to participate in the study. In cases in which it was not possible to contact the patient for any justifiable reason (loss of contact information, mainly) the data was used and a Justification of Absence of Consent was signed by the research team, ensuring the commitment to maintain confidentiality and privacy of the patients whose data and/or information was collected in the records.
Data availability
The datasets used and/or analyzed during the current study can be requested from the corresponding author on reasonable request. However, since the patients or their caregivers signed an Informed Consent Form specifying that the data will be used only for the present study, their use for another study necessarily implies a new submission to the ethics committee of the Hospital Infantil Joana de Gusmão and depends on a new approval.
References
International Classification of Functioning, Disability and Health World Health Organization Geneva ICF ii WHO Library Cataloguing-in-Publication Data International classification of functioning, disability and health: ICF. (2001).
Cappuccio, G. et al. New insights in the interpretation of array-CGH: autism spectrum disorder and positive family history for intellectual disability predict the detection of pathogenic variants. Ital. J. Pediatr. 42, 39 (2016).
Abou Jamra, R. et al. Homozygosity mapping in 64 Syrian consanguineous families with non-specific intellectual disability reveals 11 novel loci and high heterogeneity. Eur. J. Hum. Genet. 19, 1161–1166 (2011).
D’Amours, G. et al. SNP arrays: comparing diagnostic yields for four platforms in children with developmental delay. BMC Med. Genomics 7, 70 (2014).
Roselló, M. et al. Phenotype profiling of patients with intellectual disability and copy number variations. Eur. J. Paediatr. Neurol. 18, 558–566 (2014).
Wu, C.-L. et al. The effectiveness of healthy physical fitness programs on people with intellectual disabilities living in a disability institution: Six-month short-term effect. Res. Dev. Disabil. 31, 713–717 (2010).
Pereira, R. R. et al. Screening for intellectual disability using high-resolution CMA technology in a retrospective cohort from Central Brazil. PLoS One 9, e103117 (2014).
Redon, R. et al. Global variation in copy number in the human genome. 444, 444–454 (2006).
Pfundt, R. et al. Clinical performance of the CytoScan Dx Assay in diagnosing developmental delay/intellectual disability. Genet. Med. 18 (2016).
Anazi, S. et al. Clinical genomics expands the morbid genome of intellectual disability and offers a high diagnostic yield. Mol. Psychiatry 22, 615–624 (2017).
Kashevarova, A. A. et al. Array CGH analysis of a cohort of Russian patients with intellectual disability. Gene 536, 145–150 (2014).
Wu, X.-L. et al. Chromosome microarray analysis in the investigation of children with congenital heart disease. BMC Pediatr. 17, 117 (2017).
Bartnik, M. et al. Application of array comparative genomic hybridization in 256 patients with developmental delay or intellectual disability. J. Appl. Genet. 55, 125–144 (2014).
Coutton, C. et al. Array-CGH in children with mild intellectual disability: a population-based study. Eur. J. Pediatr. 174, 75–83 (2015).
Preiksaitiene, E. et al. Considering specific clinical features as evidence of pathogenic copy number variants. Journal of Applied Genetics 55, 189–196 (2014).
Lay-Son, G. et al. Chromosomal microarrays testing in children with developmental disabilities and congenital anomalies. J. Pediatr. (Rio. J). 91, 189–195 (2015).
Chen, C. P. et al. Prenatal diagnosis and molecular cytogenetic characterization of a de novo interstitial duplication of 14q (14q31.3→q32.12) associated with abnormal maternal serum biochemistry. Taiwan. J. Obstet. Gynecol. 52, 125–128 (2013).
García-Acero, M., Suárez-Obando, F. & Gómez-Gutiérrez, A. CGH analysis in Colombian patients: findings of 1374 arrays in a seven-year study. 11, 46 (2018).
Pratte-Santos, R., Ribeiro, K. H., Santos, T. A. & Cintra, T. S. Analysis of chromosomal abnormalities by CGH-array in patients with dysmorphic and intellectual disability with normal karyotype. Einstein (São Paulo) 14, 30–34 (2016).
Vianna, G. S., Medeiros, P. F. V., Alves, A. F., Silva, T. O. & Jehee, F. S. Array-CGH analysis in patients with intellectual disability and/or congenital malformations in Brazil. Genet. Mol. Res. 15 (2016).
Bi, W. et al. Comparison of chromosome analysis and chromosomal microarray analysis: what is the value of chromosome analysis in today’s genomic array era? Genet. Med. 15, 450–457 (2013).
Wang, J., Fan, H. C., Behr, B. & Quake, S. R. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell 150, 402–12 (2012).
Nowakowska, B. A. et al. Parental insertional balanced translocations are an important cause of apparently de novo CNVs in patients with developmental anomalies. Eur. J. Hum. Genet. 20, 166–70 (2012).
Borlot, F., Regan, B. M., Bassett, A. S., Stavropoulos, D. J. & Andrade, D. M. Prevalence of Pathogenic Copy Number Variation in Adults With Pediatric-Onset Epilepsy and Intellectual Disability. JAMA Neurol. 74, 1301–1311 (2017).
Cooper, G. M. et al. A copy number variation morbidity map of developmental delay. Nat. Genet. 43, 838–46 (2011).
Quintela, I. et al. Copy number variation analysis of patients with intellectual disability from North-West Spain. Gene 626, 189–199 (2017).
Heide, S. et al. Copy Number Variations Found in Patients with a Corpus Callosum Abnormality and Intellectual Disability. J. Pediatr. 185, 160–166.e1 (2017).
Shoukier, M. et al. Microduplication of 3p263 in nonsyndromic intellectual disability indicates an important role of CHL1 for normal cognitive function. Neuropediatrics 44, 268–271 (2013).
Te Weehi, L. et al. Microduplication of 3p26.3 Implicated in. Cognitive Development. Case Rep. Genet. 2014, 1–6 (2014).
Li, C., Liu, C., Zhou, B., Hu, C. & Xu, X. Novel microduplication of CHL1 gene in a patient with autism spectrum disorder: A case report and a brief literature review. Mol. Cytogenet. 9 (2016).
Fernandez, T. et al. Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am. J. Hum. Genet. 74, 1286–1293 (2004).
Hu, J. et al. CNTN6 copy number variations in 14 patients: A possible candidate gene for neurodevelopmental and neuropsychiatric disorders. J. Neurodev. Disord. 7 (2015).
Database, G. GeneCards - Human Genes|Gene Database|Gene Search. Genecards.org Available at, https://www.genecards.org/, (Accessed: 20th October 2019) (2017).
UniProtKB. Available at, https://www.uniprot.org/help/uniprotkb, (Accessed: 19th October 2019).
Tsurusaki, Y. et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat. Genet. 44, 376–8 (2012).
Hoyer, J. et al. Haploinsufficiency of ARID1B, a member of the SWI/SNF-A chromatin-remodeling complex, is a frequent cause of intellectual disability. Am. J. Hum. Genet. 90, 565–572 (2012).
Santen, G. W. E. et al. Mutations in SWI/SNF chromatin remodeling complex gene ARID1B cause Coffin-Siris syndrome. Nat. Genet. 44, 379–380 (2012).
van der Sluijs, E. P J. et al. The ARID1B spectrum in 143 patients: from nonsyndromic intellectual disability to Coffin–Siris syndrome. Genet. Med., https://doi.org/10.1038/s41436-018-0330-z (2018).
Zhang, X. et al. Cell-Type-Specific Alternative Splicing Governs Cell Fate in the Developing Cerebral Cortex. Cell 166, 1147–1162.e15 (2016).
Underwood, J. G., Boutz, P. L., Dougherty, J. D., Stoilov, P. & Black, D. L. Homologues of the Caenorhabditis elegans Fox-1 Protein Are Neuronal Splicing Regulators in Mammals. Mol. Cell. Biol. 25, 10005–10016 (2005).
Carreira-Rosario, A. et al. Repression of Pumilio Protein Expression by Rbfox1 Promotes Germ Cell Differentiation. Dev. Cell 36, 562–571 (2016).
Lal, D. et al. Rare exonic deletions of the RBFOX1 gene increase risk of idiopathic generalized epilepsy. Epilepsia 54, 265–271 (2013).
Bhalla, K. et al. The de novo chromosome 16 translocations of two patients with abnormal phenotypes (mental retardation and epilepsy) disrupt the A2BP1 gene. J. Hum. Genet. 49, 308–311 (2004).
Martin, C. L. et al. Cytogenetic and molecular characterization of A2BP1/FOX1 as a candidate gene for autism. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 144, 869–876 (2007).
Hamada, N. et al. Essential role of the nuclear isoform of RBFOX1, a candidate gene for autism spectrum disorders, in the brain development. Sci. Rep. 6 (2016).
Gehman, L. T. et al. The splicing regulator Rbfox1 (A2BP1) controls neuronal excitation in the mammalian brain. Nat. Genet. 43, 706–11 (2011).
Lee, J. A. et al. Cytoplasmic Rbfox1 Regulates the Expression of Synaptic and Autism-Related Genes. Neuron 89, 113–128 (2016).
McLysaght, A. et al. Ohnologs are overrepresented in pathogenic copy number mutations. Proc. Natl. Acad. Sci. USA 111, 361–6 (2014).
Rice, A. M. & McLysaght, A. Dosage sensitivity is a major determinant of human copy number variant pathogenicity. Nat. Commun. 8, 14366 (2017).
Qiao, Y. et al. Copy number variants (CNVs) analysis in a deeply phenotyped cohort of individuals with intellectual disability (ID). BMC Med. Genet. 15, 82 (2014).
Chaves, T. F. et al. Long contiguous stretches of homozygosity detected by chromosomal microarrays (CMA) in patients with neurodevelopmental disorders in the South of Brazil. BMC Med. Genomics 12, 50 (2019).
Rehder, C. W. et al. American College of Medical Genetics and Genomics: standards and guidelines for documenting suspected consanguinity as an incidental finding of genomic testing. Genet. Med. 15, 150–152 (2013).
Bruno, D. L. et al. Detection of cryptic pathogenic copy number variations and constitutional loss of heterozygosity using high resolution SNP microarray analysis in 117 patients referred for cytogenetic analysis and impact on clinical practice. J. Med. Genet. 46, 123–31 (2009).
Di Gregorio, E. et al. Copy number variants analysis in a cohort of isolated and syndromic developmental delay/intellectual disability reveals novel genomic disorders, position effects and candidate disease genes. Clin. Genet. 92, 415–422 (2017).
Sansović, I., Ivankov, A.-M., Bobinec, A., Kero, M. & Barišić, I. Chromosomal microarray in clinical diagnosis: a study of 337 patients with congenital anomalies and developmental delays or intellectual disability. Croat. Med. J. 58, 231–238 (2017).
Kessi, M. et al. Rare Copy Number Variations and Predictors in Children With Intellectual Disability and Epilepsy. Front. Neurol. 9, 947 (2018).
Acknowledgements
We thank all the families who have agreed to participate in this study. We thank CAPES for their financial support with a postgraduate grant to T.F.C., N.B. and L.F.O. T.F.C., N.B. and L.F.O. were supported with a scholarship from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) - Brazil. The funding body provided financial support but had no other role in the design of the study and collection, analysis, and interpretation of data or in writing of the manuscript.
Author information
Authors and Affiliations
Contributions
T.F.C. and N.B. conducted the entire study as a master’s degree working assignment, under the guidance of A.F.M. T.F.C., N.B., M.O., M.A., I.T.B. and L.F.O.: Were responsible for the design of the study, data collection, analysis and interpretation of the results, analysis of statistical data. T.F.C. and A.F.M.: Wrote the manuscript. N.B., M.O. and L.F.O.: Reviewed the manuscript. G.R.D.L., J.H.B.F., L.L.C.P. and P.B.: Are collaborating physicians in the study, requested the exams and participated with the clinical aspects of the cases. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chaves, T.F., Baretto, N., Oliveira, L.F.d. et al. Copy Number Variations in a Cohort of 420 Individuals with Neurodevelopmental Disorders From the South of Brazil. Sci Rep 9, 17776 (2019). https://doi.org/10.1038/s41598-019-54347-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-54347-z
This article is cited by
-
Genotype-phenotype associations in microtia: a systematic review
Orphanet Journal of Rare Diseases (2024)
-
A cohort study of neurodevelopmental disorders and/or congenital anomalies using high resolution chromosomal microarrays in southern Brazil highlighting the significance of ASD
Scientific Reports (2024)
-
Copy number variation analysis in 189 Romanian patients with global developmental delay/intellectual disability
Italian Journal of Pediatrics (2022)
-
Clinical evaluation of rare copy number variations identified by chromosomal microarray in a Hungarian neurodevelopmental disorder patient cohort
Molecular Cytogenetics (2022)
-
Chromosomal microarray analyses from 5778 patients with neurodevelopmental disorders and congenital anomalies in Brazil
Scientific Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
