
Abstract
We developed anticancer drug-conjugated biodegradable polymer-nanoparticle-loaded adipose-derived stem cells (AdSCs) as a tool for biodrug delivery systems for cancer therapy. Pirarubicin was conjugated in polylactic/glycolic acid (PLGA) followed by formation of nanoparticles (NPs), which were loaded with human AdSCs and cocultured. The pirarubicin-conjugated PLGA NP-loaded AdSCs (PirNP-AdSCs) were overall viable within 48 h and exhibited significantly enhanced migration activity. We confirmed that pirarubicin was gradually released into the culture medium from PirNP-AdSCs, and the conditioned medium significantly inhibited the proliferation activity and induced the apoptosis of human pancreatic cancer cells (KP1N). PirNP-AdSCs also significantly induced tumor cell apoptosis in an ex vivo culture system with KP1N-derived tumors, and there was increased invasion/migration of PirNP-AdSCs inside the tumor. Finally, we compared the therapeutic efficacy of the PirNP-AdSCs on KP1N-derived tumor growth with that of treatments of AdSCs alone, PirNPs alone or normal saline (control) in immunodeficient mice. Subcutaneous local administration of PirNP-AdSCs significantly inhibited tumor growth, inducing the apoptosis of tumor cells and vasculature compared with the other groups. The present therapeutic strategy might give rise to a novel cancer therapy minimizing the adverse side effects of anticancer drugs in patients who suffer from cancer.
Similar content being viewed by others

Introduction
Pancreatic cancer is highly malignant and is the fourth most common cause of death due to cancer in Japan and in the US1. Pancreatic cancer patients have a poor prognosis, with statistical data indicating that the 5-year relative survival rate is currently 8%. Resection is the only an opportunity for a cure, but approximately half of the patients with pancreatic cancer already have metastases at the time of diagnosis. Standard chemotherapy is ineffective to improve the poor prognosis. Even if chemotherapy is performed, adverse events appear frequently2, so adequate treatment is difficult to achieve for patients with poor prognosis. Therefore, an efficient drug delivery system (DDS) seems to be important for future chemotherapy.
Mesenchymal stem cells have multiple characteristics and have received attention for their contrasting roles in cancer cell behavior. For instance, bone marrow-derived stem cells inhibit the proliferation of Kaposi’s sarcoma cells3. In addition, mesenchymal stem cells promote the proliferation of tumor cells derived from colon cancer, breast cancer and melanoma4,5. Mesenchymal stem cells are present not only in embryonic tissues such as bone marrow, skin, and placenta but also in adipose tissue. Like bone marrow-derived stem cells, adipose-derived stem cells (AdSCs) have been reported to both inhibit and promote cancer-derived cell proliferation. AdSCs promote the tumor growth of colon, lung, breast, glioblastoma and thyroid cancer6,7,8,9. However, AdSCs also inhibit pancreatic cancer and prostate cancer with metastasis growth in vitro and in vivo10,11,12. Based on the evidence that AdSCs have the ability to be recruited to sites of tumor via an NF-kB-involved signaling pathway13, AdSCs have been used as a vehicle for carrying viruses or nanoparticles (NPs) beyond the blood-brain barrier to brain tumors14,15.
The aim of this study was to develop a novel hybrid treatment for pancreatic cancer utilizing AdSCs and anticancer drugs with a slow drug-release system. We tested the hypothesis that when anticancer-drug-NP-loaded AdSCs are applied to tumor sites, AdSCs recruited to the tumor sites would not only exhibit an inhibitory effect on pancreatic cancer cell proliferation but also inhibit cancer cell proliferation with the slow release of anticancer drugs upon PLGA degradation, inducing apoptosis. The combination of anticancer-drug-loaded PLGA NPs and AdSCs could be a novel and effective therapy against pancreatic cancer that exhibits few adverse side effects.
Materials and Methods
hAdSC isolation and cell culture
Adipose tissue was obtained from the resected abdominal scars of patients undergoing a second cesarean section. The Osaka Medical College Ethics Committee approved this study (Protocol No. 1040–01), and all methods were performed in accordance with the relevant guidelines and regulations. Informed written consent was obtained from all subjects. The hAdSCs were isolated from the adipose tissue as previously described with minor modifications16. Briefly, the adipose tissue was washed in phosphate-buffered saline (PBS), minced, and digested in 5 ml of LiberaseTM Research Grade (Sigma-Aldrich Japan K.K., Tokyo, Japan) (0.5 mg/ml in 1% bovine serum albumin (BSA)/Hanks’ balanced saline solution; Roche Diagnostics, K.K., Tokyo, Japan) for 15 minutes at 37 °C. The digested tissue was filtered through a 40-mm cell strainer (BD Falcon, Tokyo, Japan) and centrifuged at 450 RCF for 10 minutes. The supernatant containing adipocytes and debris was discarded. The pelleted cells were collected as the stroma vascular fraction (SVF), and the number of cells not stained by trypan blue and sized 5–30 mm was measured using a conventional cytometer (LUNA; Logos Biosystems, Inc., Annandale, VA). The freshly isolated SVFs were cultured in 10% fetal bovine serum (FBS)/Dulbecco’s modified Eagle’s medium (DMEM)-F12 containing antibiotics on plastic dishes at a density of 104/cm2 under conditions of 5% CO2 and 37 °C. Adherent cells were passaged between passages 3 and 6 and used as maximum hAdSCs for the experiments17.
KP1N human pancreatic cancer cells were purchased from the JCRB (Japanese Collection of Research Bioresources) Cell Bank. The cells were maintained in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin (Life Technologies, Burlington, Ontario, Canada).
Pirarubicin-conjugated PLGA (Pir-PLGA) NP
A PLGA NP was generated as described previously18 Briefly, 25 mg of PLGA (#B6017–1 50:50 DL-PLG, Corefront, Inc.) and 4 mg of pirarubicin (Sigma-Aldrich) were dissolved in 1 ml of acetone. The solution was then placed in 20 ml of a 2% polyvinyl alcohol (PVA)/distilled water mixture and irradiated with an ultrasonic wave at a power level of 1.0 (Sonicator 3000, Misonix Inc.) for 10 minutes followed by stirring for 16 h to evaporate the acetone. Then, the supernatant was collected after centrifugation at 200 RCF for 10 minutes and further centrifuged at 15000 RCF for 30 minutes with additional distilled water for washing. The washing step was repeated 3 times, and PBS was added to the pellet and stirred with a homogenizer at 10,000 rpm for 10 minutes to disperse the NPs. Finally, the solution was freeze-dried using a lyophilizer (VD-550R, TAITEC, Tokyo, Japan) for over 48 h. The freeze-dried powder was used for all the experiments. We generated rhodamine-labeled Pir-PLGA NPs the same method by mixing rhodamine with pirarubicin as a landmark of incorporated NPs to AdSCs.
Uptake of Pir-PLGA NPs in AdSCs and release pattern of pirarubicin from AdSCs
Pir-PLGA NPs (0, 10, 25, 50 and 100 µg) were coincubated with adhered AdSCs for 30 minutes, and the uptake of Pir-PLGA in the AdSCs was visualized by a fluorescence confocal microscope (TCS SP8, Leica, Inc.) Spontaneous fluorescent signals were detected from Pir-PLGA loaded in the AdSCs. To evaluate the release of pirarubicin, the Pir-PLGA NPs (500 µg) were loaded onto the AdSCs (106 cells) and maintained in Dulbecco’s modified Eagle medium/Nutrient Mixture F-12 (DMEM/F-12, no phenol red) with 0% FBS. The culture medium was changed at 8, 24, 48, 72, and 96 h after initial plating. The concentration of pirarubicin in the collected medium was quantified by fluorometry. The fluorescence was measured using an EnSpire® Multimode Plate Reader (PerkinElmer, Inc., Waltham, MA) at excitation/emission wavelengths of 470/585 nm.
Cell proliferation assay
The proliferation activity of AdSCs was examined using the Cell Counting Kit-8 (Dojindo Laboratories, Kumamoto, Japan) according to the manufacturer’s instructions. Briefly, the Pir-PLGA NPs (0, 10, 25, 50 and 100 µg) or PLGA NPs (0, 10, 25, 50 µg) were incorporated into the AdSCs (1 × 105 cells). The AdSCs were then seeded onto 96-well culture plates at a density of 1.0 × 104 cells per well and cultured in DMEM/F12 containing 10% FBS for 24 or 48 h at 37 °C. For the coculture assays, KP1N cells were cocultured with AdSCs using a TranswellTM separate culture system for 48 h. A total of 1.0 × 105 KP1N cells and 1.0 × 104 AdSCs at a ratio of 10:1 were cultured in complete 10% FBS/DMEM.
Cell migration assay
The migration activity of Pir-PLGA NP-loaded AdSCs was evaluated by a migration assay using a TranswellTM insert with a 5.0 μm pore size for 24-well plates (Sigma-Aldrich). Briefly, the Pir-PLGA NPs or PLGA NPs (0, 10, 25, 50 and 100 µg) were incorporated into the AdSCs (1.0 × 105 cells). Two different approaches were adopted. First, a plate well was filled with AdSC growth medium (DMEM/F12 600 μl) with 20% FBS. The insert chamber was used to accommodate 2.0 × 104 AdSCs in DMEM/F12 (200 μl) with 0% FBS. A second plate well was filled with tumor growth medium (DMEM 600 μl) with 10% FBS and 5.0 × 104 KP1N cells. AdSCs in DMEM/F12 (200 μl) with 10% FBS were placed in the insert chamber. The cells were allowed to migrate for 4 h or 16 h at 37 °C, immediately followed by cell fixation with 4% paraformaldehyde (PFA). The nuclei were counterstained with DAPI (Sigma). The number of AdSCs on the basolateral side was counted under a computer-assisted fluorescence/light microscope BioZero BZ-X700 (Keyence, Osaka, Japan).
Cell apoptosis assay
Pir-PLGA NPs (0, 10, 25, 50 and 100 µg) were incorporated into the AdSCs (1.0 × 105 cells), and the Pir-PLGA NP-loaded AdSCs were cultured in DMEM/F12 containing 10% FBS for 48 h at 37 °C. The nuclei were counterstained with DAPI, a terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay (In Situ Cell Detection Kit, TMR red, Roche) was performed, and the cells were immediately observed under a computer-assisted fluorescence/light microscope (BioZero BZ-X700, Keyence, Osaka, Japan). For the coculture assays, 50 µg of Pir-PLGA NPs was incorporated into the AdSCs (1.0 × 105 cells). The KP1N cells were cocultured with AdSCs using transwells. A total of 1.0 × 105 KP1N cells and 1.0 × 104 AdSCs at different ratios (10:1) were cultured in complete DMEM with 10% FBS for 48 h. The KP1N cell nuclei were counterstained with DAPI, a TUNEL assay was performed, and the cells were immediately observed under a computer-assisted fluorescence/light microscope (BioZero BZ-X700).
Animals and experimental groups
All animal procedures were performed according to the guidelines of the Osaka Medical College Animal Care and Use Committee (Approved protocol No. 28081). Female nonobese diabetic-severe combined immunodeficiency (NOD-SCID) mice (CLEA Japan) aged 6–8 weeks were used in this study. A total of 2.0 × 106 KP1N cells with 60 μL of MatrigelTM (Becton Dickinson Labware, Franklin Lakes, NJ) were injected subcutaneously using a 28-gauge needle to create an in vivo model of pancreatic cancer. The mice were assigned into the following groups: 1) Control (50 µL of PBS), 2) Pir-PLGA NP-loaded AdSC (Pir-PLGA NPs 250 µg were incorporated into 5.0 × 105 AdSCs in 50 µL of PBS), 3) AdSC (5.0 × 105 in 50 µL of PBS), and 4) Pir-PLGA NPs (250 µg in 50 µL of PBS). These treatments were performed by injection to the marginal site of the tumor 21 days after xenograft tumor transplantation. Tumor volume measurements were performed once a week using the formula length X width X depth X 0.523619.
Assessment for adsc recruitment to tumor and pancreatic cancer cell apoptosis
A total of 2.0 × 106 KP1N cells with 60 μL of MatrigelTM were injected subcutaneously into the dorsal skin of 6- to 8-week-old female NOD-SCID mice to create an in vivo model of pancreatic cancer. On day 21, the xenografts were harvested and cocultured with NP-loaded (Pir-PLGA NPs and rhodamine-conjugated PLGA NPs) AdSCs in DMEM/F12 with 10% FBS in 24-well plates for 7 days.
Immunohistochemistry
The KP1N-derived xenografts were harvested on day 42. The tumors were fixed for 6 h in 4% PFA and incubated overnight in a 15% sucrose solution. The tissues were embedded in optimal cutting temperature (OCT) compound (Sakura FineTek, Japan) and sectioned at a 6-mm thickness. Fluorescent immunostaining was performed to detect tumor apoptosis and vascularity. The TUNEL assay (DeadEndTM Fluorometric TUNEL system, Promega) was used as a marker for apoptotic cells. Isolectin B4 (ILB4) (1:100; Vector Laboratories) was used for capillary staining with the DyLight 549 streptavidin-biotin binding method. Anti-mouse CD3 and F4/80 antigen (1:100; Thermo Fisher Scientific) were used for staining inflammatory cells with a secondary antibody of Alexa Fluor 594-conjugated IgG. The nuclei were counterstained with DAPI, and the sections were mounted in aqueous mounting medium. The images were examined under a computer-assisted fluorescence/light microscope (BioZero BZ-X700, Keyence, Osaka, Japan).
Histological analysis
The KP1N-derived xenografts were harvested on day 42. The tumors were fixed for 6 h in 4% PFA and incubated overnight in a 15% sucrose solution. The tissues were embedded in OCT compound and sectioned at a 6-mm thickness. Masson’s trichrome staining was performed to evaluate tumor fibrosis. The percentage of fibrosis in the entire tumor area was calculated using ImageJTM and Adobe Photoshop CS4 (Adobe Systems, San Jose, CA, USA) software.
Statistical analysis
All values are expressed as the mean ± s.e.m. Statistical analyses were performed using PrismTM software (GraphPad Software, CA, USA). Nonparametric unpaired t-tests (Mann–Whitney U-test) were used for comparisons between two groups, and repeated-measure two-way ANOVA with Bonferroni post hoc tests were used for comparing multiple groups. P values less than 0.05 were considered statistically significant.
Results
Uptake of Pir-PLGA NPs in AdSCs and extracellular release of pirarubicin
We first generated and examined several kinds of Pir-PLGA NPs for the experiments. The amount of pirarubicin increased in the NPs in a dose-dependent manner. (Suppl. Fig. 1) In this study, we used 25 mg PLGA (50:50) and 4.0 mg pirarubicin to form the Pir-PLGA NPs. The average size of the Pir-PLGA NPs were 357.3 ± 10 nm.
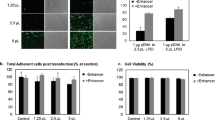
We confirmed the loading of Pir-PLGA NPs in the AdSCs under a confocal laser microscope. The Pir-PLGA NPs were excited by a laser at 560 nm wavelength, and the AdSCs were visualized in red; in this way, the dose-dependent uptake of Pir-PLGA NPs in the AdSCs could be observed. (Fig. 1a) We also confirmed the release pattern of pirarubicin by measuring its concentration in medium with Pir-PLGA NP-loaded AdSCs (500 µg/1 × 106 of AdSCs). Pirarubicin was gradually released up to 48 h after uptake of Pir-PLGA NPs in the AdSCs. Although a large number of Pir-PLGA NP-loaded AdSCs died 7 days after incubation, the attached/survived AdSCs still contained 1.92 ± 0.3% pirarubicin.

Uptake of Pir-PLGA NP and pirarubicin release from AdSCs.Pir-PLGA NPs (0, 10, 25, 50 and 100 µg) were incorporated into the AdSCs (1.0 × 105 cells). (a) Confocal microscopy was used to confirm the uptake of rhodamine-labeled Pir-PLGA NPs (red) in AdSCs. The nuclei were stained with DAPI (blue). (b) The medium was collected at 8, 24, 48, 72, and 96 h after uptake of 500 µg Pir-PLGA NPs by the AdSCs (1.0 × 106 cells). The amount of pirarubicin in the collected medium was quantified by fluorometry and is expressed as a percent of the initially loaded pirarubicin that was released in the AdSCs.
Pir-PLGA NP loading did not alter AdSC migration activity but inhibited proliferation activity
We assessed the cell functions of Pir-PLGA NP-loaded AdSCs to evaluate the influence of Pir-PLGA on the AdSCs following 48 h of incubation. The proliferation of Pir-PLGA NP-loaded AdSCs significantly declined in a dose-dependent manner (Fig. 2a), and the trend was similar to that following 16 h and 24 h of incubation. (Suppl. Fig. 1b) The apoptosis of AdSCs was induced at high doses of 50 µg < 100 µg. (Fig. 2c) The migration of AdSCs toward FBS-containing medium declined at 16 h (Fig. 2b) but increased toward KP1N cells after uptake of PLGA NPs (Fig. 2d) and even with Pir-PLGA NPs. (Fig. 2e) At a short time (4 h) after uptake of Pir-PLGA NPs, the migration of AdSCs toward the FBS-containing medium did not decline. (Suppl. Fig. 3b) In addition, the PLGA NPs did not influence the proliferation of the AdSCs (Suppl. Fig. 2a) and the migration toward the FBS-containing medium in a short time (Suppl. Fig. 3a).

Cell functional activities of Pir-PLGA NP-loaded AdSCs.NPs (0, 10, 25, 50 and 100 µg) were incorporated into the AdSCs (1.0 × 105 cells). NP 0 µg incorporated into the AdSCs is the control (Ctrl). (a) The proliferation activity of AdSCs was examined using the Cell Counting Kit-8 after 48 h. (b) The chemotactic migration of AdSCs toward FBS-containing growth medium was evaluated at 16 h. (c) Apoptosis was detected by the TUNEL assay at 48 h. The percentage of apoptotic TUNEL-positive cells was quantified. PLGA NPs (0, 10, 25, 50 and 100 µg) were incorporated into the AdSCs (1.0 × 105 cells). Chemotactic migration of PLGA NP-loaded AdSCs toward KP1N cells at 16 h (d) and Pir-PLGA NP-loaded AdSCs toward KP1N cells at 16 h (e). ns, not significant; *P < 0.05; ***P < 0.001; and ****P < 0.0001 vs. Ctrl.
Pir-PLGA NP-loaded AdSCs inhibited proliferation and induced apoptosis of KP1N cells
We tested the hypothesis that pirarubicin released from Pir-PLGA NP-loaded AdSCs would inhibit the proliferation activity of KP1N cells when cocultured in vitro. First, the Pir-PLGA NPs or PLGA NPs were incorporated into the AdSCs. Then, the KP1N cells and the AdSCs were cocultured separately. The KP1N cells were also cultured with Pir-PLGA NPs or PLGA NPs only. The proliferation of the KP1N cells was significantly inhibited at 48 h, specifically, at a dose of 50 µg Pir-PLGA NPs in 1.0 × 105 AdSCs. (Fig. 3a) The apoptosis of KP1N was examined at 48 h. The Pir-PLGA NP-loaded AdSCs (50 µg Pir-PLGA NPs in 1.0 × 105 AdSCs) and Pir-PLGA NPs induced much more apoptosis compared with AdSCs alone (0 µg Pir-PLGA NPs). (Fig. 3b,c)

Proliferation and apoptosis of KP1N cells cocultured with Pir-PLGA NP-loaded AdSCs. (a) Pir-PLGA NPs (0, 10, 25, 50 and 100 µg) were incorporated into AdSCs (1.0 × 105 cells). The KP1N cells (1 × 105) and treated AdSCs (2 × 104) were cocultured separately. The KP1N cells were also cultured with Pir-PLGA NPs or PLGA NPs only (same amount incorporated into 1.0 × 105 AdSCs for 50 µg Pir-PLGA NPs). The proliferation activity of KP1N cells was examined, and that of KP1N cells before coculture was used as control (Ctrl). The KP1N cells were cocultured for 48 h with AdSCs, Pir-PLGA NP-loaded AdSCs (50 µg to 1.0 × 105cells), or Pir-PLGA NPs only (same amount incorporated into 1.0 × 105 AdSCs for 50 µg Pir-PLGA NPs). (b) The detection of KP1N apoptosis (red) was performed by the TUNEL assay. (c) The percentage of apoptotic cells was quantified. The nuclei were counterstained with DAPI (blue). **P < 0.01; and ****P < 0.0001 vs. Ctrl.
AdSCs recruited to ex vivo cultured tumor inducing apoptosis
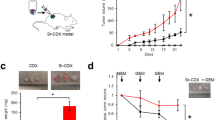
The KP1N cells were inoculated in the NOD-SCID mice subcutaneously. On day 21 after xenograft transplantation, the tumors were harvested and cultured with NP-loaded (Pir-PLGA NPs and rhodamine-conjugated PLGA NPs) AdSCs for 7 days. The AdSCs accumulated at the tumor, specifically, at a dose of 25 and 50 µg Pir-PLGA NPs in 1.0 × 105 AdSCs. (Fig. 4a,b) Most of the AdSCs were distributed to the marginal part of the tumor, but some of the AdSCs were observed evenly in the center of the tumor. Tumor apoptosis was induced along with the recruited AdSCs, specifically, at a dose of 50 µg Pir-PLGA NPs in 1.0 × 105 AdSCs. (Fig. 4c)

AdSC accumulation and apoptosis of tumors (ex vivo). The KP1N cells were transplanted subcutaneously into NOD-SCID mice. On day 21, the xenografts/tumors were harvested and cultured with 1.0 × 105 NP-loaded (0, 10, 25, 50 and 100 µg Pir-PLGA NPs and 50 µg rhodamine-conjugated PLGA NPs) AdSCs for 7 days. AdSCs that accumulated in the tumors were counted. Additionally, apoptosis of the tumor cells (green) was evaluated by the TUNEL assay. NPs 0 µg incorporated into the AdSCs is the control (Ctrl). (a) The nuclei were counterstained with DAPI (blue). The AdSCs were labeled with rhodamine (red). (b) The percentage of accumulated AdSCs in the tumor cells was quantified. (c) The percentage of apoptotic tumor cells was quantified. ns, not significant; and ****P < 0.0001 vs. Ctrl.
Pir-PLGA NP-loaded AdSCs inhibited KP1N cell-derived tumor growth
The KP1N cells were inoculated in the NOD-SCID mice subcutaneously. On day 21 after xenograft transplantation, 5.0 × 105 AdSCs (250 µg Pir-PLGA NP-loaded or none) or only Pir-PLGA NPs (250 µg) were transferred to the margin of the tumor. Tumor volume measurement was performed once a week. Tumor growth was inhibited in the order of Pir-PLGA NP-loaded AdSCs, AdSCs and Pir-PLGA NPs only. (Fig. 5a,5b) The tumors were harvested on day 21 after injection of Pir-PLGA NP-loaded AdSCs, AdSCS, or Pir-PLGA NPs only. Histological analysis performed to assess tumor fibrosis (Fig. 5c) showed that fibrosis was low and in the order of Pir-PLGA NP-loaded AdSCs, AdSCs and Pir-PLGA NPs only on the marginal and central sites. (Fig. 5d) Fluorescent immunostaining was performed to assess tumor apoptosis and vascularity (Fig. 6a,b). For apoptosis, there was no difference in the margin of the tumor. However, there was significantly more induced apoptosis at the center of the tumor in the Pir-PLGA NP-loaded AdSCs group than in the other groups. (Fig. 6c) The vascularity of the tumors was lower in the order of Pir-PLGA NP-loaded AdSCs, AdSCs and Pir-PLGA NPs only. (Fig. 6d) The apoptosis of tumor blood vessels in the AdSCs was significantly higher at the marginal site than at the center of the tumor, and that in the Pir-PLGA NP-loaded AdSCs was higher at the center than at the margin of the tumor. (Fig. 6e) In addition, we tried to confirm the presence of inflammatory cells on the tumors by staining the tumors cells with DAPI, CD3, and F4/80 to assess inflammatory cell infiltration; however, inflammatory cells were not observed. (Suppl. Fig. 4)

Effect of Pir-PLGA NP-loaded AdSCs on KP1N cell-derived tumor growth and histological assessment. The KP1N cells were transplanted subcutaneously into NOD-SCID mice, and the xenograft was developed for tumor formation on day 21. (a) The following treatments were performed on the mice, and observations were made for 21 more days. The dotted circles indicate xenograft/tumor on the dorsal side. (b) PBS (Control, Ctrl), AdSCs (5.0 × 105) alone, Pir-PLGA NPs (250 µg) alone, or Pir-PLGA NP (250 µg)-loaded AdSCs (5.0 × 105) were subcutaneously injected into the margin of the xenograft/tumor. The tumor size was measured weekly in each group and evaluated by calculating the percent of tumor size at each time point after the treatment and is expressed as growth relative to the initial xenograft/tumor. (c) The tumors were stained with Masson’s trichrome stain for assessment of fibrosis (blue). (d) The fibrosis area was quantified and is expressed as the percent of total tissue area in the marginal (left panel) and central (right panel) sites of the tumor. ns, not significant; *P < 0.05; **P < 0.01; and ****P < 0.0001 vs. Ctrl.

Immunofluorescence histological assessment of tumor apoptosis and vascularity. The tumors were harvested on day 21 after the administration of PBS (Control, Ctrl), AdSCs (5.0 × 105) alone, Pir-PLGA NPs (250 µg) alone, or Pir-PLGA NPs (250 µg)-loaded AdSCs (5.0 × 105). The tumors were stained by the TUNEL method (green) for apoptosis (a) and an antibody (ILB4) for vasculature (red) (b). The nuclei were counterstained with DAPI (blue). The marginal (upper panels) and central (lower panels) sites of the tumor were assessed. (c) The percentage of apoptotic tumor cells was quantified. (d) The percentage of ILB4-stained vascular cells was quantified. (e) The percentage of apoptotic cells out of the ILB4-positive cells was quantified. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001 vs. Ctrl.
Single treatment was more effective than multiple treatments
To compare the effect of single versus and multiple (3 repeats) treatments, we administered either one injection of 1 × 105 of Pir-PLGA NP-loaded AdSCs or multiple (3) injections of 1/3 the number (3.3 × 104) of Pir-PLGA NP-loaded AdSCs into the marginal site of the tumor on days 21, 28, and 35 after the xenograft transplantation. At first, there was a gradual difference between the single treatment and multiple treatments, and the difference became significant on day 42 in the xenografts. (Fig. 7a) The tumors were harvested on day 42 and examined histologically. (Fig. 7b) Apoptosis of the tumor (Fig. 7c) and blood vessels (Fig. 7d) was significantly higher in the marginal site than in the central site in the multiple-treatment group. In contrast, tumor and blood vessel apoptosis was significantly higher in the central site than in the marginal site in the single-treatment group. The vascularity and fibrosis in the multiple-treatment group were higher than that in the single-treatment group. (Fig. 7e,f, respectively)

Effect of single treatment versus multiple treatments on KP1N cell-derived tumor growth. The KP1N cells were subcutaneously transplanted into NOD-SCID mice, and the xenograft was developed for tumor formation on day 21. (a) The following treatments (local administrations) were performed on the mice, and observations were made for 21 more days. A single treatment with Pir-PLGA NP (250 µg)-loaded AdSCs (5.0 × 105) and multiple (three) treatments with Pir-PLGA NP (250 µg)-loaded AdSCs (1/3 of 5.0 × 105) were subcutaneously administered to the margin of the xenograft/tumor, and the size of the tumor was measured weekly. ns, not significant; *P < 0.05 vs. single treatment. (b) The tumors were stained with Masson’s trichrome stain for assessment of fibrosis (blue), the TUNEL method (green) for apoptosis and an antibody (ILB4) for vasculature (red). The nuclei were counterstained with DAPI (blue). The marginal (upper panels) and central (lower panels) sites of the tumor were assessed. PBS subcutaneously administrated to the margin of the xenograft/tumor instead of Pir-PLGA NPs-loaded AdSCs is the control (Ctrl). (c) The percentage of apoptotic tumor cells was quantified. (d) The percentage of apoptotic cells out of the ILB4-positive cells was quantified. (e) The percentage of ILB4-stained vascular cells was quantified. (f) The fibrosis area was quantified and is expressed as a percent of the total tissue area. ns, not significant; *P < 0.05; **P < 0.01; ***P < 0.001; and ****P < 0.0001 vs. Ctrl.
Discussion
In the present study, we demonstrated that Pir-PLGA NP-loaded AdSCs reduced the growth of pancreatic cancer by inhibiting KP1N proliferation and inducing apoptosis both in vitro and in vivo. PLGA protects drug molecules and provides physicochemical stability20. NPs are taken up by cells more efficiently than larger micromolecules and, therefore, could be used as an effective delivery system and have been extensively investigated21,22.
We first examined Pir-PLGA NPs made of PLGA NP and pirarubicin compounded at different proportions. When the encapsulation efficiency was high, the amount of pirarubicin appearing on the surfaces increased, and the PLGA NP negatively affected cell viability after being taken up by the AdSCs. (data not shown). In this study, we used 25 mg of Pir-PLGA NP compounded with 25 mg of PLGA (50:50) and 4.0 mg of pirarubicin for the experiments. Our in vitro results demonstrated that a high dose of Pir-PLGA NPs (50 µg Pir-PLGA NPs in 1.0 × 105 AdSCs) negatively influenced the viability of the AdSCs, while a high dose of Pir-PLGA NP-loaded AdSCs effectively inhibited cancer cell proliferation. In addition, even a high dose of Pir-PLGA NPs did not influence migration activity for at least 16 h (incubation time in a 5% CO2 incubator), suggesting that the Pir-PLGA NP-loaded AdSCs could migrate into the inside of the tumor, releasing pirarubicin, which could lead to effective anticancer therapy. In an ex vivo experiment, we also confirmed the accumulation and migration of AdSCs inside the tumor, resulting in tumor cell and vascular apoptosis. Specifically, this experiment was conducted with a dose of Pir-PLGA NPs to minimize the inhibitory effect on migration activity, which is one of the important mechanisms by which AdSCs enter the central site of the tumor. This dose of Pir-PLGA NPs also minimized the apoptosis of AdSCs (less than 5% in 48 h after uptake of Pir-PLGA NPs). Based on the results of the in vitro and ex vivo experiments, we finally decided to use a dose of 50 µg Pir-PLGA NPs in 1.0 × 105 AdSCs for the in vivo experiments. In vivo experiments, we first challenged intravascular administration of AdSCs as previous report13, that MSCs injected to left ventricular cavities accumulated to subcutaneous tumor. This thought to be ideal place of administration, but we could not confirm the accumulation of AdSCs (data not shown). This time we chose the method to inject to the marginal site of the tumor.
Immunofluorescence staining indicated that cell apoptosis at the central site of the tumor, including tumor blood vessels, was significantly higher in Pir-PLGA NP-loaded AdSCs than in the other treatment groups. As a result, the vascularity of the tumors was reduced in the Pir-PLGA NP-loaded AdSC group, suggesting that the AdSCs carried Pir-PLGA NPs to the central site, releasing pirarubicin. Indeed, the numbers of AdSCs recruited to the tumor and apoptotic tumor cells were significantly increased at the central site in the Pir-PLGA NP-loaded AdSC group. The inhibition of tumor growth by Pir-PLGA NP-loaded AdSCs is attributed to the dual effect of the released pirarubicin, which has an inhibitory effect on DNA replication and secreted anti-pancreatic cancer factors, i.e., TRAIL23, transcriptional intermediary factor 1 gamma (Tif1gamma)24, and microRNA-14525,26, and from the recruited AdSCs on proliferating/apoptotic tumor and vascular cells. Vascular cell apoptosis, which was frequently observed in the Pir-PLGA NP-loaded AdSC group, is also considered a crucial finding/mechanism by which the nutrient/blood supply for tumor growth was reduced. The antiinflammatory effect of AdSCs is also considered a possible reason why AdSCs reduce tumor growth by increasing CD4-positive regulatory T cells inhibiting IL-17 production27. However, we could not assess the reason because inflammatory cells were not observed in the tumors in any experimental groups (Suppl. Fig. 4) as NOD-SCID mice lack T cells and B cells and have reduced macrophage function28,29,30. Although there was less inflammation in the tumors, fibrosis derived from the host/mouse tissue that was required for tumor growth could be observed, and the extent of tumor fibrosis was significantly reduced in the Pir-PLGA NP-loaded AdSC group.
We also compared the effect of single treatment versus multiple treatments on the tumor growth of xenografts. In a previous study on human gastric cancer, the group that received a weekly low-dose treatment had better outcome than the group that received a high-dose treatment every 3 weeks31. In contrast, our study indicated that a single high-dose treatment was significantly more effective than multiple low-dose (1/3 of the high dose) treatments. Histological findings for these two groups showed that there was a large difference in the percentage of vascular apoptosis between the marginal site and the central site of the tumor. The multiple low-dose treatment induced tumor cell and vascular apoptosis in the marginal site but not in the central site of the tumor, suggesting that a small number of Pir-PLGA NP-loaded AdSCs were not able to enter the tumor due to their spontaneous apoptosis as a result of pirarubicin release even though they could reach the marginal site during all three treatments. In contrast, some Pir-PLGA NP-loaded AdSCs that reached the marginal site might have been able to enter the tumor even during the single treatment, inducing tumor cell and vascular apoptosis in the central site of the tumor, which effectively reduced the tumor size. In general, pirarubicin is administered to patients at a dose of 40–60 mg/body for breast cancer, gastric cancer, and ovarian cancer in clinical settings. For NOD-SCID mice32, this dose corresponds to approximately 160–245 µg/mouse. The theoretical dose of pirarubicin administered in this study was 0.891 µg (pirarubicin/PLGA NP = 3.564 µg/mg × 0.25)/mouse, which was drastically low. Thus, the burden on the body would be considered to be minimal compared with that in general systemic chemotherapy. For this reason, anticancer therapy with Pir-PLGA NP-loaded AdSCs will allow us to repeat the treatment without inducing severe side effects in patients.
This study has some limitations. First, although intravascular administration of AdSCs thought to be ideal place of administration, we chose the method to inject to the marginal site of the tumor. Second, evaluation of effect of drug-loaded AdSCs against tumor growth is only based on morphological data that no molecular data is included. Adding molecula data may reveal more interesting facts, therefore further research is desired in the future.
In conclusion, we have demonstrated that only a single local treatment with Pir-PLGA NP-loaded AdSCs effectively inhibited the growth of human pancreatic cancer cell-derived xenografts, inducing tumor cell apoptosis in mice. Our results obtained in this study may give rise to a novel, next-generation anticancer therapy not only for pancreatic cancer but also for other types of cancers.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Change history
28 September 2021
This article has been retracted. Please see the Retraction Notice for more detail: https://doi.org/10.1038/s41598-021-99148-5
References
Siegel, R. L., Miller, K. D. & Jemal, A. Cancer Statistics, 2017. CA Cancer J Clin 67, 7–30, https://doi.org/10.3322/caac.21387 (2017).
Conroy, T. et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med 364, 1817–1825, https://doi.org/10.1056/NEJMoa1011923 (2011).
Khakoo, A. Y. et al. Human mesenchymal stem cells exert potent antitumorigenic effects in a model of Kaposi’s sarcoma. J Exp Med 203, 1235–1247, https://doi.org/10.1084/jem.20051921 (2006).
Sun, B. et al. Correlation between melanoma angiogenesis and the mesenchymal stem cells and endothelial progenitor cells derived from bone marrow. Stem Cells Dev 14, 292–298, https://doi.org/10.1089/scd.2005.14.292 (2005).
Karnoub, A. E. et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449, 557–563, https://doi.org/10.1038/nature06188 (2007).
Yu, J. M., Jun, E. S., Bae, Y. C. & Jung, J. S. Mesenchymal stem cells derived from human adipose tissues favor tumor cell growth in vivo. Stem Cells Dev 17, 463–473, https://doi.org/10.1089/scd.2007.0181 (2008).
Muehlberg, F. L. et al. Tissue-resident stem cells promote breast cancer growth and metastasis. Carcinogenesis 30, 589–597, https://doi.org/10.1093/carcin/bgp036 (2009).
Kucerova, L., Altanerova, V., Matuskova, M., Tyciakova, S. & Altaner, C. Adipose tissue-derived human mesenchymal stem cells mediated prodrug cancer gene therapy. Cancer Res 67, 6304–6313, https://doi.org/10.1158/0008-5472.CAN-06-4024 (2007).
Kandil, E. et al. A novel mouse model of metastatic thyroid carcinoma using human adipose tissue-derived stromal/stem cells. Anticancer Res 33, 4213–4217 (2013).
Takahara, K. et al. microRNA-145 Mediates the Inhibitory Effect of Adipose Tissue-Derived Stromal Cells on Prostate Cancer. Stem Cells Dev 25, 1290–1298, https://doi.org/10.1089/scd.2016.0093 (2016).
Cousin, B. et al. Adult stromal cells derived from human adipose tissue provoke pancreatic cancer cell death both in vitro and in vivo. PLoS One 4, e6278, https://doi.org/10.1371/journal.pone.0006278 (2009).
Takahara, K. et al. Adipose-derived stromal cells inhibit prostate cancer cell proliferation inducing apoptosis. Biochem Biophys Res Commun 446, 1102–1107, https://doi.org/10.1016/j.bbrc.2014.03.080 (2014).
Uchibori, R. et al. NF-kappaB activity regulates mesenchymal stem cell accumulation at tumor sites. Cancer Res 73, 364–372, https://doi.org/10.1158/0008-5472.CAN-12-0088 (2013).
Josiah, D. T. et al. Adipose-derived stem cells as therapeutic delivery vehicles of an oncolytic virus for glioblastoma. Mol Ther 18, 377–385, https://doi.org/10.1038/mt.2009.265 (2010).
Huang, W. C. et al. Tumortropic adipose-derived stem cells carrying smart nanotherapeutics for targeted delivery and dual-modality therapy of orthotopic glioblastoma. J Control Release 254, 119–130, https://doi.org/10.1016/j.jconrel.2017.03.035 (2017).
Bai, X. et al. Both cultured and freshly isolated adipose tissue-derived stem cells enhance cardiac function after acute myocardial infarction. Eur Heart J 31, 489–501, https://doi.org/10.1093/eurheartj/ehp568 (2010).
Otsuki, Y. et al. W9 peptide enhanced osteogenic differentiation of human adipose-derived stem cells. Biochem Biophys Res Commun 495, 904–910, https://doi.org/10.1016/j.bbrc.2017.11.056 (2018).
Koda, S., Okumura, N., Kitano, J., Koizumi, N. & Tabata, Y. Development of Poly Lactic/Glycolic Acid (PLGA) Microspheres for Controlled Release of Rho-Associated Kinase Inhibitor. J Ophthalmol 2017, 1598218, https://doi.org/10.1155/2017/1598218 (2017).
Gleave, M. E., Bruchovsky, N., Moore, M. J. & Venner, P. Prostate cancer: 9. Treatment of advanced disease. CMAJ 160, 225–232 (1999).
Danhier, F. et al. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release 161, 505–522, https://doi.org/10.1016/j.jconrel.2012.01.043 (2012).
Stylios, G. K., Giannoudis, P. V. & Wan, T. Applications of nanotechnologies in medical practice. Injury 36(Suppl 4), S6–S13, https://doi.org/10.1016/j.injury.2005.10.011 (2005).
Brannon-Peppas, L. & Blanchette, J. O. Nanoparticle and targeted systems for cancer therapy. Adv Drug Deliv Rev 56, 1649–1659, https://doi.org/10.1016/j.addr.2004.02.014 (2004).
Grisendi, G. et al. Adipose-derived mesenchymal stem cells as stable source of tumor necrosis factor-related apoptosis-inducing ligand delivery for cancer therapy. Cancer Res 70, 3718–3729, https://doi.org/10.1158/0008-5472.CAN-09-1865 (2010).
Ligr, M. et al. Imbalanced expression of Tif1gamma inhibits pancreatic ductal epithelial cell growth. Am J Cancer Res 4, 196–210 (2014).
Khan, S. et al. MicroRNA-145 targets MUC13 and suppresses growth and invasion of pancreatic cancer. Oncotarget 5, 7599–7609, https://doi.org/10.18632/oncotarget.2281 (2014).
Han, T., Yi, X. P., Liu, B., Ke, M. J. & Li, Y. X. MicroRNA-145 suppresses cell proliferation, invasion and migration in pancreatic cancer cells by targeting NEDD9. Mol Med Rep 11, 4115–4120, https://doi.org/10.3892/mmr.2015.3294 (2015).
Larocca, R. A. et al. Adipose tissue-derived mesenchymal stem cells increase skin allograft survival and inhibit Th-17 immune response. PLoS One 8, e76396, https://doi.org/10.1371/journal.pone.0076396 (2013).
Hudson, W. A., Li, Q., Le, C. & Kersey, J. H. Xenotransplantation of human lymphoid malignancies is optimized in mice with multiple immunologic defects. Leukemia 12, 2029–2033 (1998).
Shultz, L. D. et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol 154, 180–191 (1995).
Bosma, M., Schuler, W. & Bosma, G. The scid mouse mutant. Curr Top Microbiol Immunol 137, 197–202 (1988).
Shitara, K. et al. Nab-paclitaxel versus solvent-based paclitaxel in patients with previously treated advanced gastric cancer (ABSOLUTE): an open-label, randomised, non-inferiority, phase 3 trial. Lancet Gastroenterol Hepatol 2, 277–287, https://doi.org/10.1016/S2468-1253(16)30219-9 (2017).
Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Pharmacology and Toxicology, 1-27 (2005).
Acknowledgements
The authors thank Eiko Kohbayashi and Chinatsu Shiraoka for their helpful technical support.
Author information
Authors and Affiliations
Contributions
Conception and design: M. Aoki and K. Kakimoto Development of methodology: M. Aoki and K. Kakimoto Acquisition of data (provided animals, provided facilities, etc.): M. Aoki Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): M. Aoki and K. Kakimoto Writing, review, and/or revision of the manuscript: M. Aoki Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): M. Aoki Study supervision: K. Kakimoto, M. Goto, and K. Higuchi
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article has been retracted. Please see the retraction notice for more detail:https://doi.org/10.1038/s41598-021-99148-5
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Aoki, M., Kakimoto, K., Goto, M. et al. RETRACTED ARTICLE: Novel Therapeutic Approach Using Drug-loaded Adipose-derived Stem Cells for Pancreatic Cancer. Sci Rep 9, 17971 (2019). https://doi.org/10.1038/s41598-019-53807-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53807-w
This article is cited by
-
Research update of adipose tissue-based therapies in regenerative dermatology
Stem Cell Reviews and Reports (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
