
Abstract
Orthohantaviruses, negative-sense single-strand tripartite RNA viruses, are a global public health threat. In humans, orthohantavirus infection causes hemorrhagic fever with renal syndrome or hantavirus cardiopulmonary syndrome. Whole-genome sequencing of the virus helps in identification and characterization of emerging or re-emerging viruses. Next-generation sequencing (NGS) is a potent method to sequence the viral genome, using molecular enrichment methods, from clinical specimens containing low virus titers. Hence, a comparative study on the target enrichment NGS methods is required for whole-genome sequencing of orthohantavirus in clinical samples. In this study, we used the sequence-independent, single-primer amplification, target capture, and amplicon NGS for whole-genome sequencing of Hantaan orthohantavirus (HTNV) from rodent specimens. We analyzed the coverage of the HTNV genome based on the viral RNA copy number, which is quantified by real-time quantitative PCR. Target capture and amplicon NGS demonstrated a high coverage rate of HTNV in Apodemus agrarius lung tissues containing up to 103–104 copies/μL of HTNV RNA. Furthermore, the amplicon NGS showed a 10-fold (102 copies/μL) higher sensitivity than the target capture NGS. This report provides useful insights into target enrichment NGS for whole-genome sequencing of orthohantaviruses without cultivating the viruses.
Similar content being viewed by others

Introduction
Orthohantaviruses belong to the family Hantaviridae of the order Bunyavirales and are enveloped negative-sense single-stranded RNA viruses1,2. The viral RNA genomes are segmented into large (L), medium (M), and small (S) segments. The tripartite segments encode an RNA-dependent RNA polymerase (RdRp), two envelope glycoproteins (Gn and Gc), and a nucleocapsid (N) protein. The absence of effective prevention and treatment strategies for hantavirus infections is a global public health threat3,4. In humans, infection from the Old World hantaviruses, e.g., Hantaan orthohantavirus (HTNV), Seoul orthohantavirus (SEOV), Dobrava-Belgrade orthohantavirus, and Puumala orthohantavirus, causes hemorrhagic fever with renal syndrome (HFRS), while infection from the New World hantaviruses, e.g., Sin Nombre orthohantavirus, Andes orthohantavirus, and New York virus, results in hantavirus cardiopulmonary syndrome5. Orthohantaviruses are transmitted to humans when viral infectious particles from the excreta of infected rodents are inhaled through the respiratory tract. HTNV, harbored by the striped field mouse (Apodemus agrarius), is an etiological agent for HFRS with fatality rates ranging between 1–15%6,7.
Next-generation sequencing (NGS) has been applied to various fields in virology, e.g., metagenomics of the virome, whole-genome sequencing, tracking the spread of virus, development of therapeutics, and identification of putative pathogens8,9. Whole-genome sequencing of emerging viruses is increasingly being used to identify and characterize viruses that pose a threat to human health10,11,12. However, the low abundance of viral particles in the hosts often presents an obstacle in virus identification. To diagnose and detect extremely low-titer viruses, several NGS methods have been developed, including enrichment of viral nucleic acids by using target-specific oligonucleotide probes, removing host genome sequences, purifying virus-like particles, and performing small-RNA deep sequencing13,14,15,16.
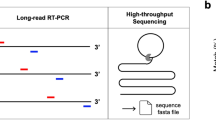
One of the most prominent techniques for random access to viral nucleic acids is sequence-independent, single-primer amplification (SISPA)17,18. Previously, it was used for sequencing HTNV isolates collected from HFRS endemic areas19. However, a significant limitation of whole-genome sequencing is the ultra-low number of copies of the target genome found in clinical or environmental specimens. To enrich the nucleic acids of interest, target-enrichment methods, including target capture or amplicon NGS, have been used to sequence viral genomes20,21,22. In target capture NGS, the samples are subjected to fragmentation, random reverse-transcription, ligation with barcoded library adapters, and polymerase chain reaction (PCR) amplification. These libraries are pooled and enriched using virus-specific probes. Recently, a novel target-enrichment deep-sequencing platform called “ViroFind”, which uses probes that cover the genome of 535 DNA or RNA viruses, was applied to the detection and analysis of viral populations in clinical samples23. Metsky et al. recovered Lassa virus genomes in low-titer clinical samples using multiple probe sets targeting 356 viral species, leading to the bio-surveillance in uncharacterized specimens24. To generate full-genome coverage, a tiling amplicon scheme is commonly used25,26. Previous studies have applied amplicon NGS to whole-genome sequencing of HTNV from patients and rodent hosts in HFRS endemic areas27. For this approach, the primer set was designed by amplifying short (150 nt) amplicons to maximize the recovery of virus genomic sequences. The whole-genome sequences of human- and rodent-derived HTNV revealed putative infection sites of HFRS patients by performing phylogeographic and epidemiological analyses. However, a comparison of the different target-enrichment NGS methods is yet to be conducted for the whole-genome sequencing of orthohantaviruses.
In this work, we prepared the RNA samples from 14A. agrarius lung tissues and enriched the viral RNA using SISPA, target capture, and amplicon methods prior to the NGS library preparation. To evaluate three NGS methodologies, we analyzed and compared the depth of coverage and the recovery of virus genomes on a basis of the viral RNA copy number per μL.
Results
Sample selection for HTNV whole-genome sequencing
A total of 161 A. agrarius were captured in HFRS-endemic areas including Gyeonggi and Gangwon provinces, Republic of Korea (ROK) between 2016 and 2017 (Fig. 1). Laboratory diagnosis examined the presence of anti-HTNV IgG and HTNV RNA by IFA and RT-PCR, respectively (Table 1, Supplementary Fig. 1). HTNV RNA loads was quantified on 14 sero- and RT-PCR-positive rodent lung tissues. The Ct values ranged from 20.8 to 32.8 regardless of the anti-HTNV IgG titer. The taxonomic identity of 14 A. agrarius was confirmed by sequencing the mitochondrial cytochrome b (Cyt b) gene. Phylogenetic analysis showed that 14 A. agrarius formed a genetic lineage with A. agrarius collected in the ROK (Supplementary Fig. 2).

A geographic map of the trapping sites for Apodemus agrarius collected in the Republic of Korea. The trapping locations of rodents are shown in this study (red circles). Paju-si, Yeoncheon-gun, and Pocheon-si are included in Gyeonggi province. Yanggu-gun is localized in Gangwon province. We used the Quantum Geographical Information System (QGIS) software V.3.4.10. to create the geographic map.
Determination of HTNV RNA copy number in A. agrarius lung tissues
HTNV RNA copy number was determined by generating a linear regression curve using a recombinant plasmid DNA, containing S segment of HTNV 76–118 (Supplementary Fig. 3). The coefficient of correlation (R2) value was 0.998 and the HTNV viral copy number of each sample was calculated. Aa16-19, Aa16-50, Aa17-8, and Aa17-49 showed Ct values ranging from 20.8–21.5 corresponding to 105 copies/μL of viral RNA in the HTNV positive A. agrarius lung tissue. Aa16-181, Aa16-185, Aa17-48, Aa17-52, and Aa17-53 contained 103 to 104 copies/μL of HTNV RNA with 24.7–27.9 of Ct values. The Ct values of Aa16-21, Aa16-22, Aa17-7, Aa17-66, and Aa17-76 were 30.2–32.8, indicating that the rodents harbored 102 copies/μL of HTNV RNA in the lung tissue.
Whole-genome sequencing of HTNV using SISPA, target capture, and amplicon NGS
To obtain the whole-genome sequence of HTNV, all samples were sequenced in the MiSeq using SISPA, target capture, and amplicon NGS (Fig. 2). SISPA NGS showed the lowest coverage of HTNV tripartite genome for the lung tissues containing HTNV RNA of 105 copies/μL; 40%, 45%, and 75% for L, M, and S segments, respectively, compared to full-length nucleotides of the prototype HTNV 76–118 (Fig. 3). The coverage of HTNV genomic sequence remarkably decreased as the copy number of viral RNA was low (103–104 to 102 copies/μL). Using target capture NGS, nearly complete genome sequence of HTNV was recovered in the samples carrying more than 103 copies/μL of HTNV RNA (L segment, 99.6%; M segment, 99.1%; S segment, 99.6%). In the samples with low viral RNA copy number of 102 copies/μL, decreases of the coverage rate were observed for L (84.9%) and M (69.4%) segments, while the coverage of S segment was slightly reduced (96.8%). Finally, amplicon NGS recovered nearly whole-genome sequences of HTNV from 105 to 102 copies/μL of the viral RNA copy numbers.

Description of different methods for next-generation sequencing (NGS) for Hantaan orthohantavirus (HTNV) in rodent lung tissues. Three methodologies of sample preparation were shown for NGS of HTNV. These methods amplified reads of viral genomes using different strategies. (a) For SISPA NGS method, RNA was reverse-transcribed using a random primer (FR26RV-N) and then cDNA was amplified using a single primer (FR20RV). (b) Target capture NGS captures viral genomes by using virus-specific probes. RNA samples were fragmented and then cDNA was synthesized using random hexamer. To enrich the target interest, first & second hybridization and capture were performed using virus-specific probes. (c) Amplicon NGS was applied to enrich viral genomes. Viral cDNA was amplified using HTNV-specific multiple primer mixture.

Coverage rate of consensus sequences for Hantaan orthohantavirus (HTNV) by viral copy number. The percentage coverage rate of consensus sequences by viral copy number among the three next-generation sequencing methods. Coverage rate was calculated by matching the consensus sequences with the sequence of HTNV 76–118 strain (GenBank accession number, NC_005222, NC_005219, NC_005218).
Comparison for coverage and depth of HTNV whole-genome sequences based on the different NGS methods
The composition of reads mapped to HTNV genomes was shown in the total reads generated by three NGS methods (Fig. 4). In the SISPA NGS, an average of 673,832 total reads were observed (Supplementary Table 1). The average number of viral sequence reads were 22 (0.003%) for L segment, 16 (0.002%) for M segment, and 33 (0.005%) for S segment. The average depth of coverage was scarcely found; one for M segment and three for S segment. In the target capture NGS, 443,731 (26.2%), 480,973 (28.4%), and 552,172 (32.6%) reads out of a total of 1,692,164 reads mapped to the reference sequence for the HTNV L, M, and S segments, respectively (Supplementary Table 2). The total read numbers and depth of coverage for HTNV were decreased corresponding to the 105 to 102 copies/μL viral RNA copy numbers. Lastly, in amplicon NGS we observed 438,666 (30.3%), 444,437 (30.7%), and 468,114 (32.3%) reads out of a total of 1,448,091 reads for HTNV L, M, and S segments, respectively (Supplementary Table 3). The viral read counts and depth of coverage were comparable at the HTNV RNA copy numbers of 105 and 103 to 104 copies/μL. However, the read counts and depth of coverage for HTNV L and M segments were reduced at viral RNA copy numbers of 102 copies/μL except for the S segment.

The composition of Hantaan orthohantavirus (HTNV) genomic reads in the total reads generated by three next-generation sequencing methods. The composition of viral reads mapped to HTNV was exhibited in the total number of reads. The total reads were produced by performing SISPA, target capture, and amplicon NGS. These reads were mapped to HTNV 76–118 tripartite genomes (GenBank accession number: L segment, NC_005222; M segment, NC_005219; S segment, NC_005218). A circle represents for total reads obtained by SISPA, target capture, and amplicon NGS methods. Red, Blue, and Green colors indicate the composition of viral reads for HTNV L, M and S segments, respectively, over the total reads. Gray color indicates non-viral reads in the total reads. The HTNV reads were shown as a percentage (%) evaluated by the ratio of viral reads over the total reads.
Phylogenetic analysis
To validate the whole-genome sequences of HTNV obtained by target enrichment NGS methods, phylogenetic trees were generated by maximum likelihood (ML) method (Fig. 5). The phylogenies of HTNV L, M, and S segments from Aa16-181, Aa16-185, and Aa17-8 formed a genetic lineage with the strains collected from Paju-si. The phylogenetic pattern of HTNV tripartite genome from Aa16-19 shared a common ancestor with the HTNV strain in Pocheon-si. Phylogenetically, HTNV L, M, and S segments from Aa16-50 grouped to the HTNV strains from Tonghyeon-ri, Yeoncheon-gun, while Aa17-48, Aa17-49, Aa17-52, and Aa17-53 grouped to the HTNV strains from Dosin-ri, Yeoncheon-gun.

Phylogenetic analysis of Hantaan orthohantavirus (HTNV) whole-genome sequences using target capture and amplicon next-generation sequencing (NGS). Phylogenetic trees were generated by the ML method using the TN93 + G, T92 + G, and T92 + G model based on (a) L segment (1–6530 nt), (b) M segment (1–3616 nt), Red characters indicate and (c) S segment (1–1696 nt), respectively. The numbers at each node are bootstrap probabilities, as determined for 1,000 iterations. Sequences obtained in target capture NGS were indicated in red. Sequences obtained in amplicon NGS were indicated in blue. Other hantaan orthohantaviruses were used as outgroups. HTNV Aa03–387 (L segment, KT934958; M segment, KT934992; S segment, KT935026), HTNV Aa05-331 (L segment, KT934962; M segment, KT934996; S segment, KT935030), HTNV Aa14-406 (L segment, KT934985; M segment, KT935019; S segment, KT935053), HTNV Aa14-412 (L segment, KT934987; M segment, KT935021; S segment, KT935055), HTNV 76-118 (L segment, NC_005222; M segment, NC_00519; S segment, NC_005218), and HTNV HV004 (L segment, JQ083393; M segment, JQ083394; S segment, JQ093395).
Discussion
Whole-genome sequences of viruses play a critical role in the rapid identification and tracking of the infection source during disease outbreaks. To take advantage of the whole-genome sequence, various NGS methodologies are utilized for the rapid and robust virus genomic sequencing prior to culturing of virus. SISPA NGS was used for characterizing RNA and DNA viruses including Astrovirus, Parvovirus, and Rotavirus28,29,30. Target capture NGS was implemented as an important tool for the genome sequencing of ZIKV (Zika virus), Ebola virus and chikungunya virus21,31,32. The complete genome sequences of ZIKV, Respiratory syncytial virus and HTNV were obtained by amplicon NGS21,27,33. However, different NGS methods remain to be comparatively evaluated for optimal whole-genome sequencing of specimens containing low viral RNA loads.
This study describes different NGS methodologies, SISPA, target capture, and amplicon NGS, for the whole-genome sequencing of HTNV directly from the host rodent lung tissues without the need for cultivating virus particles. To compare the robustness of individual methods, the whole-genome sequencing of HTNV was performed by using tissue specimens containing different viral RNA loads ranging from 102 to 105 copies/μL. Although SISPA is a representative method to gain whole-genome sequence of viruses via high-throughput sequencing, poor genome coverage rates of HTNV, even in samples containing highest RNA copy number (105) per μL, were observed in this study. The limitation for SISPA NGS was low sensitivity for obtaining the viral whole-genome sequence from the specimen containing low titer of the virus or non-isolated samples. On the basis of whole-genome sequencing of HTNV, target capture NGS enabled recovery of nearly complete genome sequences of HTNV from 105 to 103–104 copies/μL of HTNV RNA loads. At the HTNV RNA loads of 102 copies/μL, coverage rates of HTNV genome sequences remarkably decreased for L and M segments. The coverage of HTNV S segments showed over 95% recovery at this level due to short length of the genome. The whole-genome sequences of HTNV by amplicon NGS were completely recovered from 105 to 102 copies/μL of HTNV RNA loads. Several studies that used a tiling amplicon scheme showed reliable coverage rate (>95%) of the whole viral genome by long amplicons (1–2.5 kb in length) using NGS25,34,35. However, with lower virus abundance in tissue, long fragments reduced the coverage rate of genome sequence. To ensure high coverage at low copy number of HTNV, we developed the amplicon NGS using specific primer sets which generate few hundred-length short amplicons. This methodology is robust and efficient for acquiring the whole-genome sequence of HTNV and SEOV from patients and rodent samples22,27. In this study, the capacity of amplicon NGS was delineated up to 102 copies/μL of HTNV RNA loads.
Obtaining whole-genome sequences in clinical samples or in samples with low virus titers is problematic. Worobey et al. employed a “jackhammering” approach to the detection of target RNA molecules in degraded and low-titer samples36. This approach used primer pools to acquire genomes by generating a large number of small amplicons of the target genome. High-copy templates of the dengue virus genome were fully acquired with a few amplicons, whereas low-copy viral templates could be recovered using 10 overlapping amplicons37. These results demonstrate that short amplicons are easier to generate than long amplicons in low-copy viral specimens. The usage of multiplex primer sets to generate amplicons is an efficient strategy for obtaining viral genomes in low-titer specimens without the need for cultivating the viruses. However, amplicon-based methods introduce PCR artifacts due to the presence of Taq DNA polymerase through a number of PCR cycles. In addition, multiplex primers could generate amplicons for unrecognized unexpected variants38. In this study, multiplex primer sets facilitated the whole-genome sequencing of HTNV due to high nucleotide homology among virus strains; however, the efficiency of amplicon sequencing of viruses with low nucleotide homology should be evaluated.
In conclusion, we evaluated different methodologies for target enrichment NGS coverage for the complete genome sequence of HTNV from the rodent lung tissues. The amplicon NGS is the most robust method to recover nearly whole-genome sequences of HTNV from the specimen containing up to 102 copies/μL of HTNV RNA loads. Thus, this study provides useful insights into target enrichment NGS for the rapid identification and characterization of orthohantaviruses during endemic outbreaks.
Methods
Ethics statement
This study was approved in accordance with the ethical guidelines for the Korea University Institutional Animal Care and Use Committee (KU-IACUC), Korea University. Rodents trapping at US military training sites and installations was approved by US Forces Korea in accordance with regulation 40-1 (Prevention, Surveillance, and Treatment of Hemorrhagic Fever with Renal Syndrome). Rodents were euthanized by cardiac puncture under isoflurane anesthesia and the tissues were collected in accordance with the procedures approved by KU-IACUC (#2016-0049) protocol. All experiments were conducted in the biosafety level 3 facility.
Sample selection
Rodents (A. agrarius, Mus musculus, and Myodes regulus) were captured using live-capture Sherman traps (H.B. Sherman, Tallahassee, FL, USA) in Gyeonggi province, ROK between 2016 and 2017. Rodent species were identified by their morphology. Sera, lung, spleen, kidney, and liver tissues of the rodents were collected aseptically and frozen at −80 °C until use. For the whole-genomic sequencing of HTNV, the sera of the rodents were used for detecting the anti-HTNV immunoglobulin G (IgG) using indirect immunofluorescence assay (IFA) test. HTNV RNA was detected using HTNV-specific primers by RT-PCR. Lung tissues, which were positive in both sera anti-HTNV IgG and RT-PCR, from the rodents were selected and quantified for the HTNV genome copy number.
Indirect immunofluorescence antibody test
Rodent sera were diluted 1:32 in phosphate-buffered saline and examined for IgG against HTNV. The diluted sera were added to acetone-fixed HTNV-infected Vero E6 cells and the cells were incubated at 37 °C for 30 min. The cells were washed and treated with fluorescein isothiocyanate-conjugated anti-mouse IgG (ICN Pharmaceuticals, Laval, Quebec, Canada), and the cells were further incubated at 37 °C for 30 min. The cells were washed, and virus-specific fluorescence was examined by using a fluorescent microscope (Axio Scope, Zeiss, Berlin, Germany).
Mitochondrial DNA (mtDNA) analysis
Total DNA was extracted from liver tissues using TRIzol reagent solution (Life Technologies, Carlsbad, CA, USA). Rodent species was confirmed by performing mitochondrial Cyt b gene-specific PCR39.
RNA extraction and cDNA synthesis
Total RNA was extracted from the lung tissues of A. agrarius using a Hybrid R Kit (GeneAll Biotechnology, Seoul, ROK) according to the manufacturer’s specifications. cDNA was synthesized using a High Capacity RNA-to-cDNA Kit (Applied Biosystems Inc., Carlsbad, CA, USA) with random hexamers or OSM55 (5′-TAGTAGTAGACTCC-3′) primer.
Nested RT-PCR
Nested PCR was performed in a 25 μL reaction mixture containing 0.1 mM dNTP Mix, 0.625 units TaKaRa Ex Taq polymerase (Takara, Shiga, Japan), 0.4 µM of each primer, and 1.5 µL template. Oligonucleotide primer sequences for the nested PCR were G2F1 (outer): 5′-TGGGCTGCAAGTGC-3′, G2-2 (outer): 5′-ACATGCTGTACAGCCTGTGCC-3′, G2-1 (inner): 5′-TGGGCTGCAAGTGCATCAGAG-3′, and G2-4 (inner): 5′-ATGGATTACAACCCCAGCTCG-3′ for the M segment40. The PCR conditions were: initial denaturation at 94 °C for 5 min, followed by 6 cycles of denaturation at 94 °C for 30 s, annealing at 37 °C for 30 s, elongation at 72 °C for 1 min, followed by 32 cycles of denaturation at 94 °C for 30 s, annealing at 42 °C for 30 s, elongation at 72 °C for 1 min. Additionally, final elongation was done at 72 °C for 5 min. PCR products were extracted using a PCR Purification Kit (Cosmo Genetech, Seoul, ROK), and DNA sequencing performed in an Automatic Sequencer, ABI 3730XL DNA Analyzer (Applied Biosystems, USA).
Real-time quantitative PCR
RT-qPCR was performed using SYBR Green PCR Master mix (Applied Biosystems) on a Quantstudio 5 Flex Real-Time PCR System (Applied Biosystems). The primer sequences included a forward primer; 5′-TTATTGTGCTCTTCATGGTTGC-3′ and a reverse primer; 5′-CATCCCCTAAGTGGAAGTTGTC-3′ for HTNV S segment41. The PCR conditions were: 95 °C for 10 min, followed by 40 cycles of 15 s at 95 °C and 1 min at 60 °C.
Quantitation of HTNV genome copy
Viral RNA was extracted from HTNV 76–118, and cDNA was synthesized using a High Capacity RNA-to-cDNA kit (Applied Biosystems). PCR was performed in a 25 µL reaction mixture, containing 0.1 mM dNTP Mix, 0.625 units TaKaRa Ex Taq polymerase (Takara, Shiga, Japan), 0.4 µM of each primer, and 1.5 µL cDNA. Primer sequences were HTN-S-1127F (5′–CCTACCTCAGAAGGACACAATC–3′) and HTN-S-1437R (5′–ATGTTCCCATGCCCTGATATAC–3′). The amplified products were cloned into the pSTBlue-1 AccepTorTM vector (EMD Millipore Corp., Billerica, MA, USA) using T-A cloning technique with the DNA Ligation Kit (TaKaRa). The recombinant plasmid DNA was isolated using the Hybrid-Q Plasmid Preparation Kit (GeneAll Biotechnology). The concentration of recombinant plasmid DNA was measured by UV absorbance at 260 nm and 280 nm using Nano drop. Serial dilutions of the recombinant plasmid DNA standards ranging from 1 × 1010 to 1 × 103 copies/µL were amplified using SYBR Green PCR Master mix (Applied Biosystems) on a Quantstudio 5 Flex Real-Time PCR System (Applied Biosystems). The primer sequences and PCR conditions were identical to the ones used in RT-PCR. The copy number of plasmids per microgram of DNA was calculated using the total number of nucleotides in the plasmid using a described previously formula42.
Sequence-independent single-primer amplification
cDNA was generated from total RNA using FR26RV-N (5′-GCCGGAGCTCTGCAGATATCNNNNNN-3′). The reaction was performed in a 20 μL reaction mixture containing 7 μL total RNA, 2 μL 10 pM of primer, 2 μL 5X First strand buffer, 100 mM dithiothreitol, 25 mM MgCl2, 10 mM dNTPs, 0.5 μL RNaseOUT, and 0.5 μL Superscript III RTase (Life Technologies, Carlsbad, CA) in a Proplex thermocycler (Life technologies). The PCR conditions were: 25 °C for 10 min, 50 °C for 50 min, and 85 °C for 10 min. Double-stranded (ds) cDNA was synthesized using 0.2 units Klenow 3′ → 5′ exo DNA polymerase (Enzynomics, Daejeon, ROK) and1 μL RNaseH (Invitrogen, San Diego, CA). The Klenow reaction mixture was incubated at 37 °C for 1 h and 75 °C for 15 min. The ds cDNA was purified using MinElute PCR purification kit (Cat No. 28004, Qiagen, Hilden, Germany). Using the FR20RV (5′-GCCGGAGCTCTGCAGATATC-3′) primer, ds cDNA was amplified in a 50 μL reaction mixture containing 10 μL ds cDNA template, 10 pM primer, and 2X My Taq Red (Bioline, Taunton, MA). PCR conditions were: Initial denaturation at 98 °C for 30 s, followed by 38 cycles of denaturation at 98 °C for 10 s, annealing at 54 °C for 20 s, and elongation at 72 °C for 45 s.
Target capture-based enrichment
The custom-made probes (kindly provided by Dr. Gary Schroth and Dr. Stephen Gross, Illumina, San Diego, CA, USA) were designed to cover the entire HTNV tripartite genome based on the HTNV 76–118 (L segment, NC_005222; M segment, NC_005219; S segment, NC_005218) and ROKA 14-11 (L segment, KU207199; M segment, KU207203; S segment, KU207207) strains. The sequence of primers was shown in the Supplementary Data 1. Extracted RNA was fragmented at 94 °C for 10 s and each sample were enriched separately using a quarter of the reagents specified in the manufacturer’s protocol of the TruSeq RNA Access library preparation kit (Illumina). Samples were barcoded, pooled, and sequenced using the MiSeq reagent kit v2 (Illumina) on an Illumina MiSeq with a minimum of 2 × 150-bp reads.
Amplicon-based enrichment
cDNA was amplified using the primer mixture and Solg™ 2X Uh-Taq PCR Smart mix (Solgent, Daejeon, ROK) according to the manufacturer’s instruction. PCR condition and sequences of primer sets were as described previously27.
Library preparation for NGS
Libraries were prepared using TruSeq Nano DNA LT sample preparation kit (Illumina according to the manufacturer’s instructions. Double stranded cDNA was sheared using a M220 focused ultra-sonicator (Covaris, Woburn, MA, USA). The ds cDNA was size-selected, followed by evaluating the quality and concentration of the samples using an Agilent DNA 1000 chip kit or a high-sensitivity DNA chip kit on a bio-analyzer (Agilent Technologies, Santa Clara, CA, USA). The libraries were A-tailed, ligated with double indexes and adaptors, and enriched by PCR. The libraries were quantified using the Library Quantification Kit (KAPA Biosystems, Wilmington, MA, USA) on a Quantstudio 5 Flex Real-Time PCR System (Applied Biosystems). NGS sequencing was performed on a MiSeq benchtop sequencer (Illumina) with 2 × 150 base pairs using a MiSeq reagent kit v2 (Illumina).
NGS data analysis
Adaptor and index sequences of reads were trimmed, and low-quality sequences were filtered using CLC Genomics Workbench version 7.5.2 (CLC Bio, Cambridge, MA). The tripartite genome sequence of HTNV 76-118 was used in a reference mapping method. The read mapping to the reference genome sequence and the extraction of consensus sequences were performed. The genomic sequences of HTNV strains were deposited in GenBank (Supplementary Table 4).
Phylogenetic analysis
Multiple sequences of HTNV were aligned using MUSCLE algorithm in MEGA 6.043. The best fit substitution model was determined for HTNV L (1–6530 nt), M (1–3616 nt) and S segments (1–1696 nt), which were TN93 + G, T92 + G, and T92 + G, respectively. ML method was employed to generate the phylogenetic trees using MEGA 6.0. Topologies were assessed by bootstrap analysis of 1000 iterations.
Data availability
Raw fastq reads have been deposited in the NCBI SRA database (Supplementary Information).
References
Vaheri, A. et al. Uncovering the mysteries of hantavirus infections. Nat Rev Microbiol 11, 539–550 (2013).
Laenen, L. et al. Hantaviridae: Current Classification and Future Perspectives. Viruses 11 (2019).
Bi, Z., Formenty, P. B. & Roth, C. E. Hantavirus infection: a review and global update. J Infect Dev Countr 2, 003–023 (2008).
Schmaljohn, C. & Hjelle, B. Hantaviruses: a global disease problem. Emerg Infect Dis 3, 95 (1997).
Gizzi, M. et al. Another case of “European hantavirus pulmonary syndrome” with severe lung, prior to kidney, involvement, and diagnosed by viral inclusions in lung macrophages. Eur J Clin Microbiol Infect Dis 32, 1341–1345 (2013).
Lee, H. W., Lee, P. W. & Johnson, K. M. Isolation of the etiologic agent of Korean hemorrhagic fever. J Infect Dis 137, 298–308 (1978).
Song, J. W. et al. Genetic diversity of Apodemus agrarius-borne hantaan virus in Korea. Virus Genes 21, 227–232 (2000).
Radford, A. D. et al. Application of next-generation sequencing technologies in virology. J Gen Virol 93, 1853–1868 (2012).
Victoria, J. G., Kapoor, A., Dupuis, K., Schnurr, D. P. & Delwart, E. L. Rapid identification of known and new RNA viruses from animal tissues. PLoS Pathog 4, e1000163 (2008).
Smith, G. J. et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459, 1122–1125 (2009).
Gire, S. K. et al. Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345, 1369–1372 (2014).
Neverov, A. & Chumakov, K. Massively parallel sequencing for monitoring genetic consistency and quality control of live viral vaccines. Proc Natl Acad Sci USA 107, 20063–20068 (2010).
Quan, P. L., Briese, T., Palacios, G. & Lipkin, W. I. Rapid sequence-based diagnosis of viral infection. Antiviral Res 79, 1–5 (2008).
Wang, F. et al. Using Small RNA Deep Sequencing Data to Detect Human Viruses. Biomed Res Int 2016, 2596782 (2016).
Quick, J. et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. nature protocols 12, 1261 (2017).
Andersen, K. G. et al. Clinical Sequencing Uncovers Origins and Evolution of Lassa Virus. Cell 162, 738–750 (2015).
Reyes, G. R. & Kim, J. P. Sequence-independent, single-primer amplification (SISPA) of complex DNA populations. Mol Cell Probe 5, 473–481 (1991).
Djikeng, A. et al. Viral genome sequencing by random priming methods. BMC Genomics 9, 5 (2008).
Song, D. H. et al. Sequence-Independent, Single-Primer Amplification Next-Generation Sequencing of Hantaan Virus Cell Culture–Based Isolates. Am J Trop Med Hyg 96, 389–394 (2017).
Mamanova, L. et al. Target-enrichment strategies for next-generation sequencing. Nat Methods 7, 111–118 (2010).
Grubaugh, N. D. et al. Genomic epidemiology reveals multiple introductions of Zika virus into the United States. Nature 546, 401–405 (2017).
Kim, W. K. et al. Multiplex PCR-Based Next-Generation Sequencing and Global Diversity of Seoul Virus in Humans and Rats. Emerg Infect Dis 24, 249–257 (2018).
Chalkias, S. et al. ViroFind: A novel target-enrichment deep-sequencing platform reveals a complex JC virus population in the brain of PML patients. PLoS One 13, e0186945 (2018).
Metsky, H. C. et al. Capturing sequence diversity in metagenomes with comprehensive and scalable probe design. Nat Biotechnol 37, 160 (2019).
Salzberg, S. L. et al. Genome analysis linking recent European and African influenza (H5N1) viruses. Emerg Infect Dis 13, 713 (2007).
Li, K. et al. Automated degenerate PCR primer design for high-throughput sequencing improves efficiency of viral sequencing. Virol J 9, 261 (2012).
Kim, W. K. et al. Phylogeographic analysis of hemorrhagic fever with renal syndrome patients using multiplex PCR-based next generation sequencing. Sci Rep 6, 26017 (2016).
Lambden, P., Cooke, S., Caul, E. & Clarke, I. Cloning of noncultivatable human rotavirus by single primer amplification. J Virol 66, 1817–1822 (1992).
Matsui, S. M. et al. Cloning and characterization of human astrovirus immunoreactive epitopes. J Virol 67, 1712–1715 (1993).
Jones, M. S. et al. New DNA viruses identified in patients with acute viral infection syndrome. J Virol 79, 8230–8236 (2005).
Costa-da-Silva, A. L. et al. First report of naturally infected Aedes aegypti with chikungunya virus genotype ECSA in the Americas. PLoS Negl Trop Dis 11, e0005630 (2017).
Mate, S. E. et al. Molecular evidence of sexual transmission of Ebola virus. New Engl J Med 373, 2448–2454 (2015).
Rebuffo-Scheer, C. et al. Whole genome sequencing and evolutionary analysis of human respiratory syncytial virus A and B from Milwaukee, WI 1998-2010. PLoS One 6, e25468 (2011).
Yu, Q. et al. PriSM: a primer selection and matching tool for amplification and sequencing of viral genomes. Bioinformatics 27, 266–267 (2010).
Arias, A. et al. Rapid outbreak sequencing of Ebola virus in Sierra Leone identifies transmission chains linked to sporadic cases. Virus Evol 2, vew016 (2016).
Worobey, M. et al. 1970s and ‘Patient 0’ HIV-1 genomes illuminate early HIV/AIDS history in North America. Nature 539, 98–101 (2016).
Cruz, C. D., Torre, A., Troncos, G., Lambrechts, L. & Leguia, M. Targeted full-genome amplification and sequencing of dengue virus types 1-4 from South America. J Virol Methods 235, 158–167 (2016).
Cotten, M. & Koopmans, M. Next-generation sequencing and norovirus. Future Virol 11, 719–722 (2016).
Irwin, D. M., Kocher, T. D. & Wilson, A. C. Evolution of the cytochrome b gene of mammals. J Mol Evol 32, 128–144 (1991).
Song, J. W. et al. Hemorrhagic fever with renal syndrome in 4 US soldiers, South Korea, 2005. Emerg Infect Dis 15, 1833–1836 (2009).
No, J. S. et al. Detection of Hantaan virus RNA from anti-Hantaan virus IgG seronegative rodents in an area of high endemicity in Republic of Korea. Microbiol Immunol 60, 268–271 (2016).
Martinez-Martinez, M., Diez-Valcarce, M., Hernandez, M. & Rodriguez-Lazaro, D. Design and Application of Nucleic Acid Standards for Quantitative Detection of Enteric Viruses by Real-Time PCR. Food Environ Virol 3, 92–98 (2011).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol 30, 2725–2729 (2013).
Acknowledgements
We thank Mr. Charles Hong from Defense Threat Reduction Agency (DTRA) for critical discussion and experimental support. We thank Dr. Gary Schroth and Dr. Stephen Gross (Illumina) for technical supports. We thank Dr. Man-Seong Park (Korea University College of medicine) for supporting NGS facility. This work was supported by the Agency for Defense Development (UD160022ID), the Research Program To Solve Social Issues of the National Research Foundation of Korea (NRF) funded by the Ministry of Science and ICT (NRF-2017M3A9E4061992), and the Institute of Biomedical Science & Food Safety, Korea University (K1909531).
Author information
Authors and Affiliations
Contributions
J.S.N., W.-K.K. and S.C. designed the study, performed experiments, analyzed, and interpreted data, and wrote the manuscript. S.-H.L. provided captured animals. J.-A.K., D.H.S. and S.H.G. provided experimental support, D.L. analyzed the data, S.T.J. provided scientific discussion. M.R.W., G.P. provided experimental support and scientific discussion. J.-W.S. designed and supervised the study, analyzed, and interpreted data, wrote, and reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
No, J.S., Kim, WK., Cho, S. et al. Comparison of targeted next-generation sequencing for whole-genome sequencing of Hantaan orthohantavirus in Apodemus agrarius lung tissues. Sci Rep 9, 16631 (2019). https://doi.org/10.1038/s41598-019-53043-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-53043-2
This article is cited by
-
Disparate macrophage responses are linked to infection outcome of Hantan virus in humans or rodents
Nature Communications (2024)
-
Genome sequencing identifies “Limestone Canyon virus” as Montaño virus (Hantaviridae: Orthohantavirus montanoense) circulating in brush deermice in New Mexico
npj Viruses (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
