
Abstract
Liriomyza is a large genus that includes polyphagous and invasive species (L. trifolii, L. sativae, and L. huidobrensis), and oligophagous species such as L. Chinensis in China. Effective control of these invasive and oligophagous species is not easy due to the fast invasion rate, interspecific competition, and pesticide resistance. In this study, we investigated population genetics of five Liriomyza species L. trifolii, L. sativae, L. huidobrensis, L. bryoniae, and L. chinensis based on COI and EF-1a genes, and microsatellite DNA. These five Liriomyza species revealed highly conservative characteristics in the COI gene among populations collected from different geographical regions and host plants. By contrast, the mutation rate of the EF-1a gene was higher than COI, and phylogenetic tree based on EF-1a showed that haplotypes of L. trifolii and L. sativae were not distinguished well. Genetic differentiation in microsatellite loci was obvious among the five species. Our results also indicated that geographic isolation had a greater impact on genetic differentiation in L. trifolii than the host plant. Populations of L. trifolii in China showed a high to moderate level of genetic differentiation and they had divided into two groups representing the coastal areas of southern China and northern regions. The genetic diversity of the southern group was higher than the northern group. We speculated that the invasion of L. trifolii likely occurred in southern regions of China and then spread northward. Bottleneck analyses revealed that the L. trifolii population in China was in a steady growth period.
Similar content being viewed by others

Introduction
Liriomyza is one of the largest genera belonging to the subfamily Phytomyzidae, family Agromyzidae, and order Diptera, consisting 330 described species1,2,3,4,5,6,7,8,9,10,11,12. Among these, 160 species are harmful in field crops or ornamental plants13, and 23 species are of great economic significance8.
Due to the small size, rapid interspecific competition, invasion rates, and adaptability, insects are sensitive to geographic isolation, hosts and phenological niches that cause species differentiation14,15,16. It has been speculated that host specialization has resulted in many new species in polyphagous Liriomyza that are highly adaptable to environmental stress9,17,18. To determine population genetic structure and migration patterns in L. sativae, several research groups analyzed different populations in China using fragments of ITS1 and β-tubulin genes and microsatellites14,19,20. In a study of Wang21, nuclear rDNA-ITS2 and mitochondrial COI sequences were used to analyze population differentiation in several invasive leafminer populations. It has been showed that populations of L. trifolii had separated into one clade representing the United States populations and a second clade for Asia-Europe populations, and a low level of differentiation was observed in domestic populations. However, mtDNA and ITS may not be the most suitable molecular markers for genetic differentiation analysis, because mitochondrial genes are highly conserved among intraspecific populations of insects and ITS is not suitable for analysis of intrageneric populations14,15,20. Microsatellites marker is a highly polymorphic co-dominant molecular marker with many characteristics, such as low requirements for DNA quality, good repeatability, simple detection methods, etc., and therefore it has been well applied in studies on population genetic structure, genetic relationship identification, genetic map construction and gene mapping to explore the population genetics, molecular systematics and ecology15,22. But there were a few researches using microsatellite marker technology to unfold the population genetic structure in Liriomyza especially for these invaded species14.
Previous studies on the population genetic structure of Liriomyza have generally involved only a single species14, with only a few comparative studies on genetic relationships among species21. In this study, we investigated intraspecies genetic differentiation in L. trifolii and interspecies variations among five species in Liriomyza in order to understanding better the species diversity during the geographic isolation and population expansion. Five species, namely L. trifolii, L. sativae, L. huidobrensis, L. bryoniae, and L. chinensis, were collected from 38 cities in 11 provinces of China. Population genetics differentiations of Liriomyza from different regions of China and host plants were evaluated using COI, EF-1a and microsatellite polymorphisms.
Results
Genetic differentiation of populations
Haplotype and nucleotide diversity of COI in L. trifolii populations was conserved with consistent characteristics among populations from different geographical regions and host plants. The maximum haplotype number among L. trifolii populations was three, and most haplotypes had only a single base difference. L. sativae populations showed slightly more diversity, and the maximum number of haplotypes among three L. sativae populations was six. The populations of L. huidobrensis, L. bryoniae and L. chinensis showed relatively low diversity (Table S1).
Haplotype and nucleotide diversity of EF-1a was relatively high among intraspecies (e.g. L. trifolii) as compared with COI. Ten haplotypes were found in the ZZJD, HLJD and HSFQ populations of L. trifolii, and the CSJD population had the highest nucleotide diversity. The other four Liriomyza spp. also showed relatively high diversity in EF-1a (Table S2).
The average observed number of alleles (Na) in L. trifolii populations ranged from 6.625 (DGJD) to 3.37 (HLJD). The average effective number of alleles (Ne) of L. trifolii populations ranged from 4.3885 (CXJD) to 1.9154 (SQNGMZ). The observed heterozygosity (Ho) values of ten L. trifolii populations were greater than 0.5, and the highest Ho was 0.6771 in the BLJD population; the remaining nine populations had Ho values less than 0.5 and the lowest was 0.3125 in the HLJD population. L. huidobrensis, L. bryoniae and L. chinensis had a low heterozygosity. Populations of different hosts in the same geographic region (DGQC and BLJD, NNQC and NNJD, SQJDMZ and SQNGMZ) showed a great degree of similarity in Na and Ho. Most populations were deviated from the Hardy-Weinberg equilibrium (Table S3).
Phylogenetic analyses
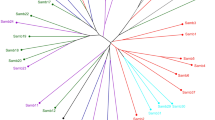
The phylogenetic tree based on COI haplotypes (Fig. 1) showed that the five Liriomyza species had an obvious interspecific differentiation. The species relationship between L. trifolii and L. sativae were the most closest, and between L. bryoniae and L. huidobrensis was closer, while the relationships of L. chinensis with each of the other four Liriomyza species were distant. The phylogenetic tree based on EF-1a haplotypes (Fig. 2) was similar as the phylogenetic tree based on COI haplotypes, but haplotypes of L. trifolii and L. sativae were not distinguished well.

BI phylogenetic tree of five Liriomyza species based on COI haplotypes. Black lines represent L. trifolii haplotypes, red lines represent L. sativae haplotypes, green line represents L. chinensis haplotype, blue line represents L. huidobrensis haplotype, orange lines represent L. bryoniae haplotypes.

BI phylogenetic tree of five Liriomyza species based on EF-1a haplotypes. Black lines represent L. trifolii haplotypes, red lines represent L. sativae haplotypes, green line represents L. chinensis haplotype, blue lines represent L. huidobrensis haplotypes, orange lines represent L. bryoniae haplotypes.
Genetic differentiation–pairwise FST analyses
Because of the obvious interspecific differentiation of COI and EF-1a in the five Liriomyza species, only the intraspecific genetic differentiations based on COI and EF-1a genes in L. trifolii populations was analyzed. The results based on COI showed that the HLJD population exhibited high genetic differentiations from other 19 populations, and the highest differentiation was found between HLJD and BLJD (Table 1). The results based on EF-1a showed that the HBJD population exhibited high genetic differentiations from other 19 populations, and the HBJD population showed the highest differentiation with the CXJD (Table 2).
In order to make clear interspecific and intraspecific nuclear genetic differentiations between five species of Liriomyza, pairwise FST scores of 25 populations (19 L. trifolii, three L. sativae, and one L. huidobrensis, L. bryoniae and L. chinensis populations) were compared based on 8 microsatellite loci (Table 3). Six pairwise FST values of L. trifolii populations were less than 0.05, and six were more than 0.25, and the other populations were between 0.05 and 0.25, indicating that most populations of L. trifolii were in a moderate level of genetic differentiation in China. The pairwise FST scores between the populations on different hosts in the same geographic region were 0.04457 for the NNQC and NNJD, 0.02928 for the DGQC and BLJD, 0.12234 for the HSFQ and HSJD, less than 0.05 for the SQJDMZ and SQNGMZ, and 0.08675 for the SQNGMZ and HNSGMZ, suggesting a lower genetic differentiation in microsatellite loci. The five Liriomyza species (especially L. trifolii vs. L. sativae) had high levels of interspecific genetic differentiation in microsatellite loci, although the species were similar in terms of morphology, niche occupation and feeding habits (Table 3).
Population genetic structure
Analysis of population genetic structure based on eight microsatellite loci
The phylogenetic tree of fifteen L. trifolii populations collected in two months in 2017 was constructed based on Nei’s genetic distances using UPGMA and the PHYLIP program. The UPGMA dendrogram (Fig. 3) showed that fifteen populations were basically clustered into two distinct main branches and four small scattered branches. Results of two population pairs NNQC/NNJD and DGQC/BLJD from different hosts in the same geographical region obviously converged to the nearest neighboring branch, which was consistent with pairwise FST analysis. However, the HSFQ/HSJD population pair did not converge. STRUCTURE analyses of the fifteen populations showed that the highest ΔK value was obtained for K = 2 (Fig. 4). Populations from coastal areas of southern China (DGQC, BLJD, ZZJD, HZQC) were assigned to one group (red portion of Fig. 4). Populations from Jiangsu and Zhejiang provinces and northern regions (CXJD, CSJD, SQJD, HSJD, HSFQ) were assigned to another group (green portion of Fig. 4).

UPGMA dendrogram between L. trifolii populations based on Nei’s genetic distances. HSFQ: Lycopersicon esculentum population in Hengshui, HDJD: Vigna unguiculata population in Handan, HZQC: Brassica chinensis population in Huizhou, NNQC: B. chinensis population in Nanning, NNJD: V. unguiculate population in Nanning, HLJD: V. unguiculate population in Hangzhou, CZJD: V. unguiculate population in Changzhou, WZJD: V. unguiculate population in Wuzhou, ZZJD: V. unguiculate population in Zhangzhou, DGQC: B. chinensis population in Dongguan, BLJD: V. unguiculate population in Dongguan, CSJD: V. unguiculate population in Changshu, HSJD: V. unguiculate population in Hengshui, SQJD: V. unguiculate population in Shangqiu, CXJD: V. unguiculate population in Huzhou.

Clustering analysis by structure for full-loci dataset between L. trifolii populations. (A) Inference of the number of genetic cluster (K) for L. trifolii populations. (B) Proportion of the genome of each individual assigned to each of the two clusters. Each individual is represented by a vertical bar.
Bottleneck test
Bottleneck analysis with populations of L. trifolii across China showed that none of these 19 L. trifolii populations exhibited heterozygosity under the stepwise mutation model (SMM), and there were only six populations (CXJD, CSJD, HSJD, HSFQ, ZZJD, WZJD) and nine populations with a statistically significant heterozygotes under the two-phase model (TPM) and the infinite allele model (IAM) (Table 3), respectively. These results indicated that the majority of L. trifolii populations did not undergo a genetic bottleneck and were in a steady growth period.
Discussion
Population genetic structure and diversity are important factors affecting the survival and adaptability of invasive species. Population genetics in many pests were studied to find out their invasion and transmission routes14,15,19,20,21,22,23. In this study, the phylogenetic tree, pairwise FST, and STRUCTURE analysis indicated that the degree of differentiation and direction of nuclear and mitochondrial genes were not completely consistent. COI in the five species of Liriomyza showed very conservative characteristics, but the mutation rate of EF-1a gene was relatively higher, and phylogenetic tree results showed that haplotypes of L. trifolii and L. sativae were not distinguished well. The results of microsatellite analysis showed that genetic distances among the five species of Liriomyza were significantly much longer than those within L. trifolii populations. In short, the five Liriomyza species showed high levels of genetic differentiation in mitochondrial and nuclear genes, and the interspecies differentiation in nuclear genes was obvious. COI and EF-1a gene were suitable molecular markers for interspecies genetic differentiation analysis and not for intraspecies of Liriomyza species, because COI as a mitochondrial gene and EF-1a as a reference gene are highly conserved among intraspecific populations of the five Liriomyza species. Microsatellites marker were suitable molecular markers for both interspecies and intraspecific genetic differentiation analysis of the five Liriomyza species, because microsatellite analysis showed both interspecies and intraspecific genetic differentiations among the five species of Liriomyza.
Spencer (1964) suggested that host specialization caused the development of many new species5. We found that geographic isolation had a greater influence on genetic differentiation within L. trifolii, which is consistent with previous results for L. Sativae14, but we did not find obvious influence of host plants on genetic differentiation in these species. We hypothesize that host plants have not yet driven reproductive isolation among populations, so the gene exchange among populations on different hosts occurs frequently.
The results of genetic differentiation and structure analysis showed that most populations of L. trifolii in China were in a high or moderate degree of genetic differentiation. Populations of L. trifolii could be divided into two groups, one from coastal areas of southern China and the other from northern China including Jiangsu and Zhejiang provinces. The genetic diversity of the southern group was higher than the other group, so the invasion of L. trifolii likely occured in southern regions of China and then spread toward northward. Bottleneck test analysis showed that the L. trifolii population in China was in a steady growth period, which was similar as L. sativae14. Genetic variation may lead to the rapid adaptation of insects to new environments and contributes to population establishment and spread. Our study has produced information on the geographical distribution of genetic variation of five Liriomyza species in China that may also help in management programs of these important pests.
Materials and Methods
Sample collection and DNA extraction
Liriomyza individuals (n = 281; Table 4) were collected and preserved in 100% ethanol at −20 °C until DNA extractions were performed. Genomic DNA was extracted from samples using the LabServ Tissue DNA Kit (Thermo Fisher Scientific, Massachusetts, USA) and then used for PCR.
Primers and microsatellite markers
The primers for mtDNA COI gene were referred to Simon et al.24. Specific primers for EF-1a gene and eight microsatellite primers were designed in this study (Supplementary Table S4). A fluorophore (FAM, ROX, HEX or TAMRA) was included at the 5’ end of each pair of microsatellite primers (Supplementary Table S5) used for genotyping. All the primers used in this experiment were synthesized by GENEWIZ Inc (Suzhou, China), and microsatellite genotyping was performed by GENEWIZ Inc.
PCR amplification and sequencing
The COI (n = 268; Supplementary Table S1) and EF-1a PCR (n = 252; Supplementary Table S2) of Liriomyza individuals (Table 4) were successfully amplified and sequenced. The amplification conditions were as follows: initial denaturation for 4 min at 94 °C, followed by 35 cycles of denaturation for 30 s at 94 °C, annealing for 30 s at 58 °C, elongation for 50 s at 72 °C, and a final extension step of 72 °C for 5 min. The microsatellite amplification of Liriomyza individuals (n = 281; Table 4) conditions were as follows: initial denaturation for 4 min at 94 °C, followed by 35 cycles of denaturation for 30 s at 94 °C, annealing for 30 s at 51–58 °C, elongation for 30 s at 72 °C, and a final extension step of 72 °C for 5 min. All amplified products were sequenced and genotyped by GENEWIZ Inc.
Data analysis of COI and EF-1a
The COI and EF-1a sequences were preliminarily aligned using the CLUSTALW program25. Haplotype diversity (h), nucleotide diversity (p), and the mean number of pairwise differences were calculated to estimate DNA polymorphism using DnaSP 5.026. Analysis of F-statistics (Fst) and genetic differentiation were performed using Arlequin v. 3.527 with 10,000 permutations. Phylogenetic relationships were deduced by Bayesian inference (BI) and maximum likelihood (ML). Phylogenetic trees were constructed using MrBayes v. 3.1.128 and a PHYML online web server29. For BI, nucleotide alignments were constructed using the MrBayes program with 20,000,000 generations and with the first 5000 discarded as burn-in. Support values for trees generated by BI were expressed as Bayesian posterior probabilities in percentages. ML analysis was performed by Mega 6.030. Tree information was visualized and edited using Treeview. The haplotype network was performed using NETWORK v. 4.631,32.
Data analysis of microsatellites
Fundamental genetic parameters were calculated for all eight loci using POPGENE v. 3.233 including the number of alleles (Na), the effective number of alleles (Ne), and observed (Ho) and expected heterozygosity (HE), as well as Nei’s genetic distance and genetic similarity. Deviation from Hardy-Weinberg equilibrium and linkage disequilibrium at each locus were calculated using GenePop v. 4.0 (http://wbiomed.curtin.edu.au/genepop/). The polymorphic information content (PIC) was calculated using Cervus 2.034. Differentiation indices (FST) were calculated using ARLEQUIN 3.527. A phylogenetic tree based on Nei’s genetic distance was constructed using the unweighted pair group with the arithmetic mean (UPGMA) method of PHYLIP v. 3.6935. Bootstrap values were calculated using 1000 replicates. To assess the population genetic structure, we used Bayesian model-based clustering analysis with STRUCTURE v. 2.3.336. We specified an initial range of potential genotype clusters (K) from 1 to 10 under the admixed model and the assumption of correlated allele frequencies among populations. For each value of K, ten runs were performed with 100,000 iterations discarded as burn-in followed by an additional 10,000 iterations. The most probable number of K values in the data was detected by comparing the log probability of the data lnP (D) for each value of K across all ten runs of Structure and by examining the standardized second-order change of lnP (D) and ΔK37. For selected K values, CLUMPP v 1.1.238 was used to align cluster membership coefficients from ten replicates of cluster analyses using the Greedy algorithm with 10,000 random input orders; the results were then graphically displayed with DISTRUCT v. 1.139. We also analyzed our data with the GENELAND package40 to further investigate the number of populations and the spatial location of genetic discontinuities between them. K was allowed to vary (1 to 10) with 100,000 MCMC iterations, and uncertainty was attached to spatial coordinates fixed to 1 km, and then the fixed modal K was obtained with the other parameters unchanged. A potentially significant heterozygosity excess (the signature of a bottleneck) was detected using a Wilcoxon signed rank test, as implemented in Bottleneck v. 1.241. When a population experiences a reduction of its effective size, it generally develops a heterozygosity excess at selectively neutral loci. Previous analyses have shown that the most useful markers for bottleneck detection are those evolving under IAM, and they provide guidelines for selecting sample sizes of individuals and loci41,42,43,44; meanwhile, the TPM is thought to more closely simulate microsatellite mutation45. Unlike the SMM, which predicts all mutations corresponding to the increment or decrement of a single base-pair repeat, the TPM predicts the occurrence of an occasional multiple base-pair repeat42. The strict SMM is obviously the most conservative model for testing for a significant heterozygosity excess caused by bottlenecks, because in some conditions it can produce a heterozygosity deficiency, and due to the heterozygosity excess it is always lower than other mutation models. Because the actual mutation model followed by our microsatellites is unknown, we ran the program Bottle neck under the IAM, SMM, and TPM to determine whether these populations recently experienced a population decline or not.
References
Frick, K. E. A synopsis of Agromyzidae leaf miners described from North America (Diptera). Proc. USNM. 108, 347–465 (1959).
Griffiths, G. C. D. Studies on boreal Agromyzidae (Diptera), V–VIII. Quaestionares Entomologicae, 35–314 (1974).
Menken, J., Trussell, J. & Larsen, U. Age and infertility. Science 233, 1389–1394 (1986).
Nowakowski, J. T. Introduction to a systematic revision of the family Agromyzidae (Diptera) with some remarks on host plant selection by these flies. Annu. Zool. Warszawa. 20, 67–183 (1962).
Spencer, K. A. The species-host relationship in the Agromyzidae (Diptera) as an aid to taxonomy. Insect Mol. Biol. 1, 101–102 (1964).
Spencer, K. A. Agromyzidae (Diptera) of economic importance. 9: Series Entomologica, ed. by E. Schimitschek, Gottingen. Bath: The Hague Publishers. 162–166 (1973).
Spencer, K. A. & Steyskal, G. C. Manual of the Agromyzidae (Diptera) of the United States. Agric. Handbook. 638, 478 (1986).
Spencer, K. A. Agromyzidae. In: Manual of Nearctic Diptera, Volume 2. Research Branch, Agriculture, Canada (1987).
Spencer, K. A. Host specialization in the world Agromyzidae (Diptera). Kluwer Academic Publishers, The Netherlands (1990).
Spencer, K. A. Flycatcher: Memoirs of an Amateur Entomologist. SOB Academic Publishing, The Hague, The Netherlands (1992).
Parrella, M. P., Jones, V. P., Youngman, R. R. & Lebeck, L. M. Effect of leaf mining and leaf stippling of Liriomyza spp. on photosynthetic rates of chrysanthemum. Ann. Entomol. Soc. Am. 78, 90–93 (1985).
Sasakawa, M. Oriental Agromyzidae (Diptera) in Bishop. Museum, Part.1. Pac. Insects. 15, 23–50 (1963).
Chen, W. L., Li, Z. Z., Gu, D. & Liu, Q. Y. Species of Liriomyza from China and description of 2 new record species (Diptera: Liriomyza). J. Southwest. Univ. 29, 154–158 (2007).
Tang, X. T., Ji, Y., Chang, Y. W. & Du, Y. Z. Population genetic structure and migration patterns of Liriomyza sativae in China: moderate subdivision and no Bridgehead effect revealed by microsatellites. B. Entomol. Res. 106, 114–123 (2016).
Tang, X. T., Tao, H. H. & Du, Y. Z. Microsatellite-based analysis of the genetic structure and diversity of Aleurocanthus spiniferus (Hemiptera: Aleyrodidae) from tea plants in China. Gene. 560, 107–113 (2015).
Blacket, M. J., Rice, A. D., Semeraro, L. & Malipatil, M. B. Dna-based identifications reveal multiple introductions of the vegetable leafminer Liriomyza sativae (Diptera: Agromyzidae) into the torres strait islands and papua new guinea. B. Entomol. Res. 105, 533–544 (2015).
Carolina, J. C. H. & Johnson, H. M. W. Host plant preference of Liriomyza sativae (Diptera: Agromyzidae) population infesting green onion in Hawaii. Environ. Entomol. 21, 1097–1102 (1992).
Bernays, E. A. & Chapman, R. E. Host-plant selection by phytophagous insects. London, Chapman & Hall (1994).
Wang, L. P., Du, Y. Z., He, W. T., Lu, W. J. & Lu, Z. Q. Sequence analysis and comparison of rDNA-ITS1 of geographical populations of Liriomyza sativae Blanchard (Diptera: Agromyzidae) from China and closely related species. Acta. Entomol. Sin. 6, 54–60 (2007).
Du, Y. Z., Wang, L. P., Lu, W. J., Zheng, F. S. & Lu, Z. Q. Differentiation of different host-populations and geo-populations of Liriomyza sativae Blanchard based on β-tubulin gene sequences. Sci. Agric. Sin. 41, 1542–1547 (2008).
Wang, S. Y., Lei, Z. R., Wang, H. H., Dong, B. X. & Ren, B. Z. The complete mitochondrial genome of the leafminer Liriomyza trifolii (Diptera: Agromyzidae). Mol. Biol. Rep. 38, 687–692 (2011).
Yang, W. Y., Tang, X. T., Cai, L., Dong, C. S. & Du, Y. Z. Isolation and characterization of nine microsatellite loci from the sycamore lace bug Corythucha ciliata (Hemiptera: Tingidae). Fla. Entomol. 97, 1070–1074 (2014).
Yang, W. Y., Tang, X. T., Ju, R. T., Zhang, Y. & Du, Y. Z. The population genetic structure of Corythucha ciliata (Hemiptera: Tingidae) provides insights into its distribution and invasiveness. Sci Rep-UK, 7, 635 (2017).
Simon, C. et al. Evolution, weighting and phylogenetics utility of mitochondrial gene sequences and compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 87, 651–701 (1994).
Chenna, R. et al. Multiple sequence alignment with the Clustal series of programs. Nucleic. Acids. Res. 31, 3497–3500 (2003).
Librado, P. & Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Ronquist, F. & Huelsenbeck, J. P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574 (2003).
Guindon, S., Lethiec, F., Duroux, P. & Gascuel, O. PHYML Online–a web server for fast maximum likelihood-based phylogenetic inference. Nucleic. Acids. Res. 33, W557–W559 (2005).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Templeton, A. R., Crandall, K. A. & Sing, C. F. A cladistic analysis of phenotypic associations with sequence types inferred from restriction endonuclease mapping and DNA sequence data: III. Cladogram estimation. Genetics 132, 619–633 (1992).
Bandelt, H. J. Networks with condorcet solutions. Eur. J. Oper. Res. 20, 314–326 (2007).
Yeh, F. C., Yang, R. C. & Boyle, T. POPGENE. Microsoft windows based freeware for population genetic analysis. Release 1.31. University of Alberta, Edmonton, Canada (1999).
Marshall, T. C. Statistical confidence for likelihood-based paternity inference in natural population. Mol. Ecol. 7, 639–655 (1998).
Felsenstein, J. Mathematics vs. evolution. Science 246, 941–942 (1989).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007).
Rosenberg, N. A. Distruct: a program for the graphical display of population structure: PROGRAM NOTE. Mol. Ecol. Notes. 4, 137–138 (2004).
Guillot, G., Mortier, F. & Estoup, A. Geneland: a computer package for landscape genetics. Mol. Ecol. Notes. 5, 712–715 (2005).
Cornuet, J. M. & Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics. 144, 2001–2014 (1996).
Peterson, A. C., Garzat, J. C., Valdes, A. M., Slatkint, M. & Freimer, N. B. Mutational processes of simple-sequence repeat loci in human populations. Proc. Natl. Acad. Sci. 91, 3166–3170 (1994).
Maruyama, T. & Fuerst, P. A. Population bottlenecks and nonequilibrium models in population genetics. II. Number of alleles in a small population that was formed by a recent bottleneck. Genetics. 111, 675–689 (1985).
Spencer, C. C., Neigel, J. E. & Leberg, P. L. Experimental evaluation of the usefulness of microsatellite DNA for detecting demographic bottlenecks. Mol. Ecol. 9, 1517–1528 (2000).
Primmer, C. R., Saino, N., Moller, A. P. & Ellegren, H. Unraveling the processes of microsatellite evolution through analysis of germ line mutations in barn swallows Hirundo rustica. Mol. Biol. Evol. 15, 1047–1054 (1998).
Acknowledgements
We express our deep gratitude to the Testing Center of Yangzhou University. This research was funded by the Basic Research Program of Agricultural Application of Suzhou (SNG201602), the Jiangsu Science and Technology Support Program (BE2014410), the Science and Technology Program of Yangzhou (YZ2014171), the Priority Academic Program Development of Jiangsu Higher Education, the National Science and Technology Support Program (2012BAD19B06) and the Yangzhou University International Academic Exchange Fund.
Author information
Authors and Affiliations
Contributions
J.Y.C., Y.W.C. and Y.Z.D. designed the experiments; J.Y.C., Y.W.C. and S.Z.Z. performed the experiments; J.Y.C., X.T.T and Y.W.C. analyzed the data and wrote the paper. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, JY., Chang, YW., Tang, XT. et al. Population genetics of Liriomyza trifolii (Diptera: Agromyzidae) and comparison with four Liriomyza species in China based on COI, EF-1a and microsatellites loci. Sci Rep 9, 17856 (2019). https://doi.org/10.1038/s41598-019-53886-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53886-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
