
Abstract
Bowel microbiota is a “metaorgan” of metabolisms on which quantitative readouts must be performed before interventions can be introduced and evaluated. The study of the effects of probiotic Clostridium butyricum MIYAIRI 588 (CBM588) on intestine transplantees indicated an increased percentage of the “other glycan degradation” pathway in 16S-rRNA-inferred metagenomes. To verify the prediction, a scoring system of carbohydrate metabolisms derived from shotgun metagenomes was developed using hidden Markov models. A significant correlation (R = 0.9, p < 0.015) between both modalities was demonstrated. An independent validation revealed a strong complementarity (R = −0.97, p < 0.002) between the scores and the abundance of “glycogen degradation” in bacteria communities. On applying the system to bacteria genomes, CBM588 had only 1 match and ranked higher than the other 8 bacteria evaluated. The gram-stain properties were significantly correlated to the scores (p < 5 × 10−4). The distributions of the scored protein domains indicated that CBM588 had a considerably higher (p < 10−5) proportion of carbohydrate-binding modules than other bacteria, which suggested the superior ability of CBM588 to access carbohydrates as a metabolic driver to the bowel microbiome. These results demonstrated the use of integrated counts of protein domains as a feasible readout for metabolic potential within bacteria genomes and human metagenomes.
Similar content being viewed by others


Introduction
Bowel microbiota are now considered “metaorgans”1 for humans in which bacteria occupy a considerable proportion2. Various functions are associated with these microbes. For example, gut immune maturation depends on colonization with a host-specific microbiota3, but an abundance of Ruminococcus gnavus is related to allergic diseases in infants4. In addition to immune interactions with hosts, these microbes contribute substantially to metabolic processes in the bowel. For example, short chain fatty acids are crucial energy sources produced by bacteria5 but are also pathologically related to the metabolic syndrome in humans6. Due to the multifaceted roles of this metaorgan, informative readouts are crucial for evaluating its metabolic potential.
Recent advances in sequencing technology7 have enabled the in-depth taxonomic profiling of gut microbiota. Signatures from 16S subunits of ribosomes have made the culture-free categorization of bacteria possible8. However no practical markers are available to quantify metabolic functions. Tools such as PICRUSt9 or Piphillin10 can be extrapolated to identify metabolic profiles by mapping the characteristic 16S sequences to known reference genomes. Alternatively, bioinformatic pipelines can directly interpret shotgun metagenomes11. However most of these pipelines require specialized programs or a series of tools to yield results that are difficult to interpret for individuals without specialized knowledge such as patients. A system that summarizes the metabolic profile of this metaorgan in formats conveyable to both nonexperts and experts is highly desirable for streamlining the use of interventions such as courses of prebiotics or probiotics and fecal material transfer.
We conducted a pilot study examining Clostridium butyricum Miyairi 588 (CBM588) in patients undergoing small bowel transplantation (SBT). Although the survival rate of patients after SBT is now 70%12 with the assistance of optimized immunosuppressants, further improvements could be made. Studies have found that bacteria diversities in bowels can confer a favorable prognosis factor in patients undergoing allogeneic hematopoietic stem cell transplantation13. Patients who do and do not reject SBTs do have different compositions of ileal microbiota14. A proactive measure to control gut microbiota could be a valuable addition to the care of SBT patients. Probiotics could be a promising option in suitable candidates. Based on these considerations and the results of 16S-based taxonomic and functional analyses, we successfully developed a scoring system that not only had favorable correlations with 16S-based reports but also offered mechanistic insights into how CBM588 drives the evolution of fecal bacteria communities in SBT recipients. The scores were based on integrated counts of carbohydrate-active protein domains after probability analyses were conducted using hidden Markov models. This system indicates the potential of protein domain–based scoring of focused metabolisms as readouts for understanding probiotic characteristics and their effects on fecal metagenomes.
Results
Taxonomic shifts of fecal microbiota associated with CBM588 ingestion
We recruited 7 patients 6 months after their small bowel transplantations (SBTs) (Table 1; 3 males and 4 females). They took oral CBM588 (1.5 × 109 CFU/day) daily for 1 month (Fig. 1A). The median age was 37 years (range, 16–59 years). Stool samples were collected before, 1 week after, and 1 month after CBM588 ingestion. Microbiota were profiled by sequencing 16S rRNA-based amplicons8 with a paired-end approach (raw read numbers are presented in Supplementary Fig. S1A). Operational taxonomic units (OTUs) were defined using a USEARCH-based pipeline15. Rarefaction curves of distinct OTUs were constructed by administering 10 random samplings for each patient (Supplementary Fig. S1B). At each depth, the averages of unique OTUs were plotted. All rarefaction curves showed saturating behaviors with an increase in read depths. To compensate for uneven read numbers among different samples, 10 randomly rarefied data sets with normalized reads in mapped OTU format were prepared from the original data sets before downstream analyses were conducted (Supplementary Fig. S2A). These 7 SBT recipients experienced no apparent infection or rejection during the study period.

Study design and taxonomic evaluations of fecal microbiota associated with CBM588 ingestion for 1 month. (A) Patients took CBM588 continuously for 1 month. Stool samples were collected before, 1 week after, and 1 month after the administration of CBM588. (B) OTU diversities at 1 month were assayed using q-parameterized Hill numbers. No OTUs were dominant after CBM588 administration, as suggested by overlapping dashed (before administration) and solid (1 month after administration) curves. α diversity (q = 0) was increased from 226.7 ± 3.6 to 282.0 ± 4.7 (SD), as averaged from 10 rarefied data sets. (C) The administration of CBM588 for 1 month caused discernible changes in the OTU profiles of all patients according to principal component analyses. Circles (before administration) and triangles (1 month after administration) of the same color were separated. (D) Percentages of 5 phyla, namely Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria, and Verrucomicrobia, shifted significantly (p < 0.05) according to mixed linear models of at least half of the rarefied data sets after the administration of CBM588 for 1 month. (E) 4 families were selected after patients received CBM588 for 1 month by intersecting the results of both support vector classification and logistic regression models among rarefied data sets. Bacteroidaceae, Enterobacteriaceae, and Veillonellaceae were evident among all rarefactions, whereas Clostridiaceae was noted in half of the data sets.
Sample diversities were assayed in units of OTU. Those at the same time point were pooled and averaged before analyses. Hill numbers16 were adopted to evaluate diversities with a parameter q (Fig. 1B and Supplementary Fig. S3A). With increasing values of q, the contributions of OTU abundances were increasingly weighted in results of Hill number-based diversities. Without weights (q = 0), Hill numbers were equal to α diversities (see Methods). All 10 rarefied data sets were evaluated and exhibited almost identical plots (Supplementary Fig. S4). After 1 week, α diversities positively built up from 226.3 ± 2.6 to 251.0 ± 3.2 (SD), representing a 10.9% increase (Supplementary Fig. S3A). The trend continued further at 1 month (Fig. 1B), increasing by 24.3% from 226.7 ± 3.6 to 282.0 ± 4.7 (SD). However, no significant differences in profiles were noted from positive-q Hill numbers after CBM588 ingestions for either 1 week or 1 month, implying the absence of dominant OTUs in samples.
Contrasts between samples were summarized using principal component analyses (PCAs). For each sample, percentages of OTUs were Hellinger-transformed before analyses were conducted17. All 10 rarefied data sets were evaluated, and few differences were noted among the plots (Supplementary Fig. S5). Supplementary Fig. S3B and Fig. 1C present representative 1-week and 1-month results, respectively. With variances of 28.80 ± 0.03% and 14.85 ± 0.03% (SD) explained by PCA leading components, we revealed that 1 week of exposure did not alter microbiota considerably from the baselines (Supplementary Fig. S3B, triangles vs. circles of the same color). At 1 month, 30.32 ± 0.08% and 18.34 ± 0.03% (SD) of total variances could be accounted for by the top 2 PCA components. CBM588 samples were separated from CBM588-naïve samples for all patients (Fig. 1C, triangles vs. circles of the same color). This time-dependent divergence suggested a CBM588-driven effect on the taxonomic profiles of gut microbiota. The inability of PCAs to identify clustering for any of the 3 time points implied considerable individual variations in microbe compositions.
Each OTU was taxonomically classified using USEARCH15 with a SILVA-based reference18. 16S rRNA reads of each sample were mapped indirectly to a SILVA taxonomy via OTU. At the phylum level (Supplementary Fig. S3C and Fig. 1D), 5 (Actinobacteria, Bacteroidetes, Firmicutes, Proteobacteria, and Verrucomicrobia) out of 10 phyla were demonstrated to have changed significantly (p < 0.05) according to mixed linear models19 (MLMs) of at least half of the rarefied data sets. Out of the 3 most abundant of these 5 phyla, Bacteroidetes increased the most (from 6.84 ± 0.03% to 25.09 ± 0.05% [SD]) after 1 month of CBM588 administration. Firmicutes and Proteobacteria decreased from 41.78 ± 0.06% to 31.47 ± 0.05% and 41.53 ± 0.04% to 34.83 ± 0.04% (SD), respectively. Although MLM did not reveal significant changes among any phyla at 1 week, some of the varying trends at 1 month were already discernible at that time point (Supplementary Fig. S3C).
To identify feature families associated with CBM588 administration, we used support vector classification20 (SVC) and logistic regression21 (LR) to determine the signature families from contrasts between naïve and 1-month data sets. There were 69 to 74 SILVA-mapped families among the 10 rarefied data sets. Only those that satisfied both SVC and LR models among at least half of the rarefied data sets were selected. We identified Bacteroidaceae, Enterobacteriaceae, and Veillonellaceae among all rarefactions, and Clostridiaceae was observed in half of the data sets (Supplementary Fig. S3D and Fig. 1E). At 1 month, the abundance of Bacteroidaceae and Veillonellaceae increased from 4.00 ± 0.02% to 21.43 ± 0.06% and 10.42 ± 0.03% to 11.55 ± 0.04% (SD), respectively (Fig. 1E). By contrast, the abundance of Enterobacteriaceae and Clostridiaceae decreased from 41.20 ± 0.04% to 34.16 ± 0.04% and 6.86 ± 0.01% to 2.66 ± 0.02% (SD), respectively. Similar trends were already observable at 1 week (Supplementary Fig. S3D).
Functional shifts of fecal metagenome associated with CBM588 ingestion
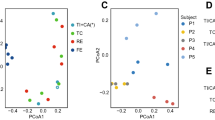
To investigate metagenome functions from 16S rRNA data sets, all 10 rarefaction data sets were subjected to cross-reference analyses to KEGG pathways22 using Piphillin10. Variations in associated KEGG pathways were further analyzed using PCA. All rarefied data sets yielded similar plots (Supplementary Fig. S6), with the 1-month administration of CBM588 causing significant shifts in the plots for most patients (Fig. 2A, triangles vs. circles of the same color). The first 2 components explained 63.02 ± 0.10% and 22.82 ± 0.10% (SD) of total variances, respectively. To select feature pathways that were most associated with the 1-month ingestion of CBM588, SVC20 and LR21 models were tested against all rarefied data sets. Only those pathways chosen by both models would be accepted. In each rarefaction, 297 to 301 KEGG pathways were observed; however, only 1 pathway, ko00511 or “other glycan degradation,” was constantly distinct from the others. Figure 1B shows radar plots with model coefficients as pointing hands toward 301 KEGG pathways. The only selected pathway was ko00511 with coefficients of 9.6 ± 0.1 (SD) and 20.0 ± 0.2 (SD) for SVD and LR models, respectively. The corresponding percentages of ko00511 among the samples of all rarefied data sets are displayed in Fig. 2C. Most patients exhibited an upward trend for this “other glycan degradation” pathway with CBM588 ingestion.

Assessments of fecal metagenome functions with 16S-rRNA. (A) PCAs were conducted on profiles of KEGG functional pathways, as inferred by Piphillin with 16S rRNA sequences. Samples before (circles) and 1 month after (triangles) CBM588 ingestion were plotted. Each patient was designated a specific color. In most patients, apparent shifts after 1 month of exposure were noted. (B) Radar plots of 10 rarefactions are prepared with pointing hands as model coefficients from support vector classification and logistic regression, respectively. ko00511 was one and the only pathway agreed by both models. (C) Percentages of the ko00511 pathway among 10 rarefied data sets of all samples collected before or 1 month after CBM588 ingestion are plotted. Red lines connect the same patients. Variations between rarefied data sets were minimal. Nearly all patients had upward trends for the ko00511 pathway.
To verify these findings, 6 samples from 3 patients (P2, P3, and P5) with distinct profiles of ko00511 pathway percentages (Fig. 2C) were subjected to the shotgun sequencing of metagenomes. Numbers of raw reads for each sample are listed in Supplementary Fig. S1C. We assumed that the quantities of protein domains involved in carbohydrate processing could serve as indicators of metabolic potential in fecal metagenomes. We employed the dbCAN database23 to analyze 585 hidden Markov models (HMMs) of carbohydrate-active domains. Reads from shotgun sequencing were routed to a pipeline to define domain fractions per million amino acids per 250 nucleotides (DFPMAA250) for every dbCAN-defined HMM (Supplementary Fig. S2B). The total number of identified domain fractions on a given HMM after normalizations are conducted is DFPMAA250 of the given domain. The addition of DFPMAA250 across all HMMs was used to estimate overall carbohydrate processing capabilities, (i.e., ∑DFPMAA250). Ten rarefied data sets at depths across 3 logs were tested for the robustness of ∑DFPMAA250 (Fig. 3A), which revealed a stable trend, especially among depths with read numbers above or equal to 62,367.

Assessments of fecal metagenome functions with DFPMAA250 scores. (A) ∑ DFPMAA250 values were stable across 3 logs of sequencing depths, as illustrated increasingly from 6,236 to 5,613,112 read numbers. All samples were assayed with 10 rarefied data sets at each depth with error bars in SE. (B) ∑DFPMAA250 values showed a significant correlation (R = 0.90, p < 0.015) with the ko00511 pathway percentages in 16S rRNA-inferred KEGG functional profiles. (C) Patients showed an upward trend of ∑DFPMAA250 values after 1 month of CBM588 ingestion. Notably, the CBM588 genome had higher ∑DFPMAA250 scores (red line) than all human fecal metagenomes. (D) Diversities of DFPMAA250 across all carbohydrate-active protein domains were evaluated with Hill numbers from averages of 10 rarefactions. At q = 0, Hill numbers equal counts of identified domains. The values stepped up from 284 to 311, 294 to 300, and 251 to 277 for P2, P3, and P5, respectively. Mild increases of abundance-weighted diversities were found, especially for P2 and P5 in the parameter range between 0.0 to 1.0 on the q axis. (E) Square-rooted abundance (mean ± SE) of “glycogen degradation I” by HUMAnN2 is plotted. Complementarity to corresponding ∑DFPMAA250 scores was noted. (F) A strong negative correlation (R = −0.97, p < 0.002) between square-rooted quantities of “glycogen degradation I” by HUMAnN2 and summed DFPMAA250 scores was evident.
We evaluated if estimates of ∑DFPMAA250 (Fig. 3A) could exhibit favorable correlations with Piphillin-reported10 percentages of the KEGG ko00511 pathway22 (Fig. 2C), both of which were averaged from 10 rarefactions. The Pearson coefficient between ∑DFPMAA250 and the ko00511 percentages was up to 0.90, which indicated a significant correlation (p < 0.015; Fig. 3B). All 3 patients had higher ∑DFPMAA250 values after CBM588 ingestion (p < 0.005 after bootstrapping was conducted 10,000 times) (Fig. 3C). The availability of individual DFPMAA250 estimates for each HMM enabled the profiling of diversities of carbohydrate-active domains in fecal metagenomes. Hill numbers with varying parameters up to 3.0 were plotted against averages of 10 rarefactions (Fig. 3D), in which higher parameter q values attached more weight to quantitatively dominant domains. Diversities were increased for all 3 patients, especially in the range between 0.0 and 1.0. α diversity, or the zero-q Hill number which equals counts of non-zero domains, increased with CBM588 ingestion from 284 to 311, 294 to 300, and 251 to 277 for P2, P3, and P5, respectively.
To validate above observations, we used HUMAnN224 and MetaCyc25 to evaluate the same data sets of shotgun metagenomes (Supplementary Fig. S1C). HUMAnN2 is based on an enhanced search upon known reference genomes, and MetaCyc provides a different catalog of metabolic pathways from KEGG. Analyses were repeated 5 times each with randomly selected 5 million paired reads per sample from original shotgun data sets (Supplementary Fig. S1C). MetaCyc defines “Glycan Degradation” Class with 11 instance pathways. Among the 10 prokaryote-relevant pathways only “glycogen degradation I” was identified by HUMAnN2 in all rarefied data sets. This pathway breaks down intracellular glycogen when carbon sources are limiting26. Figure 3E shows the pathway abundance of “glycogen degradation I” in square-rooted units. The values were found to complement ∑DFPMAA250 scores (Fig. 3C). The Pearson coefficient between both modalities of assessment was up to −0.97, indicating a strong negative correlation (p < 0.002; Fig. 3F).
Distinct properties of CBM588 in carbohydrate metabolism
We applied the same DFPMAA250 pipeline to shotgun sequences of the CBM588 genome (Supplementary Fig. S2B except Bowtie 2 filters). Notably, ∑DFPMAA250 estimated a distinct value for CBM588 at 66.37 ± 0.13 (SEM), which was higher than any of human samples (Fig. 3C). Because human feces would yield averages from all bowel bacteria, we suspected that most bacteria would carry lower values of ∑DFPMAA250 estimates in their genomes. From the data set of Köser et al.27, we found the shotgun genome sequences of several strains of bacteria (Table 2). To compensate for artificial underestimations of ∑DFPMAA250 due to shorter read lengths (Supplementary Fig. S7A), only those bacteria with average paired read lengths longer than 187.6 bp were subjected to DFPMAA250 calculation (Table 2 and Supplementary Fig. S7B). Unsurprisingly 8 out of the 9 evaluated bacteria had lower ∑DFPMAA250 values than CBM588, with 1 being on the same level (ATCC BAA-334; Fig. 4A). All Gram-positive bacteria were found to have higher estimates of ∑DFPMAA250 values than Gram-negative ones (61.39 ± 2.76 vs. 31.64 ± 1.60; SEM; p < 5 × 10−4 according to t-test; Fig. 4A and Table 2).

Distinct properties of DFPMAA250 scores for CBM588. (A) The summed DFPMAA250 values (∑DFPMAA250) across all carbohydrate-active protein domains of the CBM588 genome was higher than most evaluated bacteria strains. Only 1 strain had a similar value. The gram-positive (blue bars) bacteria had considerably higher estimates than the gram-negative (red bars) bacteria (61.39 ± 2.76 vs. 31.64 ± 1.60 [SEM]; p < 5 × 10−4 according to the Student’s t-test). (B) The DFPMAA250-based heatmap showed a similar distribution pattern for human samples. Different bacteria, however, were equipped with different spectra of carbohydrate-active protein domains encoded in genomes. Domain categories include auxiliary activities (AAs), carbohydrate-binding modules (CBMs), carbohydrate esterases (CEs), glycoside hydrolases (GHs), glycosyltransferases (GTs), polysaccharide lyases (PLs), and others. (C) Up to 45% of the CBM588 carbohydrate-active domains were in the category of CBM, which was considerably higher (p < 10−5) than the average of the other 9 bacteria (25.42% ± 1.45%, SEM). (D) CBM588 and ATCC BAA-334 were the two strains with highest CBMs diversities as shown in Hill number profiles. CBM588 had a steeper curve slope, indicating an unevener distribution of CBMs relative abundances than ATCC BAA-334.
The DFPMAA250-based heatmap revealed a similar pattern for all human samples (Fig. 4B), implying a universal repertoire requirement for carbohydrate-active protein domains. Instead the uses and abundances of these protein domains varied considerably among different bacteria (Fig. 4B). We categorized scored counts of families of carbohydrate-active domains28 among the 10 strains of bacteria studied, including auxiliary activities (AAs), carbohydrate-binding modules (CBMs), carbohydrate esterases (CEs), glycoside hydrolases (GHs), glycosyltransferases (GTs) and polysaccharide lyases (PLs; Fig. 4C). For CBM588, up to 45% of the genome-coded carbohydrate-active domains were in the CBM category, which differed significantly (p < 10−5) from the other 9 bacteria (25.42% ± 1.45%, SEM). Further characterization of CBMs diversities with Hill numbers (Fig. 4D) revealed that CBM588 and ATCC BAA-334 were the two strains with highest values. CBM588, however, had a declining curve at a sharper slope, suggesting an unevener distribution of CBMs relative abundances than ATCC BAA-334.
Discussion
DFPMAA250 estimates the potentials of carbohydrate metabolisms within bacteria genomes and human metagenomes by integrating counts of relevant protein domains. The underlying assumption is the enzyme amount is the key determinant of chemical reactions within a complex system such as human bowels regardless of the availability of reactants. A system based on a similar logic can successfully predict metabolomic turnover in oceans29. With the increasing number of interventions into bowel microbiota for medical benefits, a quantitative base would be required to make logical decisions and predictions for this metaorgan. Our study demonstrates a potential framework for this purpose. Other metabolisms could be formatted similarly. A set of essential scores would provide a valuable reference for clinicians to assess and evaluate bowel microbiomes in the same manner as creatinine for the kidney and transaminases for the liver. Scored summaries would also be easy for non-experts like patients to follow.
Construction of metabolic models between hosts and microbes have been pioneered by many scientists (reviewed by Heinken et al.30). One category of approaches is “top-down” to find correlations among metabolites by statistical analyses. The other category is “bottom-up” to build metabolic networks with units and coefficients of known pathways plus functional annotations from reference genomes. Designs of “top-down” could help identify novel correlations but often lack mechanistic insights; approaches of “bottom-up” would provide astonishing precision but often take tons of time to calculate. Instead our design can be considered as a middle stop between both ends. Scientists can use domain-based scores as correlation targets to make hypotheses of mechanisms and to design experiments with known properties of the given domains. Involved proteins can be even cloned from nucleotide sequences as mapped to the given HMMs in the data sets. Pathways can be constructed accordingly. These are all advantages not readily available from previous approaches.
Values of ∑DFPMAA250 were sensitive to mean read lengths but correctable up to a threshold of approximately 187.6 bp (Supplementary Fig. S7A). This was likely due to an inherent criterion set by dbCAN23, which requires covered fractions of HMMs by aligned reads to be higher than 0.3. With 90% of the known protein domains smaller than 200 amino acids31, mean read lengths at 187.6 bp would likely be sufficient for calculating DFPMAA250 values. Although this would set a limit on available choices of sequencing platforms, the demonstrated insensitivity to sequencing depths (Fig. 3A) could adequately compensate for this disadvantage. With rapid improvements on read lengths from the platform manufacturers, more sequence sets ready for the DFPMAA250 pipeline would be expected.
Enhancements of carbohydrate processing capabilities in fecal metagenomes after CBM588 ingestions were supported by increased ∑DFPMAA250 scores (Fig. 3C). These results are compatible with previous findings that Clostridia is associated with glycan degradation potential32. Because the abundance of the Clostridiaceae family decreased in our study (Fig. 1E), CBM588 had a low likelihood of having a direct contribution by mass effect. Instead, we observed that CBM588 not only carried a higher overall value of ∑DFPMAA250 than other bacteria (Fig. 4A) but also had a significant proportion of carbohydrate-active protein domains in the carbohydrate-binding module category (Fig. 4C). It is likely that CBM588 indirectly diversifies the microbe community by offering access to more glycan varieties. In addition, the increased Bacteroidetes abundance (Fig. 1D) could positively reinforce the glycan-metabolizing potential of the microbiome33,34. Independent validation with HUMAnN224 and MetaCyc25 identified a negative correlation (Fig. 3F) between pathway abundance of “glycogen degradation I” and ∑DFPMAA250 scores. This finding suggests that microbiomes carrying higher values of ∑DFPMAA250 can compensate the requirements of “glycogen degradation” in the microbe community. In other words, with diversified availability of carbons via enhanced metabolic potentials as implied by high ∑DFPMAA250 scores, bacteria can thrive with less dependence upon glycogen degradation. It would be interesting to determine if simultaneous additions of glycan-rich foods with CBM588 could elicit any synergistic effects.
In this study, we used CBM588 instead of the more common Lactobacilli-based probiotics because its safety is well established35. Reports of its efficacy against enterohemorrhagic Escherichia coli O157:H736 and Clostridium difficile37 also increased our confidence in its use in immunocompromised patients. In a rat model following SBT, Price et al. found that rejection and graft-versus-host disease are associated with shifts in gut microbiota toward potentially pathogenic organisms38, which can be ameliorated using probiotics39. In humans, Oh et al. indicated that the presence of the Enterobacteriaceae family significantly increases during episodes of rejection after SBT14. Bacterial compositions could also be affected by the presence of ileostomy and the availability of oxygen after a transplantation40. In our study, the most notable alterations were decreases in Enterobacteriaceae and increases in Bacteroidaceae after the 1-month ingestion of CBM588 in patients (Fig. 1E). With the known association between the Enterobacteriaceae family and graft rejection14, our results would support the use of CBM588 to improve the survival of small intestine allografts. The decrease in Enterobacteriaceae per se would in addition imply a lower likelihood of infection by potential pathogens in the family. Risks of rejection and infection might accordingly be minimized from the use of CBM588. Although number of patients in our cohort was limited, our data did reveal the time-dependent divergence of microbiota and concordant change of carbohydrate metabolism after CBM588 ingestion. Further large-scale investigations are warranted. The developed scoring system detailed herein could readily provide objective and quantitative readouts to facilitate the establishment of clinical reasoning behind adopting probiotics in the care of SBT recipients.
Methods
Sample collection and ethics approval
All patients received SBT 6 months before enrolment into the study (Table 1). Attendees took CBM588 (1.5 × 109 CFU/day) daily for 1 month (Fig. 1A). Stool and/or blood samples were collected before, 1 week, and 1 month after CBM588 ingestion. The experimental protocol was approved by the Institutional Review Board of Far Eastern Memorial Hospital, New Taipei City. The study was conducted in accordance with the relevant guidelines. Informed consent was obtained from patients directly or from their parents if attendees were younger than 18 years old.
Software and hardware
All analyses were performed on a 2013 Mac Pro equipped with 3.7-GHz Quad-Core Intel Xeon E5, 64 GB of memory, and 2 AMD FirePro D700 6 GB graphics cards. Inputs and outputs of various specialized packages were glued with Python 2.7 scripts.
Next-generation sequencing
All sequencing libraries including 16S rRNA, shotgun metagenomes, and CBM588 genome were constructed and sequenced using commercial tools. The 16S libraries were sequenced on Illumina MiSeq as 2 × 300 bp paired-end readings, whereas the shotgun fecal metagenomes and CBM588 genome were determined on Illumina NextSeq as 2 × 150 bp paired-end readings.
Operational taxonomic units
Raw reads (Supplementary Fig. S1A) carrying primer sequences were trimmed using a self-developed script. Results were paired using PEAR41 and filtered using USEARCH15. Pooled sequences were used to define operational taxonomic units (OTUs) by USEARCH with default criteria. Saturation curves of distinct OTUs (Supplementary Fig. S1B) were plotted as means of 10 random selections with increasing numbers of paired reads. Ten rarefied subsets of reads in the format of mapped OTUs were prepared by giving each the same number of reads before downstream analyses were conducted.
Diversities in Hill numbers
Hill numbers define a diversity profile with a parameter q formulated as follows, where S is “species equivalent,” f represents frequency of “species equivalent,” and q denotes the parameter.
With increasing values of q, more weights are given to the more abundant species equivalent. In this study, OTUs (Fig. 1B, Supplementary Figs S3A, and S4), or dbCAN-defined hidden Markov models (HMMs)23 (Figs 3D and 4D) were used as species equivalents. At q = 0, species equivalents were counted without considering their normalized frequencies. We employed this zero-parameterized Hill number as the definition for α diversity42. At q = 1, counts were proportional to their normalized frequencies (i.e., Shannon diversity), whereas at q = 2, only dominant species equivalents were counted. A deeper slope of the curve represents an unevener distribution of relative abundances of species equivalents.
Principal component analysis
Decomposition with principal component analysis (PCA) was performed with scikit-learn in Python43. Values were Hellinger-transformed before analyses17. OTUs and KEGG-defined pathways22 were used as variables to decompose 16S rRNA and metagenome functions, respectively.
Selection of signature phyla and families
OTUs were taxonomically classified using USEARCH15 and a SILVA-based reference18. 16S rRNA reads were given the same taxonomic designations as the associated OTUs. Mixed linear models19 (MLMs) were adopted to identify signature phyla which best discriminated CBM588 effects among at least half of the rarefied data sets. Support vector classification20 (SVC) and logistic regression21 (LR) were used to select signature families linked to CBM588 exposure. Only those families that were in agreement in both SVC and LR models among at least half of the rarefied data sets were taken. Parameters were optimized by leave-one-out cross-validations.
16S rRNA-inferred metagenome functions
Piphillin10 was used to extrapolate metagenomic functions to KEGG pathways22 with 16S rRNA sequences. In addition to PCA analyses, KEGG pathways were also subjected to signature selections with SVC20 and LR21 models. Only those picked by both models among at least half of the rarefied data sets would be accepted. Model parameters were optimized with leave-one-out cross-validations.
Domain fractions for reads of shotgun metagenomes and bacteria genomes
Raw reads (Supplementary Fig. S1C) were paired using PEAR41 with quality control defaults. Bowtie 244 was used to filter out human sequences for reads of human origin. Genes were predicted using FragGeneScan45. CAZy-associated28 HMMs were downloaded from dbCAN23 for HMMER scanning46 upon PEAR-assembled reads, where unassembled reads were excluded. “hmmsearch” with Z-value adjustment to 585 was used instead of “hmmscan” to increase scanning efficiency. Calculations of domain fractions are specified below.
Domain fractions per million amino acids 250
Domain fractions were determined by hmmscan-parser.sh as downloaded from dbCAN23, but minimal changes to switch parameters were made to reflect the use of “hmmsearch” instead of “hmmscan.” Other criteria, including p values and a minimal domain fraction of 0.3 as defined by dbCAN, were not altered. For a given domain, summed domain fractions normalized by counts of amino acid inputs and 250 base pairs were designated as “domain fractions per million amino acids per 250 nucleotides” or DFPMAA250 (Supplementary Fig. S2B). Python scripts for the demonstration purpose is available in the Supplementary Information.
HUMAnN2 validation
Evaluations were repeated 5 times against the MetaCyc25 database. Each run was conducted upon a rarefied data set of 5 million randomly picked paired reads of each sample from the original shotgun data sets (Supplementary Fig. S1C). There are 11 instance pathways in the MetaCyc25 “Glycan Degradation” Class, including (1,4)-β-D-xylan degradation, cellulose degradation I, chondroitin sulfate degradation, dermatan sulfate degradation, glycogen degradation I, glycogen degradation II, homogalacturonan degradation, L-arabinan degradation, pectin degradation I, starch degradation I, and xyloglucan degradation I. Abundance of “glycogen degradation II” was discarded because the pathway is restricted to eukaryotes.
CBM588 genome sequencing
The construction of libraries and next-generation sequencing were contracted to commercial service providers. Raw reads (Supplementary Fig. S1D) were assembled by SPAdes47 into contigs and scaffolds to confirm the probiotic identity by in silico polymerase chain reaction48 to find corresponding 16S sequences with 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′ and 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′ primers (Supplementary Fig. S1E). For calculations of DFPMAA250 raw reads were processed as fecal shotgun metagenomes without contig or scaffold assembling.
DFPMAA250 for bacteria genomes
Shotgun genomes of bacteria were found from the data sets of Köser et al.27, whereas the CBM588 genome was prepared as described in the previous section. Shorter versions of CBM588 shotgun sequences (Supplementary Fig. S7) were simulated by using random trimmings of 5′ and 3′ ends of paired full-length reads. Only those bacteria with mean lengths of paired reads of over 187.6 bp were subjected to DFPMAA250 analyses. Gene predictions and protein domain scanning were conducted in the same manner for fecal shotgun metagenomes but without the use of Bowtie 2 filters (Supplementary Fig. S2B).
Data availability
All raw sequencing data generated in this study have been submitted to the European Nucleotide Archive (https://www.ebi.ac.uk/ena) under accession number PRJEB27661.
References
Alexandre, A. et al. JRC F7 - Knowledge for Health and Consumer Safety, The Human Gut Microbiota: Overview and analysis of the current scientific knowledge and possible impact on healthcare and well-being. Publications Office of the European Union, Luxembourg, EUR - Scientific and Technical Research Reports, https://doi.org/10.2760/17381 (2018).
Stephen, A. M. & Cummings, J. H. The microbial contribution to human faecal mass. J Med Microbiol 13, 45–56, https://doi.org/10.1099/00222615-13-1-45 (1980).
Chung, H. et al. Gut immune maturation depends on colonization with a host-specific microbiota. Cell 149, 1578–1593, https://doi.org/10.1016/j.cell.2012.04.037 (2012).
Chua, H. H. et al. Intestinal Dysbiosis Featuring Abundance of Ruminococcus gnavus Associates With Allergic Diseases in Infants. Gastroenterology 154, 154–167, https://doi.org/10.1053/j.gastro.2017.09.006 (2018).
Wong, J. M., de Souza, R., Kendall, C. W., Emam, A. & Jenkins, D. J. Colonic health: fermentation and short chain fatty acids. J Clin Gastroenterol 40, 235–243 (2006).
Li, X., Shimizu, Y. & Kimura, I. Gut microbial metabolite short-chain fatty acids and obesity. Biosci Microbiota Food Health 36, 135–140, https://doi.org/10.12938/bmfh.17-010 (2017).
Weinstock, G. M. Genomic approaches to studying the human microbiota. Nature 489, 250–256, https://doi.org/10.1038/nature11553 (2012).
Olsen, G. J., Lane, D. J., Giovannoni, S. J., Pace, N. R. & Stahl, D. A. Microbial ecology and evolution: a ribosomal RNA approach. Annu Rev Microbiol 40, 337–365, https://doi.org/10.1146/annurev.mi.40.100186.002005 (1986).
Langille, M. G. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31, 814–821, https://doi.org/10.1038/nbt.2676 (2013).
Iwai, S. et al. Piphillin: Improved Prediction of Metagenomic Content by Direct Inference from Human Microbiomes. PLoS One 11, e0166104, https://doi.org/10.1371/journal.pone.0166104 (2016).
Quince, C., Walker, A. W., Simpson, J. T., Loman, N. J. & Segata, N. Shotgun metagenomics, from sampling to analysis. Nat Biotechnol 35, 833–844, https://doi.org/10.1038/nbt.3935 (2017).
Sudan, D. The current state of intestine transplantation: indications, techniques, outcomes and challenges. Am J Transplant 14, 1976–1984, https://doi.org/10.1111/ajt.12812 (2014).
Taur, Y. et al. The effects of intestinal tract bacterial diversity on mortality following allogeneic hematopoietic stem cell transplantation. Blood 124, 1174–1182, https://doi.org/10.1182/blood-2014-02-554725 (2014).
Oh, P. L. et al. Characterization of the ileal microbiota in rejecting and nonrejecting recipients of small bowel transplants. Am J Transplant 12, 753–762, https://doi.org/10.1111/j.1600-6143.2011.03860.x (2012).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, https://doi.org/10.1093/bioinformatics/btq461 (2010).
Hill, M. O. Diversity and evenness: a unifying notation and its consequences. Ecology 54, 427–432 (1973).
Legendre, P. & Gallagher, E. D. Ecologically meaningful transformations for ordination of species data. Oecologia 129, 271–280 (2001).
Quast, C. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41, D590–596, https://doi.org/10.1093/nar/gks1219 (2013).
Lindstrom, M. J. & Bates, D. M. Newton—Raphson and EM algorithms for linear mixed-effects models for repeated-measures data. Journal of the American Statistical Association 83, 1014–1022 (1988).
Chang, C.-C. & Lin, C.-J. LIBSVM: A library for support vector machines. ACM transactions on intelligent systems and technology (TIST) 2, 27 (2011).
Fan, R.-E., Chang, K.-W., Hsieh, C.-J., Wang, X.-R. & Lin, C.-J. LIBLINEAR: A library for large linear classification. Journal of machine learning research 9, 1871–1874 (2008).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30 (2000).
Yin, Y. et al. dbCAN: a web resource for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 40, W445–451, https://doi.org/10.1093/nar/gks479 (2012).
Franzosa, E. A. et al. Species-level functional profiling of metagenomes and metatranscriptomes. Nature methods 15, 962 (2018).
Caspi, R. et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic acids research 42, D459–D471 (2013).
Wilson, W. A. et al. Regulation of glycogen metabolism in yeast and bacteria. FEMS microbiology reviews 34, 952–985 (2010).
Koser, C. U. et al. Rapid single-colony whole-genome sequencing of bacterial pathogens. J Antimicrob Chemother 69, 1275–1281, https://doi.org/10.1093/jac/dkt494 (2014).
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M. & Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res 42, D490–495, https://doi.org/10.1093/nar/gkt1178 (2014).
Larsen, P. E. et al. Predicted Relative Metabolomic Turnover (PRMT): determining metabolic turnover from a coastal marine metagenomic dataset. Microb Inform Exp 1, 4, https://doi.org/10.1186/2042-5783-1-4 (2011).
Heinken, A. & Thiele, I. Systems biology of host–microbe metabolomics. Wiley Interdisciplinary Reviews: Systems Biology and Medicine 7, 195–219 (2015).
Xu, D. & Nussinov, R. Favorable domain size in proteins. Folding and Design 3, 11–17 (1998).
Eilam, O. et al. Glycan degradation (GlyDeR) analysis predicts mammalian gut microbiota abundance and host diet-specific adaptations. MBio 5, e01526–01514 (2014).
Marcobal, A. et al. Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe 10, 507–514, https://doi.org/10.1016/j.chom.2011.10.007 (2011).
Sonnenburg, E. D. et al. Specificity of polysaccharide use in intestinal bacteroides species determines diet-induced microbiota alterations. Cell 141, 1241–1252, https://doi.org/10.1016/j.cell.2010.05.005 (2010).
Isa, K. et al. Safety assessment of the Clostridium butyricum MIYAIRI 588(R) probiotic strain including evaluation of antimicrobial sensitivity and presence of Clostridium toxin genes in vitro and teratogenicity in vivo. Hum Exp Toxicol 35, 818–832, https://doi.org/10.1177/0960327115607372 (2016).
Takahashi, M. et al. The effect of probiotic treatment with Clostridium butyricum on enterohemorrhagic Escherichia coli O157:H7 infection in mice. FEMS Immunol Med Microbiol 41, 219–226, https://doi.org/10.1016/j.femsim.2004.03.010 (2004).
Woo, T. D. et al. Inhibition of the cytotoxic effect of Clostridium difficile in vitro by Clostridium butyricum MIYAIRI 588 strain. J Med Microbiol 60, 1617–1625, https://doi.org/10.1099/jmm.0.033423-0 (2011).
Price, B. A. et al. The effect of rejection and graft-versus-host disease on small intestinal microflora and bacterial translocation after rat small bowel transplantation. Transplantation 56, 1072–1076 (1993).
Zhou, H. J., Yin, L., Chen, C. Q., Shi, M. M. & Zhang, M. J. Administration of probiotics reduces bacterial translocation after intestinal transplantation in rats. Transplant Proc 42, 4643–4647, https://doi.org/10.1016/j.transproceed.2010.09.171 (2010).
Hartman, A. L. et al. Human gut microbiome adopts an alternative state following small bowel transplantation. Proc Natl Acad Sci USA 106, 17187–17192, https://doi.org/10.1073/pnas.0904847106 (2009).
Zhang, J., Kobert, K., Flouri, T. & Stamatakis, A. PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620, https://doi.org/10.1093/bioinformatics/btt593 (2014).
McCoy, C. O. & Matsen, F. At Abundance-weighted phylogenetic diversity measures distinguish microbial community states and are robust to sampling depth. PeerJ 1, e157, https://doi.org/10.7717/peerj.157 (2013).
Pedregosa, F. et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359, https://doi.org/10.1038/nmeth.1923 (2012).
Rho, M., Tang, H. & Ye, Y. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res 38, e191, https://doi.org/10.1093/nar/gkq747 (2010).
Eddy, S. R. Accelerated Profile HMM Searches. PLoS Comput Biol 7, e1002195, https://doi.org/10.1371/journal.pcbi.1002195 (2011).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455–477, https://doi.org/10.1089/cmb.2012.0021 (2012).
Kalendar, R., Khassenov, B., Ramankulov, Y., Samuilova, O. & Ivanov, K. I. FastPCR: An in silico tool for fast primer and probe design and advanced sequence analysis. Genomics 109, 312–319, https://doi.org/10.1016/j.ygeno.2017.05.005 (2017).
Acknowledgements
We are indebted to Wilson Cheng for their laboratory work and to Chien-Hao Chen for assistance with statistical analyses. We also thank Hui-Chuan Chiu for sharing their expertise in CBM588 culturing. This work was supported by research grants from the Far Eastern Memorial Hospital (FEMH-2015-C-023, FEMH-2017-C-035 and FEMH-2018-C-019) and Ministry of Science and Technology (105-2628B-400-001-MY3).
Author information
Authors and Affiliations
Contributions
H.H.L. did sequence analyses and drafting of the manuscript. Y.C.L., C.S.C., Y.C., Y.H.N. and P.F.C. designed and conducted this study. K.L., Y.H.C. and C.H.Y. did DNA extraction and part of library preparation.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, HH., Lin, YC., Chung, CS. et al. Integrated Counts of Carbohydrate-Active Protein Domains as Metabolic Readouts to Distinguish Probiotic Biology and Human Fecal Metagenomes. Sci Rep 9, 16836 (2019). https://doi.org/10.1038/s41598-019-53173-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53173-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
