
Abstract
Four new derivatives of ketoconazole (Ke) were synthesized: diphenylphosphane (KeP), and phosphane chalcogenides: oxide (KeOP), sulphide (KeSP) and selenide (KeSeP). These compounds proved to be promising antifungal compounds towards Saccharomyces cerevisiae and Candida albicans, especially in synergy with fluconazole. Simulations of docking to the cytochrome P450 14α-demethylase (azoles’ primary molecular target) proved that the new Ke derivatives are capable of inhibiting this enzyme by binding to the active site. Cytotoxicity towards hACSs (human adipose-derived stromal cells) of the individual compounds was studied and the IC50 values were higher than the MIC50 for C. albicans and S. cerevisiae. KeP and KeOP increased the level of the p21 gene transcript but did not change the level of p53 gene transcript, a major regulator of apoptosis, and decreased the mitochondrial membrane potential. Taken together, the results advocate that the new ketoconazole derivatives have a similar mechanism of action and block the lanosterol 14α-demethylase and thus inhibit the production of ergosterol in C. albicans membranes.
Similar content being viewed by others

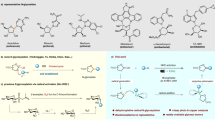

Introduction
In recent times, the emergence of pathogenic fungi resistant to commonly used antifungal drugs has been occurring at unprecedented rates1. Antifungal resistance is especially a problem with Candida infections, and Candida albicans is the predominant cause of invasive fungal infections in humans2, responsible for ~15% of nosocomial sepsis3. Invasive candidiases are mortal in 50–80% rates4,5, reaching ~70% even among patients undergoing antifungal therapy6,7. Thus, the search for new antifungals and alternative therapies has become increasingly important. To tackle this problem from a medicinal chemistry perspective, and considering that a modification of existing drugs is more straightforward than developing a new class of therapeutic agents, we chose ketoconazole (Ke), a synthetic imidazole antifungal drug approved by FDA in 19818, as a starting molecule for our present study. Ke was, for the following decade, the only antifungal available for oral treatment of systemic fungal infections caused by pathogenic yeasts. Its mechanism of action is well established. Similarly to other azole based drugs, its primary molecular target is cytochrome P450 14-alpha-demethylase (P45014DM)9,10,11,12,13,14,15, acting as a competitive inhibitor. Ke binds the Fe atom of cytochrome P450 through the N atom of its imidazole ring. Still in the 1980’s the interest on Ke expanded to other areas, namely as a potential anticancer agent, including in combinational therapy16,17, treatment of prostatic cancer18,19,20,21 and Cushing’s syndrome22,23. Moreover, it was shown that Ke could inhibit growing of several malignant and cancer cell lines24, inducing apoptosis through a p53 dependent pathway25 or inducing G0/G1 arrest by triggering mitophagy through down-regulation of COX-2 (cyclooxygenase-2)26.
Nowadays, due to all these diverse effects, Ke is regaining strong interest and new ketoconazole derivatives27,28,29 or complexes with several metal ions30,31,32 are being extensively studied. In this work, we decided to follow the pathway of modifying the existing drug and to investigate the properties of aminomethylphosphane derivatives of ketoconazole. Although a long time has passed since the first reports33, aminomethylphosphanes (or α-aminophosphanes) have been not considered interesting as potential drugs or components of biologically active metal complexes until the last decade. Recently, however, due to their flexibility and diversity, combined with the ease of synthesis, they started to gain a great attention. These compounds can bear an almost infinite variety of substituents with different hydrophilicities and steric demands. The potential presence of auxiliary coordinating atoms allows them to act as mono-, bi- or multidentate ligands. Our team has been working with this class of ligands and their Cu(I), Pt(II) and Ru(II) complexes and, in many cases, the properties of the derivatized molecules and their complexes are more interesting than the parent ones. For example, we worked with trisaminomethylphosphanes derived from morpholine and thiomorpholine (see for example refs34,35,36,37) and a variety on N4-substituted piperazines34,35,38,39,40,41, as well as with monoaminomethyldiphenylphosphanes40,41,42, including a derivative of a model dipeptide43 and the derivatives of selected fluoroquinolones: ciprofloxacin, norfloxacin44,45,46, lomefloxacin47 and sparfloxacin48,49,50. Some of the compounds listed above showed interesting antimicrobial and/or anti-tumour activity.
Herein we present the synthesis and characteristics of the diphenylphosphanomethyl derivative of ketoconazole and its chalcogenide derivatives (oxide, sulphide and selenide) in order to understand better the influence of a diphenylphosphanometyl(chalcogenide) moiety on the activity of the ketoconazole molecule.
Results and Discussion
Synthesis and characteristics of the compounds
KeP (Fig. 1) was synthesized from deacylated ketoconazole (KedA) in a modified Mannich condensation reaction of hydroxymethylphosphanes with amines33. Briefly, we added KedA to the solution of PPh2CH2OH obtained in situ from PPh2(CH2OH)2Cl by adding the excess of NEt3. Chalcogenides were synthesized in a reaction of KeP with a stoichiometric amount of H2O2, resublimed sulphur or metallic selenium in the ultrasound bath34,44,45,48. To confirm the assumed structures and check the purity of the products we employed mass spectrometry and elemental analysis as well as NMR spectroscopy.

Molecular schemes of ketoconazole (Ke) and its derivatives.
Mass spectra (Fig. S2 in ESI) confirmed the structures of the synthesized compounds. In the positive mass spectrum of the Ke molecule the most intense signals are [M + H]+ (46%) with the most intense peak at 531.2 m/z, [M + Na]+ (100%) at 553.1 and [2 M + Na]+ (63%) at 1083.5 showing strong tendency to form dimers under the experiment conditions. Phosphane KeP undergoes a strong fragmentation. The phosphane signal [M + Na]+ at 709.2 was very weak (15%) and the most intense fragmentation signals found and identified were at 489.2 [KedA + H]+ (6%) at 489.2 and [KedA + CH2]+ (100%) at 501.2. For the chalcogenide derivatives we observed a strongly increased stability of the molecules. In the case of KeOP the strongest signals were at 703.2 and 725.2 corresponding to [M + H]+ (6%) and [M + Na]+ (100%) ions respectively. A weak tendency to form dimers was reflected in the presence of the [2 M + Na]+ signal (5%) at 1429.4. Mass spectra of KeSP and KeSeP are very similar to one another. For both compounds we observed a small amount of the fragmentation products with dominating [KedA + CH2]+ at 501.2 with 13% or 19% intensity for KeSP and KeSeP, respectively. For KeSP the molecular signals were of [M + H]+ (29%) at 719.2 and [M + Na]+ (100%) at 741.2. The dimers were observed as the intense signals of [2 M + H]+ (51%) at 1439.4 and [2 M + Na]+ (32%) at 1461.4. For KeSeP the molecular signals were of [M + H]+ (48%) at 767.1 and [M + Na]+ (100%) at 789.1. The intensity of the signals of the dimers [2 M + H]+ (98%) at 1533.3 and [2 M + Na]+ (30%) at 1555.3 indicates that the tendency for dimer formation of the selenide derivative is the strongest among the compounds studied.
We measured the 1H, 13C{1H} and 31P{1H} (when applicable) NMR spectra of ketoconazole and its derivatives in CDCl3. Using two-dimensional COSY, HMQC and HMBC spectra we were able to assign unambiguously all the signals to the corresponding atoms in Ke molecule (see Figs S4–S10 and Table S1 in ESI) as well as in its derivatives (Table S2). As expected, 1H NMR spectrum of Ke is very complex and shows a set of characteristic patterns due to a presence of three different aromatic rings, racemic mixture of the chiral 2,2,4-trisubstituted 1,3-dioxolane ring and acetylated piperazine ring. It should be mentioned that the piperazine moiety in the Ke molecule shows four independent CH2 signals (carbon singlets and corresponding proton multiplets) indicating a strong binding of the acetyl group leading to the restrained rotation of the N-C bond.
Formation of KeP from PPh2(CH2OH) and KedA and further formation on the chalcogenides and the completion of the reaction is best observed by recording 31P{1H} spectra (see Tables 1, S2). In CDCl3 solution, PPh2(CH2OH) phosphorus atom exhibits a singlet at −10.03 ppm. Formation of the KeP molecule shifts this signal upfield to −27.39 ppm. Subsequent formation of chalcogenides transfers the electron density from the P atom towards a substituent, deshielding the phosphorus atom. Chemical shift for KeOP was 27.18, for KeSP was 35.15 and for KeSeP the chemical shift was 25.93 ppm with the 1J satellite coupling to 77Se equal to 721.5 Hz. All these values are typical for the diphenylphosphanomethyl ligands with N4-substituted piperazine ring attached to the P atom via methylene group and do not depend on N4 substituent40,45. In 1H and 13C{1H} NMR spectra, phosphane formation from KedA results in the new signals of methylene group being a link between KedA and -PPh2 moieties. Both proton and carbon signals are doublets with relatively small coupling constants to the phosphorus atom. This process is reflected also in the carbon signals of neighboring CH2 groups (position 2,6) in the piperazine ring, where a P-coupled doublet with 3J(P-C) around 10 Hz is observed. As for the tris(aminomethyl)phosphanes34 and the aminomethyldiphenylphosphanes40,45, formation of the chalcogenide derivatives induces dramatic changes in the signals of the groups directly bound to the P atom. This is best seen in the carbon signal of -CH2-P group, where formation of the P = X bond not only shifts the signal, but also significantly alters the values of 1J(C-P) coupling constant: from 3.6 Hz for KeP to 88.1, 73.6 and 66.3 Hz for KeOP, KeSP and KeSeP, respectively. These changes are almost repeated in the signals of the phenyl ring carbon atoms directly bound to the P atom (C(i)).
Activity against yeast Saccharomyces cerevisiae
For the preliminary studies of the biological activity of the studied compound we chose S. cerevisiae, which is generally considered a non-pathogenic organism, but currently recognized as a potentially important agent causing opportunistic infections against immunocompromised patients51. In order to assess whether all the Ke derivatives share the mechanism of action with the parent compound, or if they might present different or additional modes of action, the inhibitory concentration MIC50 (Table 2) was determined on the basis of the dose-response curves (Fig. S11 in ESI) in a 48 h assay against S. cerevisiae wt strain BY4741 and the deletion mutant erg6Δ, which lacks the gene coding for the S-adenosylmethionine: 24-methyltransferase52, accumulating cholesta-5,7,24-trienol and zymosterol instead of ergosterol53. There are several studies reporting that the ERG6 gene deletion may increase the resistance of S. cerevisiae cells to azole compounds54,55,56. Thus, we first studied Ke, and confirmed this behavior, as a MIC50 of 0.865 μM was determined against wt cells, whereas this compound showed no activity against the mutant cells. The value determined for the wt strain is in very good agreement with previous studies carried out under similar condition (48 h, 30 °C) (e.g.57).
Regarding the Ke derivatives, they exhibit clearly distinct behavior from the parent compound, and notably, they were all active against the resistant strain to similar or even greater extent as they were against the wt strain (except KeSP, which was also inactive against wt cells). These results strongly suggest that the Ke derivatives may present different or additional modes of action underlining their antifungal activity, or override mechanism of drug resistance operative against Ke, since not only they are active against yeast cells that are resistant to azole compounds, and shown to be Ke resistant in this study, but also the behavior in relation both to the parent compound and between the two strains can change. For KeP, the wt cells are less sensitive (MIC50 9.023 μM) than for Ke, while erg6Δ cells are very sensitive, presenting a MIC50 value (0.654 μM) which is even smaller than the one found for the parent compound against the wt. KeSeP and KeOP are active against both strains with similar MIC50 values (Table 2). Finally, as mentioned above, KeSP is inactive against both S. cerevisiae strains. KeOP, on the other hand, presents very high activity (MIC50 values on the nM range) against both wt and erg6Δ cells.
The lack of activity of KeSP is somewhat surprising, as it contrasts markedly with the behavior exhibited by the two other calchogenide derivatives, KeOP and KeSeP. We have, thus, hypothesized that this compound could be more efficiently exported by plasma membrane pumps. To test this hypothesis, we measured the activity of the compounds against a strain that lacks all the major ABC transporters in S. cerevisiae responsible for drug export, which is in general very susceptible to most xenobiotic compounds58. As control, we have also used the respective wt strain, W303. As with BY4741, W303 cells are susceptible to Ke and its derivatives, except for KeSP. This suggests that the inactivity of this compound against S. cerevisiae is a general trend, as it was observed in two different genetic backgrounds. On another hand, the MIC50 values are always considerably higher for W303 cells than for BY4741. This result is not unexpected, since it has been previously reported that, when grown in YPD medium, as in this study, BY4741 cells are more sensitive to Hygromycin B than W303, and they also differ in alkali-metal cation tolerance and plasma membrane potential, and in other important physiological parameters, as consequence of their different genome59,60. Notwithstanding, it is noteworthy that for both wt strains; the order of MIC50 values for the different compounds tested is almost the same, the only difference being between KeSeP and KeP, which are the second and third most active against BY4741 and in reverse order for W303 cells.
Regarding the strain lacking the major plasma membrane drug exporters, AD1-8, it is clear that all the compounds show much higher antifungal activity. The most striking result is the one for KeSP which displays a MIC50 value that is similar to KeP and KeSeP, and is more than 3 orders of magnitude lower than against the W303 cells, the wt with the same genetic bacground. The most active derivative is, again, KeOP. These results strongly suggest that the inactivity of KeSP is due to a very efficient efflux of this compound by S. cerevisiae.
Candida albicans viability
Minimal inhibitory concentration (MIC)
Fungistatic (MIC50) effect of diphenylphosphanomethyl derivatives of Ke was evaluated in comparison to Ke and a triazole antifungal – fluconazole (Flc) (Table 3). Flc is the most commonly prescribed antifungal azole drug61 but approximately 16-fold less active in vitro than Ke62. Ke based phosphanes were less active than Ke; however, two (KeP, KeSP, KeSeP) or eight (KeOP) times more active than Flc towards C. albicans CAF2-1 strain, referential in our study.
High resistance of C. albicans towards azole based agents is related to the development of a multi-drug resistance (MDR) phenotype found among clinical C. albicans isolates, which connects with overproduction of efflux transporters located in the plasma membrane (Cdr1 and Cdr2, belonging to ATP-binding cassettes (ABCs) and Mdr1, belonging to major facilitator superfamily (MFS))63. In order to determine the role of MDR efflux pumps in resistance towards Ke derivatives, we used three pairs of C. albicans strains - CAF2-1 and DSY1050; B3 and B4; Gu4 and Gu5. The mutant with no functional CDR1, CDR2 and MDR1 genes (DSY1050) was four (KeP, KeOP, KeSeP) or eight (KeSP) times more susceptible than its parental strain (CAF2-1). C. albicans B3 and B4 strains were isolated from a patient before and after fluconazole administration, respectively. Azole-resistance of B4 results from overexpression of MDR1, therefore two (KeOP, KeSP, KeSeP) and eight (Ke, KeP) times higher resistance of B4 than B3 indicates a possible role of Mdr1p in Ke derivatives efflux. High levels of Mdr1p are related to the development of resistance towards voriconazole (Vor) and Flc64,65,66,67 but to lesser extent for other azoles, e.g. Ke68,69,70,71.
C. albicans Gu4 and Gu5 strains were also isolated from a patient before and after fluconazole administration, respectively. Contrary to B4, azole-resistance of Gu5 origins in overexpression of CDR1 and CDR2. Cdr1p plays a major role in antifungal resistance in C. albicans cells by exporting most non-related xenobiotics outside fungal cells72. KeSP and KeSeP displayed no inhibitory effect on Gu5 strain and this strain was two and sixteen times more resistant than Gu4 strain towards KeOP and KeP, respectively. This result indicates a strong involvement of Cdr1p and Cdr2p in Ke derivatives resistance and that Ke derivatives are possibly substrates for Cdr1/Cdr2 pumps.
KeOP/KeP synergism with fluconazole
In case of candidiasis treatment, a synergistic combination of clinically used azoles with other drugs is highly desirable73,74. One of the approaches is to inhibit activity of MDR efflux pumps by administration of competitive pump substrates among azole drugs75. Thus, we combined Flc with KeP or KeOP, since both Ke derivatives had most promising MIC50 values. KeP (0.39 uM) and KeOP (1.56 uM) reduced MIC50 of Flc in the range from 6.25 uM to 1.56 uM and when combined with higher (3.25 and 6.25 uM) Flc concentrations lead to fungicidal effect (Fig. 2 and Table S3 in ESI). Since azoles are fungistatic2,76, biocidal combination of KeP/KeOP with Flc is promising, especially, because simultaneous administration of different azoles may either lead to synergistic, subadditive or antagonistic effects77.

KeP and KeOP synergism with fluconazole (Flc). Viability [%] of C. albicans CAF2-1. Statistical analysis was performed comparing viability at corresponding Flc concentrations with/without KeOP/KeP (*0.01 < P < 0.05; **0.001 < P < 0.01; ***P < 0.0001). ±SD, n = 3.
Human adipose-derived stromal cells (hACSs) studies
Half maximal inhibitory concentration (IC50) assessment
In order to assess the activity of the ketoconazole and its derivatives against human cells, IC50 values for Ke, KeP, KeOP as well as KeSeP for hASCs (human adipose-derived stromal cells) were obtained from the dose-response curves that were prepared using TOX-8 Assay after a 24-hours stimulation (Fig. 3).

hACSs viability in the presence of different azole compounds. Percentage of cell survival is plotted against the logarithm of treatment concentrations. Calculated IC50 values are: 120.8 for Ke, 37.65 for KeP, 50.76 for KeOP and 109.8 μM for KeSeP. It was not possible to determine the IC50 values for Flc and KeSP.
The cytotoxicity assay indicated KeP (IC50 = 37.65 µM) as the most active compound. The cytotoxicity of KeOP (IC50 = 120.8 µM) and KeSeP (IC50 = 109.8 µM) were significantly lower, and the lowest cytotoxic effect was found for Flc and KeSP (Fig. 3), which were not cytotoxic in the tested concentration range. These data stand in good agreement with other results, that show a relatively small cytotoxicity of Flc78. In this study, the IC50 value for Ke is 120.8 µM. However, contradictory data regarding cytotoxicity of Ke has been reported78, which might be due to the use of different cell sources. It was previously shown by Benkoő et al. that the IC50 value of Ke for whole population of human bone marrow cells was equal to 6,27 mg/L (11.8 µM). Moreover, the IC50 of Ke was almost 3-fold higher for murine bone marrow cells in comparison to the human cells78. In turn, Haegler et al. showed that in HepG2 cells (liver hepatocellular carcinoma cell line), Ke cytotoxic effects started to be noticed at 50 µM. In contrast, in HepaRG cells, cytotoxicity started at 100 µM79.
Apoptosis level of hASCs after 24-hours stimulation with Ke, KeP and KeOP (20 µM)
Apoptosis is characterized by cell shrinkage, alternations in the cell membrane and mitochondria, nuclear condensation, as well as DNA fragmentation and protein degradation by caspases80. It was shown that BCL-2 transcript protects cells from apoptosis by associating with mitochondria and inhibiting their release of cytochrome c, whereas BAX promotes apoptosis. Moreover, p53 gene expression is associated with induction of apoptosis80,81. Interestingly, p21 transcript plays a dual role in cell death and survival, i.e., it inhibits apoptosis through induction of cell cycle arrest and DNA repair, but p21-arrested cells may undergo apoptosis following activation of other pro-apoptotic genes81. In this study, we observed significant (p < 0.001) elevated expression of p21 transcript in cells challenged with Ke, KeP or KeOP when compared to the control group. Moreover, the relative expression levels of both BAX and BCL-2 were higher in cells treated with Ke, KeP or KeOP, but the differences were not significant. Interestingly, none of the compounds increased the expression of a master regulator of apoptosis, which is a p53 transcript (Fig. 4). Furthermore, no significant changes in the expression of p53 gene were observed when compared to control cells. This observation might suggest that the tested dosage of Ke, KeP and KeOP, which was 20 µM, did not induce apoptosis in hASC cultures. which might be a positive feature light for their future potential clinical application in antifungal therapy.

Apoptosis level of hACSs treated with 20 µM Ke, KeP and KeOP for 24 hours. Results expressed as mean ± SD. Statistical significance indicated as three asterisks ***p < 0.001.
Mitochondria network and F-actin in hASCs
Cell morphology was evaluated using MitoRed and Phalloidin Atto 590 staining after 24 h stimulation with Ke, KeP and KeOP (20 µM). MitoRed is a vital dye which interacts with functional mitochondria due to their high membrane mitochondrial potential. Decreased fluorescence of MitoRed stain has been associated with reduced mitochondrial membrane potential82. Patalano et al. showed that Ke treatment induces depolarization of mitochondria which leads to the activation of apoptosis in mouse corticotroph tumour cell line (AtT20-D16)83. Moreover, Haegler et al. showed that Ke decreases the mitochondrial potential in HepG2 cells, which was associated with decreased mitochondrial DNA synthesis and accumulation of mitochondrial superoxide that induce apoptosis84. In this work, the mitochondrial staining indicates the lowest mitochondrial membrane potential in KeP treated cells, as well as the lowest number of mitochondria. (Fig. 5). This might suggest a reduced antioxidative capacity. In turn, phalloidin staining showed a multilayer growth pattern in cells treated with Ke and KeOP. These cells where characterized by close contact with a relatively dense growth pattern, while hASCs treated with KeP were characterized by a loose growth pattern. It can be concluded that the compounds tested, i.e. Ke, KeP and KeOP, affect in different ways the morphological features of progenitor cells. Moreover, KeP seems to affect negatively cellular morphology and mitochondrial activity.

Visualization of mitochondrial network and F-actin of hASCs treated with Ke, KeP and KeOP (20 µM; 24 h) using MitoRed and Phalloidin Atto 590 staining, respectively. Scale bar: 50 µm.
Docking to CYP51 of C. albicans
As it has been said in the introduction, primary molecular target of Ke is cytochrome P450 14-alpha- demethylase. Mechanism of action is pretty simple. Ke, like other azoles, acts as an competitive inhibitor binding cytochrome Fe atom by N atom from imidazole ring and therefore blocking the enzyme active site. It was proven by several X-ray structures of the enzyme with different azoles. For Ke there one can find a structure of human CYP51 inhibited by the Ke molecule13,14, and only recently a structure of fungal CYP51 from C. albicans in complex with posaconazole was determined15. To check if the presented here derivatives can also target this enzyme we performed Molecular Docking using AutoDock Vina software85.
The docking results together with the best-fit structures are summarized in Fig. 6. The reference structure is the CYP51-posaconazole complex and the location of posaconazole serves to determine the correct binding mode. Posaconazole itself was also subjected to the docking to validate the docking procedure. Interestingly, while the correct overall binding mode is found for posaconazole, the triazole ring is rotated so that the Fe-N distance is much larger than in the experimental crystal structure (3.96 Å vs. 2.08 Å). However, the calculated binding affinity for posaconazole is the second strongest among the molecules studied, next only to KeP. For all five Ke derivatives, the proper location was found by the docking algorithm, which is a good indication of their binding affinity and the proper shape of the binding site cavity. The search space was large enough (cubic box, a = 80 Å) not to force the ligand into the cavity. The best-fit structures are always located in the binding site and oriented properly (that is, the heme iron atom is coordinated by the N atom in the imidazole ring of a given derivative), with the two important exceptions of KeSP and KeSeP, for which the sulfur or selenium atoms are oriented towards the heme iron. The resulting structure is “head to tail” with respect to posaconazole and the remaining Ke derivatives. This might arise from the stronger preference of the Fe-S(Se) interaction over the Fe-N contact, or from an increased volume of the P-S(Se) fragment. The matter of possible Fe-S(Se) coordination will be further investigated experimentally; nevertheless, both KeSP and KeSeP are also located in the correct cavity. Taking into account the approximate nature of the docking score function, it is not possible to determine whether the investigated Ke derivatives bind more or less strongly to the protein than Ke itself, but no dramatic changes were noted, suggesting that binding affinities among the analyzed Ke derivatives should be of similar order.

Docking results: heme of the C. albicans CYP51 and the best-fit docked compounds; posaconazole position from the original crystal structure15 is also indicated (as a black wire). (A) docked posaconazole, (B) Ke, KeP, KeOP, C: KeSP, KeSeP.
For KeP and KeOP, the ligands oriented as Ke, an analysis of the vicinity of the phosphorus atom was carried out. For KeP, the P atom interacts via its lone pair with the Pro230 ring (the closest distance P-C(Pro230) is 3.811 Å). Moreover, the P atom is also in potential contact with the phenyl ring of Phe233 (the closest P-C(Phe233) distance is 4.766 Å). In the case of KeOP, the direct environment around the ligand is somewhat different: Pro230, Phe233, Phe235 and Phe380 form a hydrophobic cage surrounding the –PPh2 phenyl rings of the ligand, while the P = O moiety is in contact with the Tyr64 phenol group (the closest (KeOP)O…C(Tyr64) distance is 3.313 Å, and the O…O(phenol,Tyr64) distance is 4.667 Å). These values must be taken as a rough estimation, due to the aforementioned nature of the docking scoring function, but they represent a possible diversity of bonding between the protein and the phosphane moiety of the Ke derivatives. In particular, the possibility of additional stabilization by interactions with a hydrophobic cage could add to the binding strength of KeP.
Conclusions
Derivatization of ketoconale with diphenylphosphanomethyl moiety gave an opportunity to develop a set of similar molecules differing only by the direct environment of the phosphorus atom. Four new derivatives of ketoconazole (Ke) were synthesized: diphenylphosphane (KeP) and its chalcogenides: oxide (KeOP), sulphide (KeSP) and selenide (KeSeP). Their structures were analyzed using NMR spectroscopy and mass spectrometry.
The derivatives of ketoconazole synthesized and tested in this work, especially KeP and KeOP, proved to be promising antifungal compounds. A strong antifungal activity, in some occasions surpassing largely the parent compound, was shown for selected strains of S. cerevisiae and also in the detailed study for C. albicans, especially in synergy with fluconazole. All compounds tested are substrates for pumps expelling drugs (mainly Cdr1 and Cdr2) from C. albicans cells, and simultaneous administration of fluconazole allowed a significant reduction in concentrations of ketoconazole derivatives tested and low survival of the fungus. Simulations of docking of the derivatives to the cytochrome P450 14-α-demethylase (the azoles’ primary molecular target) did not show large differences between their binding strength, also in comparison with ketoconazole, proving that they are capable of inhibiting this enzyme. While KeP and KeOP, like Ke, form a bond between nitrogen atom from the azole ring and the heme iron atom, for KeSP and KeSeP, a reversed placement in the hydrophobic cage was the preferred one. This opens a possibility of a different/secondary action mechanism for these compounds and will be studied in detail.
In what concerns cytotoxicity towards hACSs, the individual compounds had displayed some activity, except for KeSP which was not toxic. Two ketoconazole derivatives were further studied (KeP and KeOP) and in both cases we observed an increased level of the p21 gene transcript, but no changes in the level of p53 transcript. A decreased mitochondrial membrane potential was also apparent. This may indicate that all the new ketoconazole derivatives have a similar mechanism of action and block the lanosterol 14α-demethylase and thus inhibit the production of ergosterol in C. albicans membranes. The IC50 value in the cytotoxicity assay with human cells was significantly higher than the MIC50 for C. albicans, especially when the compounds were used with fluconazole. Therefore, we conclude that phosphane derivatives of ketoconazole are a promising group of compounds with potential use in the treatment of mycoses.
Taking into account all our results, we conclude that Ke based phosphane is a very interesting ligand and enables a significant expansion of possible metal complexes bearing the Ke moiety. Currently, we are working on copper(I) and ruthenium(II) complexes with KeP. Moreover, we are going to study phosphanes bearing more than one Ke moieties.
Experimental Part
Syntheses
Reagents and solvents used for synthesis were purchased from Sigma-Aldrich, Poland and used without further purifications. All syntheses were performed under inert (N2) atmosphere using standard Schlenk techniques. Deacylated ketoconazole (KedA; 1-4-[2-(2,4-Dichlorophenyl)-2-imidazol-1-ylmethyl-[1,3]-dioxolan-4-ylmethoxy]phenyl-piperazine; M = 489.4 g/mol) and PPh2(CH2OH)2Cl (M = 282.7 g/mol) were prepared following literature procedures86,87.
Ph2PCH2-Ke (KeP)
Triethylamine (3 mL) was added drop wise to a solution containing 0.3207 g of PPh2(CH2OH)2Cl (1.134 mmol) in 10 mL of MeOH in an ice bath. After stirring for 30 min, an MeOH solution (15 mL) of KedA (0.5550 g; 1.134 mmol) was added. The mixture was stirred for 1 h at room temperature and its volume was reduced by 1/3 of initial volume under a stream of nitrogen. Then 100 mL of water was slowly added. The obtained white solid was filtered, washed five times with water and dried under reduced pressure for 24 h. Yield 61%. Anal. Calc. for C37H37Cl2N4O3P (M = 687.6 g/mol): C, 64.63; H, 5.42; N, 8.15. Found: C, 64.34; H, 5.45; N, 8.11%. MS ESI (+):489.2 [KedA + H]+ (6%), 501.2 [KedA + CH2]+ (100%), 709.2 [M + Na]+ (15%).
Ph2P(O)CH2-Ke (KeOP)
KeOP was obtained in a dichloromethane-acetonitrile mixture (1:1) (10 mL) through the reaction of KeP (0.1081 g; 0.157 mmol) placed in an ice bath with an equimolar amount of H2O2 (30% solution in water: 16 μL; 0.157 mmol). After stirring for 30 min, the solvent was evaporated under vacuum. The remaining white solid was washed twice with water and dried for 24 h under vacuum to remove any occluded solvent molecules. Yield 94%. Anal. Calc. for C37H37Cl2N4O3P (M = 703.6 g/mol): C, 62.79; H, 5.33; N, 7.92. Found: C, 62.83; H, 5.33; N, 7.92%. MS ESI (+):703.2 [M + H]+ (6%), 725.2 [M + Na]+ (100%), [2 M + Na]+ 1429.4 (5%).
Ph2P(S)CH2-Ke (KeSP)
To KeP (0.0983 g; 0.143 mmol) in chloroform (15 mL) an equimolar amount of resublimed sulfur (0.00457 g; 0.143 mmol) was added. The mixture was placed in an ultrasonic bath for 1 h at room temperature. The yellowish solid obtained after removing the solvent was redissolved in a minimal volume of methanol and precipitated with large amounts of water. The solid obtained was filtered and dried for 24 h under vacuum. Yield 68%. Anal. Calc. for C37H37Cl2N4O3PS (M = 719.7 g/mol): C, 61.75; H, 5.18; N, 7.78. Found: C, 61.40; H, 5.22; N, 7.74%. MS ESI (+): 501.2 [KedA + CH2]+ (13%), 719.2 [M + H]+ (29%), 741.2 [M + Na]+ (100%) 1439.4 [2 M + H]+ (51%), 1461.4 [2 M + Na]+ (32%).
Ph2P(Se)CH2-Ke (KeSeP)
An equimolar mixture of KeP (0.1026 g; 0.149 mmol) and metallic selenium (0.01178 g; 0.149 mmol) in chloroform (15 mL) was placed in an ultrasonic bath until the traces of solid Se disappeared (~1 h). The yellowish solid obtained after removing the solvent was redissolved in a minimal volume of methanol and precipitated with a large amount of water. The solid obtained was filtered and dried for 24 h under vacuum. Yield 73%. Anal. Calc. for C37H37Cl2N4O3PSe (M = 766.6 g/mol): C, 57.97; H, 4.87; N, 7.31. Found: C, 57.66; H, 4.90; N, 7.31%. MS ESI (+):501.2 [KedA + CH2]+ (19%), 767.1 [M + H]+ (48%), 789.1 [M + Na]+ (100%), 1533.3 [2 M + H]+ (98%), 1555.3 [2 M + Na]+ (30%).
Methods
Elemental analyses were performed on a Vario EL3 CHN analyzer for C, H, and N, and they were within ±0.3% of the theoretical values. NMR spectra were recorded on a Bruker AMX 500 spectrometer (at 298 K) with traces of solvent as an internal reference for 1H (CDCl3: 7.27 ppm) and 13C spectra (CDCl3: 77.0 ppm) and 85% H3PO4 in H2O as an external standard for 31P. All the NMR data are presented in ESI (Tables S1, S2) Mass spectra were recorded on a Bruker Daltonics micrOTOF-Q mass spectrometer equipped with an electrospray ionization (ESI) source and operated in positive ion mode.
Saccharomyces cerevisiae
The following strains of S. cerevisiae were used in this study: BY4741 (wild type towards erg6Δ, ACC Y00000; MATa; his3Δ1; leu2Δ0; met15Δ0; ura3Δ0), erg6Δ (ACC Y00568; BY4741 isogenic with YML008c::kanMX4), W303 (wild type towards AD1-8; MATα; leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15 AD1-8 (W303 isogenic with pdr1-3, ura3, his1, yor1∆::hisG, snq2∆::hisG, pdr5∆::hisG, pdr10∆::hisG, pdr11∆:: hisG, ycf1∆::hisG, pdr3∆::hisG, pdr15∆::hisG). BY4741 and erg6Δ were acquired from EUROSCARF, Frankfurt, Germany; W303 and AD1-8 were kind gifts from Prof. A. Goffeau, Chaire Internationale Blaise Pascal, Lab. Genet. Mol., École Normale Supérieure, Paris, and Prof. M. Ghislain, Université Catholique de Louvain, Faculté des sciences Agronomiques, Louvain-la-Neuve, Belgium. Visible turbid overnight cultures of S. cerevisiae strains were prepared and diluted to A600 ≈ 0.1 in fresh Yeast Extract – Peptone - Dextrose (YPD) medium. The cultures were transferred to 96 microtiter well plates containing aliquots of serially diluted antifungal drugs (Ke and its derivatives: KeP, KeOP, KeSP, KeSeP), prepared from DMS stock solutions and drug-free controls. The yeast cultures were statically incubated at 30 °C for 48 hours with compounds88. The dose-response curves were analysed with GraphPad Prism 5 software (San Diego, USA) or a microplate reader ASYS UVM (Biogenet). All values presented are the mean of at least three independent experiments.
Candida albicans
C. albicans strains and growth conditions
The C. albicans CAF2-1 (Genotype/Description: ura3Δ::imm434/URA389) and DSY1050 (Genotype/Description: cdr1Δ::hisG/cdr1Δ::hisG/cdr2Δ::hisG/cdr2Δ::hisG/mdr1Δ::hisG-URA3-hisG/mdr1Δ::hisG90) strains used in this study were generous gift from D. Sanglard (Lausanne, Switzerland)91,92 whereas C. albicans B3 and Gu4 (Genotype/Description: clinical isolates fluconazole sensitive)93), B4 (Genotype/Description: Clinical isolate fluconazole resistant due to the overexpression of MDR193) and Gu5 (Genotype/Description: Clinical isolate fluconazole resistant due to the overexpression of CDR1 and CDR293) were a generous gift from prof. S. Milewski (Gdańsk, Poland)94 and prof. J. Morschhäuser (Wurzburg, Germany)93. All strains were grown at 37 °C on Sabouraud medium (Biomerieux).
Minimal inhibitory concentration (MIC) determination
The antifungal activity was determined on the 96-well polystyrene microtiter plates (Sarstedt) according to Clinical and Laboratory Standards Institute M27-A395. The different strains were incubated with antifungal compounds (fluconazole (Flc) (Sigma-Aldrich), Ke and Ke derivatives) in the range of concentrations 0.01–200 μM. The compounds were dissolved in DMSO (10 mM) and then serially diluted in RPMI-1640 (ThermoFisher) medium to obtain the final concentration range. Inocula of all strains were prepared as suspensions with turbidity of 0.5 McFarland standard in sterile 0.85% saline and diluted in RPMI-1640. After inoculation, each well contained appr. 5 × 102 CFU/mL. The plates were cultivated for 48 h at 37 °C. Optical density was measured at A490nm using a microplate reader ASYS UVM 340 (Biogenet). The concentrations which resulted in ≥50% growth inhibition were determined as MIC50.
KeOP/KeP synergism with fluconazole
The synergistic effects of KeOP/KeP with Flc were determined using the M27-A3 method with modifications. Flc and KeOP/KeP stock solutions in DMSO (10 mM) were (alone and simultaneously) diluted in RPMI-1640 medium to obtain final concentrations of1/8; 1/4; 1/2; 1 × MIC50 towards C. albicans CAF2-1 strain in microdilution sterile plates (Sarstedt). Statistical significance analysis was performed using Student’s t-test (binomial, unpaired) comparing viability in the presence of corresponding Flc concentrations with/without KeOP/KeP.
Human Adipose Tissue-derived Stem Cells (hASCs)
Unless indicated otherwise, all reagents used in this study were purchased from Sigma-Aldrich (Poland).
Cells isolation and culture
Human Adipose Tissue-derived Stem Cells (hASCs) were isolated from subcutaneous adipose tissue fragments harvested from non-diabetic female donors (age range 33–38, n = 2) during standard surgical procedure of total hip arthroplasty. Immediately after collection of tissue, the samples were placed in sterile tissue-transport medium (Hank’s Balanced Salt Solution (HBSS) supplemented with 1% Penicillin/Streptomycin/Amphotericin B solution (P/S/A). Mesenchymal stromal cell isolation was performed using enzymatic digestion of extracellular matrix with collagenase type I. Before digestion, tissue specimens were washed twice with HBSS and incubated in collagenase type I solution (1 mg/mL) at 37 °C for 40 min. The digest was subsequently centrifuged (1200 × g; 10 minutes; room temperature) and the cell pellet was resuspended in Dulbecco’s modified Eagle’s medium (DMEM) with Nutrient F-12 Ham, 10% of Fetal Bovine Serum (FBS), and 1% of P/S/A, transferred to a culture flask, and kept in culture under optimal conditions (37 °C in a humidified atmosphere of 5% CO2). The medium was refreshed every 2–3 days. At 80–90% confluence, the cells were passaged using trypsin solution (TrypLE Express, Life Technologies, California, USA). At passage 3, hASCs were collected and cellular phenotype was confirmed by the high expression of markers CD44, CD29, CD73, CD105 and low expression of CD45. Moreover, the trilineage differentiation capacity of ASCs was confirmed as previously described96.
Half maximal inhibitory concentration (IC50) determination
The IC50 values were determined using in vitro Toxicology TOX-8 Assay Kit (resazurin based) according to the manufacturer’s protocol. For this assay, cells were seeded in a 96-well plate at a density of 5 × 103 cells/well. The cells were pre-incubated in drug-free complete medium for 24 h before adding various concentrations of the compounds to be investigated. Stock solutions of the compounds were prepared in DMSO following serial dilutions in DMEM with Nutrient F-12 Ham complete medium. The cells exposure period was 24 h. After that time media were replaced with 10% v/v resazurin dye solution in fresh complete culture medium. Incubation was carried out for 2 h at 37 °C in the CO2 incubator. Reduction of the dye was measured spectrophotometrically at 600 nm and 690 nm reference wavelength (Epoch, Biotek). Absorbance measurements allowed the determination of viable treated cells relative to untreated controls using the inflection point of a dose-response graph. IC50 values were estimated as duplicates of triplicate readings in two independent experiments.
Evaluation of cellular apoptosis
Cellular apoptosis level was assessed using quantitative reverse transcriptase real-time polymerase chain reaction (qRT-PCR). First, the cells were seeded in a 24-well plate at a concentration of 3 × 104 cells/well in DMEM with Nutrient F-12 Ham complete medium and left in the CO2 incubator overnight to allow cells to attach. The compounds (Ke, KeP and KeOP) were diluted in the fresh complete medium to a final concentration of 20 μM. After 24 h of challenge with the compounds, the cells were homogenized with TRI Reagent and total RNA was isolated by phenol-chloroform extraction and ethanol precipitation97. RNA purity and quantity were measured at a wavelength of 260 and 280 nm (Epoch, Biotek). Genomic DNA was removed with DNase I, RNase-free (1 U/L) (Thermo Scientific, Poland) and cDNA was synthesized using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific, Poland). 150 ng of total RNA served as a template for a single reaction using T100 Thermo Cycler (Bio-Rad, USA). The expression levels of p53 tumor suppressor, cyclin dependent kinase inhibitor 1A (p21), apoptosis regulators BAX and BCL-2 were determined. qRT-PCR was performed on a CFX ConnectTM Real-Time PCR Detection System (Bio-Rad) using SensiFast SYBR & Fluorescein Kit (Bioline, Cincinnati, OH, USA) and primer pairs as listed in Table S4 in ESI. 500 nM of specific primers and 1 µL of cDNA in a 10 µL final volume were applied in each reaction. The quantitative expression of the genes was calculated by the 2−ΔΔCT method using GAPDH as housekeeping genes.
Statistical analysis was performed with GraphPad Prism 5 software (San Diego, USA). All data values are presented as mean ± SD, measured in triplicates or more. Statistical significance between groups was determined using two-way ANOVA grouped analysis.
Confocal microscopy imaging
The mitochondrial network was visualized using MitoRed staining. After 24-hours treatment with either Ke, KeP or KeOP, the media were removed and replaced with MitoRed dye in 1:1000 dilution in fresh complete media. Cells were incubated for 30 minutes at 37 °C, then washed with Phosphate Buffered Saline (PBS), fixed with 4% paraformaldehyde (PFA) solution, washed three times and nuclei were counterstained with diamidino-2-phenylindole (DAPI).
In order to perform F-actin imaging, cells were fixed with 4% PFA solution, washed with PBS and then permeabilized using 0.5% TX-100 (incubation 15 minutes, room temperature). The cells were rinsed and incubated with Phalloidin-Atto 590 solution in PBS (dilution 1:1000) at 37 °C for 30 minutes in the dark. After washing, the cells were counterstained with DAPI.
Cells were observed and images were taken using confocal microscopy performed with a Leica TCS-SP8 laser scanning confocal microscope. Data were further processed with the use of ImageJ software.
Docking studies
Docking calculations were performed using the X-ray structure of C. albicans CYP51 in complex with posaconazole15 (PDB code 5FSA) as the macromolecular host, and the set of ketoconazole derivatives as the ligands. Posaconazole was also included in the set as a validation test. The DFT-optimized structures (the Gaussian 16, Rev. B.01 program98 with ωB97XD functional99 and the 6-311 + G(d,p) basis set) served as initial ligand structures. The macromolecule consisted of the protein chain A, together with the heme cofactor, but without posaconazole. The docking of the ligands was carried out using a cubic box with an 80 Å edge, centered on the iron atom of the heme cofactor, as the search space; up to nine structures for each search were retained for further analysis. For two of the ligands (KeSP, KeSeP), docking runs were also repeated with a smaller box (a = 40 Å). A docking run for each ligand was replicated three times with different random seeds, and generally the results were consistent between the replicas. Preprocessing of the structures and visualization of the results was carried out with AutoDockTools program of the MGLTools package100 and UCSF Chimera 1.11.2_b41376 graphic program101, while the docking itself was performed with the AutoDock Vina software ver. 1.1.285.
References
Fisher, M. C., Hawkins, N. J., Sanglard, D. & Gurr, S. J. Worldwide emergence of resistance to antifungal drugs challenges human health and food security. Science 360, 739–742, https://doi.org/10.1126/science.aap7999 (2018).
Krishnasamy, L., Krishnakumar, S., Kumaramanickavel, G. & Saikumar, C. Molecular mechanisms of antifungal drug resistance in Candida species. J. Clin. Diagnostic Res. 12, DE01–DE06, https://doi.org/10.7860/JCDR/2018/36218.11961 (2018).
Lohse, M. B., Gulati, M., Johnson, A. D. & Nobile, C. J. Development and regulation of single–and multi–species Candida albicans biofilms. Nat. Rev. Microbiol. 16, 19–31, https://doi.org/10.1038/nrmicro.2017.107 (2018).
Whaley, S. G. et al. Azole Antifungal Resistance in Candida albicans and Emerging Non–albicans Candida Species. Front Microbiol. 12, A2173, https://doi.org/10.3389/fmicb.2016.02173 (2017).
Gunsalus, K. T. & Kumamoto, C. A. Transcriptional Profiling of Candida albicans in the Host. Methods Mol Biol. 1356, 17–29, https://doi.org/10.1007/978-1-4939-3052-4_2 (2016).
Salci, T. P., Negri, M., Abadio, A. K. R., Svidzinski, T. I. E. & Kioshima, É. S. Targeting Candida spp. to develop antifungal agents. Drug Discov. Today 23, 802–814, https://doi.org/10.1016/j.drudis.2018.01.003 (2018).
Rodriguez, L. et al. A multi–centric study of Candida bloodstream infection in Lima–Callao, Peru: species distribution, antifungal resistance and clinical outcomes. PLoS One 12, e0175172, https://doi.org/10.1371/journal.pone.0175172 (2017).
Gupta, A. K., Daigle, D. & Foley, K. A. Drug safety assessment of oral formulations of ketoconazole. Exp. Opin. Drug Saf. 14, 325–334, https://doi.org/10.1517/14740338.2015.983071 (2015).
Tyle, H. V. J. Ketoconazole. Mechanism of action, spectrum of activity, pharmacokinetics, drug interactions, adverse reactions and therapeutic use. Pharmacotherapy 4, 343–73, https://doi.org/10.1002/j.1875-9114.1984.tb03398.x (1984).
Borelli, D. et al. Ketoconazole, an oral antifungal: laboratory and clinical assessment of imidazole drugs. Postgrad. Med. J. 55, 657–661, https://doi.org/10.1136/pgmj.55.647.657 (1979).
Loose, D. S., Kan, P. B., Hirst, M. A., Marcus, R. A. & Feldman, D. Ketoconazole blocks adrenal steroidogenesis by inhibiting cytochrome P450–dependent enzymes. J. Clin. Invest. 71, 1495–1499, https://doi.org/10.1172/JCI110903 (1983).
Shimokawa, O. et al. Relationship between MIC and Minimum Sterol 14–Demethylation–Inhibitory Concentration as a Factor in Evaluating Activities of Azoles against Various Fungal Species. J. Clin. Microbiol. 43, 5547–5549, https://doi.org/10.1128/JCM.43.11.5547-5549.2005 (2005).
Strushkevich, N., Usanov, S. A. & Park, H. W. Structural basis of human CYP51 inhibition by antifungal azoles. J. Mol.Biol. 397, 1067–1078, https://doi.org/10.1016/j.jmb.2010.01.075 (2010).
Strushkevich, N. et al. Structural Genomics Consortium (SGC) “Crystal structure of human lanosterol 14alpha–demethylase (CYP51) in complex with ketoconazole. J. Mol. Biol. 397, 1067–1078 10.2210/pdb3LD6/pdb entry: 3LD6 (2010).
RCSB PDB (PDB ID: 5FSA), https://www.rcsb.org/structure/5fsa; https://doi.org/10.2210/pdb5FSA/pdb) (accessed Nov 1, 2018).
Naftalovich, S., Yefenof, E. & Eilam, Y. Antitumor effects of ketoconazole and trifluoperazine in murine T–cell lymphomas. Cancer. Chemother. Pharmacol. 28, 384–390, https://doi.org/10.1007/BF00685694 (1991).
Lambert, A., Mitchell, R. & Robertson, W. R. The effect of ketoconazole on adrenal and testicular steroidogenesis in vitro. Biochem. Pharmacol. 35, 3999–4004, https://doi.org/10.1016/0006-2952(86)90018-3 (1986).
Patel, V., Liaw, B. & Oh, W. The role of ketoconazole in current prostate cancer care. Nature Rev. Urol. 15, 643–651, https://doi.org/10.1038/s41585-018-0077-y (2018).
Pont, A. Long–term experience with high dose ketoconazole therapy in patients with stage D2 prostatic carcinoma. J. Urol. 137, 902–904, https://doi.org/10.1016/S0022-5347(17)44290-X (1987).
Trachtenberg, J. Ketoconazole therapy in advanced prostatic cancer. J. Urol. 132, 61–63, https://doi.org/10.1016/S0022-5347(17)49464-X (1989).
Williams, G. Ketoconazole for prostate cancer. Lancet. 324, 696, https://doi.org/10.1016/S0140-6736(84)91253-4 (1984).
Contreras, P. et al. Adrenal rest tumor of the liver causing Cushing’ s syndrome: treatment with ketoconazole preceding an apparent surgical cure. J. Clin. Endocrinol. Metab. 60, 21–28, https://doi.org/10.1210/jcem-60-1-21 (1985).
Committee for Medicinal Products for Human Use (CHMP), Assessment report Ketoconazole HRA, Procedure No. EMEA/H/C/003906/0000, https://www.ema.europa.eu/documents/assessment–report/ketoconazole–hra–epar–public–assessment–report_en.pdf (accessed Nov. 1. 2018).
Rochlitz, C. F., Damon, L. E., Russi, M. B., Geddes, A. & Cadman, E. C. Cytotoxicity of ketoconazole in malignant cell lines. Cancer Chemother. Pharmacol. 21, 319–322, https://doi.org/10.1007/BF00264198 (1988).
Ho, Y. S. et al. Ketoconazole–induced apoptosis through P53–dependent pathway in human colorectal and hepatocellular carcinoma cell lines. Toxicol. Appl. Pharmacol. 153, 39–47, https://doi.org/10.1006/taap.1998.8467 (1998).
Chen, R. J. et al. Ketoconazole induces G0/G1 arrest in human colorectal and hepatocellular carcinoma cell lines. Toxicol. Appl. Pharmacol. 169, 132–141, https://doi.org/10.1006/taap.2000.9062 (2000).
Das, B. C. et al. “Synthesis of novel ketoconazole derivatives as inhibitors of the human Pregnane X Receptor (PXR, NR1I2, also termed SXR, PAR). Bioorg. Med. Chem. Let. 18, 3974–3977, https://doi.org/10.1016/j.bmcl.2008.06.018 (2008).
ManiBhasar, S. & Das, C. Ketoconazole–derivative antagonists of human pregnane x receptor. U.S. Patent 8669260 (2011).
Pirson, P., Leclef, B. & Trouet, A. Activity of ketoconazole derivatives against Leishmania mexicana amazonensis within mouse peritoneal macrophages. Ann. Trop. Med. Parasitol. 84, 133–139, https://doi.org/10.1080/00034983.1990.11812446 (1990).
Rieber, M. S., Anzellotti, A., Sánchez Delgado, R. A. & Rieber, M. Tumor apoptosis induced by ruthenium(II) ketoconazole is enhanced in nonsusceptible carcinoma by monoclonal antibody to EGF receptor. Int. J. Cancer 112, 376–384, https://doi.org/10.1002/ijc.20415 (2004).
Robles–Escajeda., E., Martínez, A., Varela–Ramirez, A., Sánchez–Delgado, R. A. & Aguilera, R. J. Analysis of the cytotoxic effects of ruthenium – ketoconazole and ruthenium – clotrimazole complexes on cancer cells. Cell Biol. Toxic. 29, 431–43, https://doi.org/10.1007/s10565-013-9264-z (2013).
Gagini, T. et al. Metal–azole fungistatic drug complexes as anti–Sporothrix spp. Agents. New J. Chem. 42, 13641–13650, https://doi.org/10.1039/C8NJ01544A (2018).
Coates, H. & Hoye, P. A. T. Tris–aminomethylphosphines. U.S. Patent 3035053 (1962).
Starosta, R., Bażanów, B. & Barszczewski, W. Chalcogenides of the aminomethylphosphines derived from 1–methylpiperazine, 1–ethylpiperazine and morpholine: NMR, DFT and structural studies for determination of electronic and steric properties of the phosphines. Dalton Trans. 39, 7547–7555, https://doi.org/10.1039/c0dt00037j (2010).
Starosta, R. et al. Biological activity and structure dependet properties of cuprous iodide complexes with phenanthrolines and water soluble tris (aminomethyl) phosphanes. J. Inorg. Biochem. 105, 1102–1108, https://doi.org/10.1016/j.jinorgbio.2011.05.007 (2011).
Starosta, R. et al. Copper(I) (pseudo)halide complexes with neocuproine and aminomethylphosphines derived from morpholine and thiomorpholine –in vitro cytotoxic and antimicrobial activity and the interactions with DNA and serum albumins. Chem. Biol. Drug Des. 82, 579–586, https://doi.org/10.1111/cbdd.12187 (2013).
Starosta, R. et al. A novel copper(I) complex, [CuI(2,2′–biquinoline)P(CH2N(CH2CH2)2O)3]–synthesis, characterization and comparative studies on biological activity. Polyhedron 50, 481–489, https://doi.org/10.1016/j.poly.2012.11.033 (2013).
Starosta, R., Komarnicka, U. K. & Puchalska, M. Luminescent copper(I) (pseudo)halide complexes with neocuproine and a novel bulky tris (aminomethyl) phosphine derived from 2–piperazinopyridine. J. Lumin. 143, 137–144, https://doi.org/10.1016/j.jlumin.2013.04.050 (2013).
Starosta, R., Komarnicka, U. K. & Puchalska, M. Solid state luminescence of Cul and CuNCS complexes with phenanthrolines and a new tris (aminomethyl) phosphine derived from N–methyl–2–phenylethanamine. J. Lumin. 145, 430–437, https://doi.org/10.1016/j.jlumin.2013.07.015 (2014).
Płotek, M. et al. New ruthenium(II) coordination compounds possessing bidentate aminomethylphosphane ligands: synthesis, characterization and preliminary biological study in vitro. Dalton Trans. 44, 13969–13978, https://doi.org/10.1039/c5dt01119a (2015).
Płotek, M. et al. Unexpected formation of [Ru(η5–C5H5)(PH{CH2N(CH2CH2)2O}2)(PPh3)2]BF4 – the first “piano–stool” ruthenium complex bearing a secondary aminomethylphosphane ligand. RSC Advances 5, 2952–2955, https://doi.org/10.1039/C4RA13037E (2015).
Płotek, M. et al. Ruthenium(II) piano stool coordination compounds with aminomethylphosphanes: synthesis, characterization and preliminary biological study in vitro. J. Inorg. Biochem. 170, 178–187, https://doi.org/10.1016/j.jinorgbio.2017.02.017 (2017).
Komarnicka, U. K., Kozieł, S., Starosta, R. & Kyzioł, A. Selective Cu(I) complex with phosphine–peptide (SarGly) conjugate contra breast cancer: synthesis, spectroscopic characterization and insight into cytotoxic action. J. Inorg. Biochem. 186, 162–175, https://doi.org/10.1016/j.jinorgbio.2018.06.009 (2018).
Bykowska, A. et al. Synthesis, properties and biological activity of a novel phosphines ligand derived from ciprofloxacin. Polyhedron 60, 23–29, https://doi.org/10.1016/j.poly.2013.04.059 (2013).
Bykowska, A. et al. Phosphine derivatives of ciprofloxacin and norfloxacin, a new class of potential therapeutic agents. New J. Chem. 38, 1062–1071, https://doi.org/10.1039/c3nj01243c (2014).
Bykowska, A., Starosta, R., Jezierska, J. & Jeżowska–Bojczuk, M. Coordination versatility of phosphine derivatives of fluoroquinolones. New CuI and CuII complexes and their interactions with DNA. RSC Advances 5, 80804–80815, https://doi.org/10.1039/c5ra07483e (2015).
Komarnicka, U. K. et al. New copper(I) complexes bearing lomefloxacin motif: spectroscopic properties, in vitro cytotoxicity and interactions with DNA and human serum albumin. J. Inorg. Biochem. 165, 25–35, https://doi.org/10.1016/j.jinorgbio.2016.09.015 (2016).
Komarnicka, U. K., Starosta, R., Kyzioł, A. & Jeżowska–Bojczuk, M. Copper(I) complexes with phosphine derived from sparfloxacin. Part I – structures, spectroscopic properties and cytotoxicity. Dalton Trans. 44, 12688–12699, https://doi.org/10.1039/c5dt01146a (2015).
Komarnicka, U. K. et al. Phosphine derivatives of sparfloxacin– synthesis, structures and in vitro activity. J. Mol. Struct. 1096, 55–63, https://doi.org/10.1016/j.molstruc.2015.04.044 (2015).
Komarnicka, U. K. et al. Copper(I) complexes with phosphine derived from sparfloxacin. Part II: a first insight into the cytotoxic action mode. Dalton Trans. 45, 5052–5063, https://doi.org/10.1039/C5DT04011F) (2016).
Pérez–Torrado, R. & Querol, A. Opportunistic Strains of Saccharomyces cerevisiae: A Potential Risk Sold in Food Products. Front. Microbiol. 6, 1522, https://doi.org/10.3389/fmicb.2015.01522 (2016).
Gaber, R. F., Copple, D. M., Kennedy, B. K., Vidal, M. & Bard, M. The yeast gene ERG6 is required for normal membrane function but is not essential for biosynthesis of the cell–cycle–sparking sterol. Mol. Cell. Biol. 9, 3447–3456, https://doi.org/10.1128/mcb.9.8.3447 (1989).
Guan, X. L. et al. Functional Interactions between Sphingolipids and Sterols in Biological Membranes Regulating Cell Physiology. Mol. Biol. Cell. 20, 2083–2095, https://doi.org/10.1091/mbc.E08-11-1126 (2009).
Anderson, J. B., et al Mode of selection and experimental evolution of antifungal drug resistance in Saccharomyces cerevisiae. Genetics 163, 1287–1298, (PubMed 12702675) (2003).
Kapitzky, L. et al. Cross–species chemogenomic profiling reveals evolutionarily conserved drug mode of action. Mol. Syst. Biol. 6, 451, https://doi.org/10.1038/msb.2010.107 (2010).
Konecna, A., Toth Hervay, N., Valachovic, M. & Gbelska, Y. ERG6 gene deletion modifies Kluyveromyces lactis susceptibility to various growth inhibitors. Yeast 33, 621–632, https://doi.org/10.1002/yea.3212 (2016).
Parker, J. E. et al. Differential Azole Antifungal Efficacies Contrasted Using a Saccharomyces cerevisiae Strain Humanized for Sterol 14α–Demethylase at the Homologous Locus. Antimicrob Agents Chemother. 52, 3597–3603, https://doi.org/10.1128/AAC.00517-08 (2008).
Holmes, A. R. et al. Targeting efflux pumps to overcome antifungal drug resistance. Future Med. Chem. 8, 1485–1501, https://doi.org/10.4155/fmc-2016-0050 (2016).
Petrezselyova, S., Zahradka, J. & Sychrova, H. Saccharomyces cerevisiae BY4741 and W303-1A laboratory strains differ in salt tolerance. Fungal. Biology 114, 144–150, https://doi.org/10.1016/j.funbio.2009.11.002 (2010).
Ralser, M. et al. The Saccharomyces cerevisiae W303-K6001 cross-platform genome sequence: insights into ancestry and physiology of a laboratory mutt Open. Biology 2, 120093, https://doi.org/10.1098/rsob.120093 (2012).
Whaley, S. G. et al. The RTA3 gene, encoding a putative lipid translocase, influences the susceptibility of Candida albicans to fluconazole. Antimicrob. Agents Chemother. 60, 6060–6066, https://doi.org/10.1128/AAC.00732-16 (2016).
Rogers, T. E. & Galgiani, J. N. Activity of fluconazole (UK 49,858) and ketoconazole against Candida albicans in vitro and in vivo. Antimicrob. Agents Chemother. 30, 418–422, https://doi.org/10.1128/aac.30.3.418 (1986).
Suchodolski, J., Feder–Kubis, J. & Krasowska, A. Antifungal activity of ionic liquids based on (−)–menthol: a mechanism study. Microbiol. Res. 197, 56–64, https://doi.org/10.1016/j.micres.2016.12.008 (2017).
Reboutier, D. et al. Combination of different molecular mechanisms leading to fluconazole resistance in a Candida lusitaniae clinical isolate. Diagn. Microbiol. Infect. Dis. 63, 188–193, https://doi.org/10.1016/j.diagmicrobio.2008.10.019 (2009).
Dunkel, N., Blass, J., Rogers, P. D. & Morschhäuser, J. Mutations in the multi–drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole–resistant Candida albicans strains. Mol. Microbiol. 69, 827–840, https://doi.org/10.1111/j.1365-2958.2008.06309.x (2008).
Silva, A. P. et al. Transcriptional profiling of azole–resistant Candida parapsilosis strains. Antimicrob. Agents Chemother. 55, 3546–3556, https://doi.org/10.1128/AAC.01127-10 (2011).
Cheng, S., Clancy, C. J., Nguyen, K. T., Clapp, W. & Nguyen, M. H. A Candida albicans petite mutant strain with uncoupled oxidative phosphorylation overexpresses MDR1 and has diminished susceptibility to fluconazole and voriconazole. Antimicrob. Agents Chemother. 51, 1855–1858, https://doi.org/10.1128/AAC.00182-07 (2007).
Cowen, L. E. et al. Evolution of drug resistance in experimental populations of Candida albicans. J. Bacteriol. 182, 1515–1522, https://doi.org/10.1128/jb.182.6.1515-1522.2000 (2000).
Li, X. et al. Changes in susceptibility to posaconazole in clinical isolates of Candida albicans. J. Antimicrob. Chemother. 53, 74–80, https://doi.org/10.1093/jac/dkh027 (2004).
Looi, C. Y. et al. Increased expression and hotspot mutations of the multidrug efflux transporter, CDR1 in azole–resistant Candida albicans isolates from vaginitis patients. FEMS Microbiol. Lett. 249, 283–289, https://doi.org/10.1016/j.femsle.2005.06.036 (2005).
Sanglard, D. & Coste, A. T. Activity of isavuconazole and other azoles against Candida clinical isolates and yeast model systems with known azole resistance mechanisms. Antimicrob. Agents Chemother. 60, 229–238, https://doi.org/10.1128/AAC.02157-15 (2016).
Prasad, R., Balzi, E., Banerjee, A. & Khandelwal, N. K. All about CDR transporters: Past, Present and Future. Yeast 36, 223–233, https://doi.org/10.1002/yea.3356 (2019).
Perlin, D. S. Echinocandin Resistance in Candida. Clin. Infect. Dis. 61(Suppl 6), S612–7, https://doi.org/10.1093/cid/civ791 (2015).
Fiori, A. & Van Dijck, P. Potent synergistic effect of doxycycline with fluconazole against Candida albicans is mediated by interference with iron homeostasis. Antimicrob. Agents Chemother. 56, 3785–3796, https://doi.org/10.1128/AAC.06017-11 (2012).
Singh, S., Fatima, Z., Ahmad, K. & Hameed, S. Fungicidal action of geraniol against Candida albicans is potentiated by abrogated CaCdr1p drug efflux and fluconazole synergism. PloS one 13, e0203079, https://doi.org/10.1371/journal.pone.0203079 (2018).
Ben–Ami, R. Treatment of Invasive Candidiasis: A Narrative Review. J. Fungi (Basel) 4, E97, https://doi.org/10.3390/jof4030097 (2018).
Maesaki, S., Kohno, S., Kaku, M., Koga, H. & Hara, K. Effects of antifungal agent combinations administered simultaneously and sequentially against Aspergillus fumigatus. Antimicrob. Agents Chemother. 38, 2843–2845, https://doi.org/10.1128/AAC.38.12.2843 (1994).
Nagarkatti, M., Rieder, S., Vakharia, A. & Nagarkatti, D. P. S. Evaluation of apoptosis in immunotoxicity testing. Methods Mol. Biol. 598, 241–257, https://doi.org/10.1007/978-1-60761-401-2_18 (2009).
Buggins, A. G. & Pepper, C. J. The role of BCl–2 family proteis in chronic lumphotic. Leuk. Res. 34, 837–842, https://doi.org/10.1016/j.leukres.2010.03.011 (2010).
Gottlieb, T. & Oren, M. M. P53 and apoptosis. Cancer Biol. 8, 359–368, https://doi.org/10.1006/scbi.1998.0098 (1998).
Abbas, T. & Dutta, A. P21 in cancer: intricate networks and multiple activities. Nat. Rev. Cancer 9, 400–414, https://doi.org/10.1038/nrc2657 (2009).
Quintanilla, R. A., Youngnam, N. J., von Bernhardi, R. & Johnson, G. V. W. Mitochondrial permeability transition pore induces mitochondria injury in Huntington disease. Mol. Neurodegener. 8, 45, https://doi.org/10.1186/1750-1326-8-45 (2013).
Patalano, R., et al Ketoconazole induces inhibition of cell viability and apoptosis in an ACTH–secreting tumour cell line model. Endocrine Abstracts 49, EP927, https://doi.org/10.1530/endoabs.49.EP927 (2017).
Haegler, P., Joerin, L., Krähenbühl, S. & Bouitbir, J. Hepatocellular Toxicity of Imidazole and Triazole. Antimycotic Agents. Toxicol. Sci. 2017, 1–13, https://doi.org/10.1093/toxsci/kfx029 (2017).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J. Comput. Chem. 31, 455–461, https://doi.org/10.1002/jcc.21334 (2010).
Ryu, J. C., Lee, K. J. & Lee, S. H. Synthesis of Ketoconazole Derivatives. Bull. Korean Chem. Soc. 24, 460–466, https://doi.org/10.5012/bkcs.2003.24.4.460 (2003).
Fawcett, J., Hoye, P. A. T., Kemmitt, R. D. W., Law, D. J. & Russell, D. R. Synthesis of bis(phosphinomethyl)amines via bis(hydroxymethyl)phosphonium salts. Isolation of 9,9–bis(hydroxymethyl)–9–phosphoniabicyclo[3.3.1]nonane hydrogensulfate and chloride salts, and the crystal structures of [PPh2(CH2OH)2]+Cl− and [(C6H11)2PCH2]2NCHMePh. J. Chem. Soc., Dalton Trans. 1993, 2563–2568, https://doi.org/10.1039/DT9930002563 (1993).
Bojsen, R., Regenberg, B. & Folkesson, A. Saccharomyces cerevisiae biofilm tolerance towards systemic antifungals depends on growth phase. BMC microbiology 14, 305, https://doi.org/10.1186/s12866-014-0305-4 (2014).
Fonzi, W. & Irwin, M. Isogenic strain construction and gene mapping in Candida albicans. Genetics 134, 717–728, https://doi.org/10.1046/j.1439-0507.1999.00498.x (1993).
Mukherjee, P. & Chandra, J. Mechanism of fluconazole resistance in Candida albicans biofilms: phase–specific role of efflux pumps and membrane sterols. Infect. Immun. 71, 4333–4340, https://doi.org/10.1128/IAI.71.8.4333-4340.2003 (2003).
Sanglard, D. et al. Mechanisms of resistance to azole antifungal agents in Candida albicans isolates from AIDS patients involve specific multidrug transporters. Antimicrob. Agents Chemother. 39, 2378–2386, https://doi.org/10.1128/AAC.39.11.2378 (1995).
Sanglard, D. & Ischer, F. Susceptibilities of Candida albicans multidrug transporter mutants to various antifungal agents and other metabolic inhibitors. Antimicrob. Agents Chemother. 40, 2300–2305, https://doi.org/10.1128/AAC.40.10.2300 (1996).
Franz, R., Ruhnke, M. & Morschhäuser, J. Molecular aspects of fluconazole resistance development in Candida albicans. Mycoses 42, 453–458, https://doi.org/10.1046/j.1439-0507.1999.00498.x (1999).
Wakieć, R. et al. Voriconazole and multidrug resistance in Candida albicans. Mycoses 50, 109–115, https://doi.org/10.1111/j.1439-0507.2006.01327.x (2007).
Reference method for broth dilution antifungal susceptibility testing of yeast. Approved Standard, 3rd ed. M27–A3 28. Clinical and Laboratory Standards Institute, ISBN 1–56238–666–2 (Wayne, PA. 2008).
Marycz, K. et al. Low–frequency, low magnitude vibrations (LFLM) enhances chondrogenci differentiation potential of human adipose derived mesenchymal stromal stem cells (hASCs). PeerJ. 4, e1637, https://doi.org/10.7717/peerj.1637 (2016).
Chomczynski, P. & Sacchi, N. Single–step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction. Anal. Biochem. 162, 156–159, https://doi.org/10.1006/abio.1987.9999 (1987).
Gaussian 16, Revision B.01, Frisch, M. J. et al. Gaussian, Inc., Wallingford CT, 2016.
Chai, J. –D. & Head–Gordon, M. Long–range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 10, 6615–6620, https://doi.org/10.1039/B810189B (2008).
Morris, G. M. et al. Autodock4 and AutoDockTools4: automated docking with selective receptor flexiblity. J. Comput. Chem. 16, 2785–2791, https://doi.org/10.1002/jcc.21256 (2009).
Pettersen, E. F. et al. UCSF Chimera – A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612, https://doi.org/10.1002/jcc.20084 (2004).
Acknowledgements
The authors gratefully acknowledge financial support from the National Science Centre, Poland, NCN Grant no. 2016/23/B/NZ1/01928 (A.K., J.S.) and Grant no. UMO-2016/22/M/ST4/00226 (A.J., J.J.P.) as well as from FCT, I.P., Portugal through the funding to the research unit (UID/Multi/00612/2019), research grants PTDC/BBB-BQB/6071/2014 (R.F.M.A.) and F.C.S. PhD fellowship SFRH/BD/108031/2015. The DFT calculations have been carried out in Wroclaw Centre for Networking and Supercomputing (http://www.wcss.wroc.pl), Grant No. 140.
Author information
Authors and Affiliations
Contributions
R.S., R.A. and A.K. designed the scientific rationale; R.A. designed S. cerevisiae related experiments; F.S. performed S. cerevisiae related experiments, analyzed data and generated figures; A.K. designed C. albicans related experiments; J.S. performed C. albicans related experiments, analyzed data and generated figures; A.J. and J.P. performed molecular docking experiments, analyzed data and generated figures; R.S. administrated the project, performed syntheses and spectroscopic characterization of the compounds, analysed the data and generated figures; K.M. designed hASCs related experiments; M.A. performed hASCs related experiments, analyzed data and generated figures; R.S., R.A., K.M., A.K., J.S., J.P. and A.J. wrote the manuscript. The manuscript was reviewed by all Authors prior to submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
de Almeida, R.F.M., Santos, F.C., Marycz, K. et al. New diphenylphosphane derivatives of ketoconazole are promising antifungal agents. Sci Rep 9, 16214 (2019). https://doi.org/10.1038/s41598-019-52525-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52525-7
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
