
Abstract
Acyl carrier proteins (ACPs) are essential to the production of fatty acids. In some species of marine bacteria, ACPs are arranged into tandem repeats joined by peptide linkers, an arrangement that results in high fatty acid yields. By contrast, Escherichia coli, a relatively low producer of fatty acids, uses a single-domain ACP. In this work, we have engineered the native E. coli ACP into tandem di- and tri-domain constructs joined by a naturally occurring peptide linker from the PUFA synthase of Photobacterium profundum. The size of these tandem fused ACPs was determined by size exclusion chromatography to be higher (21 kDa, 36 kDa and 141 kDa) than expected based on the amino acid sequence (12 kDa, 24 kDa and 37 kDa, respectively) suggesting the formation of a flexible extended conformation. Structural studies using small-angle X-ray scattering (SAXS), confirmed this conformational flexibility. The thermal stability for the di- and tri-domain constructs was similar to that of the unfused ACP, indicating a lack of interaction between domains. Lastly, E. coli cultures harboring tandem ACPs produced up to 1.6 times more fatty acids than wild-type ACP, demonstrating the viability of ACP fusion as a method to enhance fatty acid yield in bacteria.
Similar content being viewed by others

Introduction
Acyl carrier proteins (ACPs) are central to the production of fatty acids in all organisms1. They play a key role in the shuttling of substrates and intermediates during the successive rounds of malonate condensation and the subsequent modifications by ketoreductase, dehydratase and enoyl reductase enzymes that result in a full-length fatty acid2. At all times during the biosynthesis of a fatty acid, each starting material and intermediate is covalently linked to an ACP which carries the substrate from one active site to the next. ACPs are also important and serve homologous functions in other metabolic pathways such as the biosynthesis of polyketides by polyketide synthases (PKSs) and the biosynthesis of non-ribosomal peptides by non-ribosomal peptide synthases (NRPSs), all enzymes that employ a molecular logic similar to that of fatty acid biosynthesis3.
While ACPs typically function as independent proteins in most prokaryotes, there are some species that contain multiple covalently linked ACP domains in tandem4,5. The presence of tandem ACP domains has been reported in synthase systems involved in the production of several important natural products, for example, the polyunsaturated fatty acids (PUFA) as well as some polyketides6,7,8. Many PUFA synthases contain between 2 and 9 tandem ACP domains8,9,10. For instance, the PUFA synthase of Photobacterium profundum contains 5 covalently linked ACP domains4,5 (Fig. 1a). It has also been shown that the PKS that makes mupirocin in Pseudomonas fluorescens also contains tandem duplications of ACP, although most known PKSs contain a single ACP in each elongation module11,12,13.

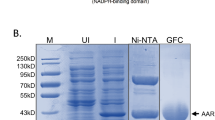
Construction of di and tri-domain tandem ACPs. To make polyunsaturated fatty acids some species of bacteria employ modular multi-enzymes known as PUFA synthases. (a) Although the PUFA synthase from P. profundum is shown here, the tandem arrangement of ACPs is a conserved feature of PUFA synthases. (b) To mimic the tandem architecture observed of natural systems we linked the native E. coli ACP using the natural peptide linker from the P. profundum PUFA synthase. (c) The ACP1, ACP2 and ACP3 were produced in E. coli, purified to homogeneity and analyzed by SDS -PAGE (each against a set of size markers). (d) The size of each construct was ascertained by size exclusion chromatography.
The advantages of this tandem ACP arrangement in synthase systems have been reported for both the biosynthesis of fatty acids and polyketide antibiotics. In PUFA synthases, the yields of PUFA increase proportionally with the number of functional ACPs. A previous study showed that ACP domains are functionally equivalent regardless of their physical location and that production enhancement results from the proximity of ACPs in the arrangement14. Other studies have confirmed this result by showing an increase in PUFA yields with the insertion of either inactivated or active ACP domains from the pfaA multidomain proteins. The pfaA subunits which harbor these ACP domains and their linkers have been shown to be exchangeable between organisms15. Previous work by our group elucidated the solution structure of tandem ACPs from P. profundum, demonstrating that they have an elongated beads-on-a-string arrangement with flexible linkers with little or no interaction between domains9.
Like most ACPs, the ACP from Escherichia coli is a stand-alone monomer, which has been demonstrated to be toxic to E. coli cells when overexpressed16. Thus, strategies to increase fatty acid production by overexpression of ACP may well end up decreasing the fatty acid yields due to cellular toxicity. To circumvent some of the toxicity issues associated with the overexpression of ACP, we engineered artificially linked tandem ACP proteins as a means to enhance fatty acid yields in E. coli. We used a naturally occurring peptide linker from the tandem ACP multidomain PUFA synthase from P. profundum to link the E. coli ACP into di-domain (ACP2) and tri-domain (ACP3) fragments (Fig. 1b). Our results show the feasibility of enhancing fatty acid production by genetically fusing ACP domains together into multi-domain proteins.
Results
Protein purification and yield
First, the ACP2 and ACP3 fragments were made by overlap PCR, using primers that were complementary to both the E. coli gene encoding ACP and to the P. profundum gene encoding the peptide that links the ACPs from the PUFA synthase (Fig. S1). The resulting DNA fragments, ACP2 and ACP3, were cloned into a pET100TOPO vector and expressed in E. coli BL21(DE3) cells. We purified the proteins by a combination of nickel-affinity purification and size exclusion chromatography, resulting in highly pure elution fractions with yields of 15, 24, and 18 mg of pure protein per liter of culture, for ACP1, ACP2, and ACP3, respectively.
Size exclusion chromatography of fused ACPs
The relative size of the mono- di- and tri-domain proteins was determined using size exclusion chromatography. The retention time of the ACP protein was compared with the retention times of protein standards of known molecular weight. Chromatograms showed retention times corresponding to molecular weights of 21.1 kDa for ACP1, 36.7 kDa for ACP2, and 141.4 kDa for ACP3 when using a high salt buffer (Data not shown). The same analysis was conducted using a lower salt buffer to better reflect the low-salt conditions required for analyzing sample both by SAXS and by circular dichroism (CD). The size exclusion chromatogram resulted in retention times consistent with molecular weights of 30.0 kDa for ACP1, 95.3 kDa for ACP2, and 261.5 kDa for ACP3 (Fig. 1d). Under all conditions tested, the estimated molecular weights were found to be consistently higher than what would be expected based on the amino acid sequence of these proteins (Table 1). This observed increase in the apparent molecular weight was not the result of intermolecular disulfide bonds since neither ACP nor the amino acid linker contain cysteine residues within their amino acid sequence.
Solution structures of fused ACPs by SAXS
To show that the engineered ACP constructs adopted a multi-domain arrangement, we gathered structural information by small-angle X-Ray scattering (SAXS). Figure 2a shows the scattering profiles for the three ACPs constructs. Indirect Fourier transformation of the scattering data yields a pair distribution function P(r), which is a real space representation of the scattering data and provides an approximation of the dimension and shape of a protein in solution. In this case, the ACP1 protein has a P(r) function that is relatively symmetric and bell-shaped, whereas the more flexible ACP2 and ACP3 have an extended tail with multiple shoulders and oscillations17,18. The P(r) functions also revealed gyration radii of 23.5 Å for ACP1, 36.6 Å for ACP2, and 48.0 Å for ACP3, all consistent with a flexible multi-domain arrangement (Fig. 2b). The maximum diameter of the proteins (Dmax) also increased as ACP domains were added, with Dmax values of 100 Å for ACP1, 155 Å for ACP2, and 170 Å for ACP3. In terms of the protein shapes, the ACP1 P(r) function suggest a somewhat elongated structure, while those of ACP2 and ACP3 show new P(r) contours with two distinct peaks, a hallmark of flexible or dumbbell-like particles, consistent beads-on-a-string arrangements of domains (Fig. 2d–f).

Solution structures of tandem ACPs by SAXS. The structure of ACP as a multidomain flexible tandem arrangement was confirmed by small-angle X-ray scattering (a) SAXS/WAXS curves were generated from the X-ray scattering data, and (b) a corresponding P(r) function was generated for each construct, from which a radius of gyration was obtained. (c) Further confirmation of the flexible arrangement in multi ACPs came from the thermal unfolding monitored at 222 nm between 20–95 °C. There was no significant difference in thermal unfolding between the constructs (40.9 °C ± 1.8 °C) indicating that ACP domains are not interacting. The SAXS data was used to generate three-dimensional structural models using DAMMIF within the ATSAS software package. The structures of (d) ACP1, (e) ACP2, and (f) ACP3 together with their corresponding homology model made using iTasser server. ACP domains are highlighted in blue, the amino acid linkers in green, and the His-tag in yellow.
Thermal stability assays of fused ACPs
As a confirmation of the multi-domain nature of the artificially generated fragments, we measured the thermal denaturation using circular dichroism (CD) spectroscopy for all ACP constructs in this study. Results show that there is little difference in thermal stability between the three ACP constructs, suggesting that ACP linkage does not significantly impact stability, an observation consistent with domains that are structurally independent with little or no contact between ACP domains. The melting temperature (Tm) values were 42.3, 38.9, and 41.6 °C for ACP1, ACP2, and ACP3, respectively in an unfolding that was irreversible (Fig. 2c).
Effect of fused ACPs on fatty acid production
Cultures of E. coli expressing the different ACP constructs were grown at three different temperatures and their fatty acid content was analyzed by gas chromatography coupled to mass spectrometry (GC/MS). The growth rate of E. coli was unaffected by ACP expression at all temperatures, indicating that any effect on fatty acid production is not due to general metabolic impairment (Fig. 3). Higher fatty acid yields were obtained at the lowest temperature of 15 °C, followed by the cultures grown in 22 °C and 37 °C, respectively (Fig. 4). Cultures expressing the ACP3 at 15 °C showed higher fatty acid yield than cultures expressing ACP2 or ACP1, where the total FA yield was 42.3 mg/L (±3.1) for ACP1, 55.8 mg/L (±8.9) for ACP2 and 70.2 mg/L (±9.1) for ACP3. This corresponds to a significant 1.6-fold enhancement for ACP3 over control ACP1. This enhancement was only observed at the lowest temperature, but not at 22 °C or 37 °C, a finding not totally unexpected since fatty acid production in E. coli is known to be suppressed at higher temperatures16.

Effect of ACP expression on E. coli growth. Liquid cultures of E. coli. expressing either ACP1, ACP2 or ACP3 were grown at three different temperatures: (a) 37 °C, (b) 22 °C and (c) 15 °C. As expected, higher temperatures correlated with faster rates but the expression of ACP had no effect on growth rate. All experiments were carried out in triplicates and the error bars represent the standard deviation.

Total fatty acid yields of E. coli cultures expressing tandem ACPs. Fatty acids were extracted from the cultures and analyzed by GC/MS. (a) Cultures grown at 37 °C and 22 °C showed no significant effect of ACP expression on the production of fatty acids. At 15 °C, however, there was a significant effect of ACP on fatty acid production. (b) This increase FA production at 15 °C is primarily due to the production of saturated fatty acids 16:0 and 18:0. Cultures harboring ACP produced 42.3 ± 4.5 mg/L; for ACP2 the yield increased to 55.8 ± 12.6 mg/L, and the highest yield was observed for ACP3 at 70.2 ± 12.9 °C. Data are shown as means ± SEM and p -values were calculated using the Student’s t-test, p < 0.05. All samples were run as biological triplicates.
Discussion and Conclusion
The central role of ACP in the biosynthesis of fatty acids has invited explorations into their possible use to enhance fatty acid production in cultured bacteria. However, attempts to overexpress recombinant native ACP in E. coli have resulted in impaired cell growth, primarily by the inhibition of glycerol-3-phosphate acyl transferase16. A natural mechanism to control or enhance ACP availability is employed by PUFA-producing bacteria that enhance their levels of fatty acid production not by overexpressing ACP, but rather by harboring multiple copies of ACP along the same polypeptide in tandem arrangements14,15. In this work, we explored the technical feasibility of forcing the native E. coli ACP to form di-domain and tri-domain polypeptides: ACP-ACP (ACP2) and ACP-ACP-ACP (ACP3). The ACPs were fused together using a natural linker peptide sequence corresponding the spacer region between tandem ACP domains in the PfaA protein from Photobacterium profundum, a PUFA-producing organism. The growth curves of E. coli expressing ACP1, ACP2 and ACP3, reveal no toxicity for any of the ACP constructs, perhaps because the cultures were not induced with IPTG and thus, the ACPs were not overexpressed for this experiment.
Previous work by other groups had shown that the presence of multiple ACP domains in a single polypeptide enhances fatty acid production. In Shewanella japonica and other organisms, the production of eicosapentaenoic acid is proportional to the number of active ACP domains in the tandem arrangement. Conversely, the elimination of domains from the tandem arrangement results in a decrease in fatty acid production14,15. In this work, with the expression of the fused tandem ACPs, we observed a modest but significant gain in the production of fatty acids: 42 mg/L in cells expressing ACP1 to 70 mg/L in cells expressing ACP3, with no change in growth rate at 15 °C. This enhancement in fatty acid production was statistically insignificant at 22 °C and 37 °C. This temperature dependence was not totally unexpected since bacteria tend to decrease fatty acid production at higher temperatures. Thus it was expected that any effect on fatty acid production by ACP would be more pronounced at the lower temperature19.
Solution structures of the multi-ACP constructs show that they form an extended and highly flexible structure with little or no contact between domains17,20. A previous solution structure of the naturally occurring tandem ACPs from P. profundum revealed an elongated arrangement of quasi-independent domains, like beads on a string9. Similarly, the artificially fused tandem ACPs in this study also show a similar arrangement of independent domains. The thermal denaturation measured by circular dichroism shows that ACP1, ACP2 and ACP3 all have similar melting temperatures (Tm ACP1 = 42.3 °C vs. Tm ACP2 = 38.9 °C vs. Tm ACP3 = 41.6 °C). The fact that the presence of additional ACP domains does not further stabilize structures is consistent with a beads-on-a-string arrangement with little or no contact with neighboring or proximate ACP domains. Similarly, the size exclusion chromatography profiles for ACP1, ACP2 and ACP3 all suggest a structure that is larger than would be expected by the sequence (141 kDa vs 37 kDa by sequence for ACP3), which is consistent with an extended or a highly flexible arrangement of ACP domains9. The formation of higher-order oligomers by ACP1, ACP2 and ACP3 can be ruled out based on the SAXS-derived structures which show essentially monomeric extended and flexible structures (Fig. 2d–f).
In conclusion, we have shown that the incorporation of ACP domains into multi-ACP constructs can have a positive effect on fatty acid yield, while circumventing possible toxicity issues associated with the overexpression of ACP in E. coli. The multi-domain nature of the engineered constructs was confirmed using multiple biophysical methods including SAXS, circular dichroism and size exclusion chromatography. With the current interest in the production of microbially derived oils and materials, this strategy could be widely applicable to many microbial hosts used for the production of fatty acids and biodiesel.
Methods
Primer design of acpP
DNA sequence for E. coli DH10B acpP (Ref. sequence NC_000913.3:11516151151851) was obtained from NCBI. The linker region corresponded to amino acids 1511–1538 of the pfaA gene from P. profundum (Accession No. CAG19871.1). The primers for the amplification of acpP contained overhangs of additional sequence that are complementary to either the 5′ or 3′ terminus of the linker sequence (Table S1). The reverse primers for the amplification of acpP contained overhangs complementary the 5′ terminus of the linker sequence, whereas forward primers contained overhangs complementary to the 3′ terminus of the linker sequence. The same principle applied for the amplification of the pfaA linker. All primers were purchased from the RCMI Core Lab at UPR Medical Sciences Campus after verification of secondary structure and primer dimer formation using the online Thermo Scientific Multiple Primer Analyzer tool (www.thermoscientificbio.com/webtools/multipleprimer).
Construction of gene fusions and cloning
A general scheme for the generation of fused tandem acpPs by overlap PCR is presented in Supplementary Fig. 1. All PCRs were done using PfuUltra II Fusion HS DNA Polymerase (Agilent). All PCR reactions were preceded by a denaturation step at 95 °C for two minutes and finished with a final extension step at 68 °C for 3 minutes. For the amplification of the DNA corresponding to the linker region of pfaA, 100 ng of plasmid DNA containing the ACP domains from P. profundum were mixed with primers 9 and 10. DNA was denatured at 95 °C for 1 minute, followed by annealing at 45.8 °C for 1 minute and finished with an extension 68 °C for 1 minute. For the amplification of acpP, approximately 2 ng of genomic DNA from E. coli DH10B cells were mixed with primers 1 and 4, using the same PCR parameters as for the tandem constructs. Three basic building blocks were constructed to either extend or terminate each tandem gene: an initiation unit (Table S2), an elongation unit (Table S4), and a termination unit (Table S3). The sequential PCR reactions were performed as outlined in Table S5. PCRs were run for 30 cycles without primers, and then 15 more cycles with primers. Individual PCR reaction products were separated by electrophoresis on an agarose gel (2%) and purified using the QIAQuick Gel Extraction Kit (Qiagen).
Protein expression and purification
E. coli BL21(DE3) - Codon Plus RIL cells (Invitrogen) were transformed with each plasmid and grown in liquid Luria-Bertani medium supplemented with 0.4% glycerol, 1% glucose, which contained kanamycin (100 mg/L) and chloramphenicol (25 mg/ L). Cultures were grown at 37 °C, 250 rpm until OD600 = 0.2–0.3, at which point the temperature was lowered to 22 °C. Protein expression was induced with 1 mM IPTG once the OD600 reached 0.5–0.6. After 4 hours, the cells were harvested by centrifugation at 4 °C and 11000 × g on a Sorvall Lynx 4000 Centrifuge using a Fiberlite™ F14-14 × 50cy Fixed-Angle Rotor (Thermo). Samples were stored at −20 °C overnight. Pellets were resuspended in lysis buffer (20 mM Tris, 500 mM NaCl, 1 mM DTT, 20% glycerol, pH 7.8) in the presence of lysozyme (10 mg/ml), DNAse (1 mg/ml), 2X protease inhibitor cocktail (Pierce) and sonicated. The lysates were collected by centrifugation (11000 × g, 4 °C, 30 min). The soluble lysates were poured through a column filled with Ni-Sepharose (Sigma) that had been equilibrated with the corresponding buffer +5 mM imidazole, and washed twice with the same buffer +10 mM imidazole. His-tagged proteins were eluted in the in the corresponding buffer containing 200 mM imidazole. Afterwards, fractions were infused into a size exclusion chromatography Superdex 200 Increase 10/300 GL column (GE Healthcare) equilibrated in the NiNTA lysis buffer minus glycerol, and eluted at a flowrate of 0.8 mL/min. Purities of elution fractions were analyzed by SDS-PAGE. Protein yields were calculated using Nanodrop A280 quantification and dividing total milligrams of protein per volume of culture.
Bacterial growth curves
After inocculation for protein production, 150 μL of the inoculated media were transferred to a Falcon 96 flat bottom plate and growth curves at 37 and 22 °C were generated using a Synergy H1 plate reader by monitoring absorbance at 600 nm as a function of time. Equipment configuration was as follows: normal read speed with a 100 msec delay, continuous orbital shake at a frequency of 237 cpm. The total runtime was 27 hours with a 10 minute interval between reads. Due to equipment limitations, the samples analyzed at 15 °C were grown inside a shaker incubator at the same agitation speed and the absorbance was then measured inside the plate reader. Because of the delayed growth, the total runtime was 96 hours.
Determination of molecular weights by size exclusion chromatography
The purified proteins were exchanged into a low-salt system (20 mM NaH2PO4, 50 mM NaCl, pH 7.8) and run as in the purification step. Each run with the proteins was preceded by a run with a mixture of standard proteins (GE Healthcare) [aprotinin (6,500 Da), ribonuclease (13,700 Da), ovalbumin (44,000 Da), conalbumin (75,000 Da), aldolase (158,000 Da), ferritin (440,000 Da)] to generate a Kav vs. logMW curve, where Kav = (Ve − Vo)/(Vc − Vo). Proteins were eluted at a flowrate of 0.8 mL/min, and the elution volumes were determined using the Unicorn software integration function. The resulting standard curve was used to estimate the molecular weight for our proteins.
Thermal stability determination by circular dichroism
Proteins were exchanged into 20 mM NaH2PO4, 50 mM NaCl, pH 7.8 using size exclusion chromatography. All samples came from new aliquots that had not been previously thawed and refrozen. Samples were diluted to 0.1 mg/mL and placed in a Jasco 1 mm quartz cuvette. Thermal denaturation was measured by monitoring the CD signal at 218 nm as a function of temperature in the range of 20–95 °C (1 °C/min) in a Jasco-1500 spectropolarimeter using the Interval Temperature Scan Measurement program. Precipitation was observed at the end of the run in every sample. Parameters for Tm scan were: CD scale 200mdeg/1.0dOD, D.I.T 1 s, bandwidth 1 nm. CD units (mdeg) were converted to θMRE units using the equation θMRE = (deg*MRW)/(10*c*b), where MRW is the mean residue weight (Da), c is the enzyme concentration (g/mL), and b is the cell pathlength (cm). Denaturation analysis was performed using the built-in Thermal Denaturation Analysis Program.
Solution and in silico structures using small angle X-Ray scattering and I-tasser
Proteins were exchanged into 20 mM NaH2PO4, 50 mM NaCl, pH 7.8 and concentrated between 1–3 mg/mL. X-ray scattering measurements were conducted at the life sciences X-ray scattering (LiX) beamline at the National Synchrotron Light Source II at Brookhaven National Laboratory, Brookhaven, NY. About 10 μl of the enzymes and buffer solutions were continuously flowed through a capillary and exposed to the X-ray beam for 5 s. The measurements were carried out in triplicate. Data processing was performed using an automated Python-based package developed at LiX. Overall, the two-dimensional scattering patterns from protein solutions were first recorded on 3 detectors simultaneously and then converted into one-dimensional scattering profiles. The SAXS/WAXS data were then merged, averaged, and buffer subtracted to obtain relative scattering intensity (I) as a function of the momentum transfer vector, q (q = (4πsinθ)/λ), where λ is the beam wavelength, and θ is the scattering angle. The resulting combined and merged scattering pattern was analyzed using PRIMUS, GNOM, and DAMMIF of the ATSAS software package21,22,23. The Rg value derived from the Guinier analysis corresponded well to the Rg obtained through the indirect transform algorithm in GNOM. Theoretical three-dimensional models of the ACPs were generated using the I-TASSER algorithm, which builds protein models from primary sequences by combining remote homologs with ab initio calculations24. Pymol was used for graphical analysis and figure generation.
Extraction and modification of fatty acids for quantification
Cultures were grown in triplicate in LB media using the same procedure as before at 37, 22, and 15 °C, in the absence and presence of 1 mM IPTG. After a total of 27 h, cells were harvested, frozen at −80 °C, and lyophilized. Fatty acids were extracted using the Bligh-Dyer method25. Briefly, dried pellets were dissolved in NaCl, chloroform, MeOH, and vortexed. The organic phase was isolated after centrifugation and concentrated under N2. Finally, the concentrated fatty acids were resuspended in a mixture of chloroform:MeOH (2:1). To convert fatty acids to their respective methyl esters, the extracted fatty acids in the chloroform:MeOH mixture were reacted with methanolic HCl and refluxed for 2 h under constant stirring and heating. Methyl heneiocosanoate was added as an internal standard prior to the addition of methanolic HCl. Fatty acids were isolated by extraction with hexane, dried under N2, and resuspended in 200 μL of hexane.
Quantification of fatty acids and statistical analysis
Fatty acid profiling was performed using a GC/MS-QP2010 (Shimadzu) equipped with a fused-silica FAMEWAX capillary column (30 m × 0.32 mm i.d. × 0.25 μm film thickness) (Restek) and an auto-injector (Shimadzu, AOC-20i). Samples were injected in the split mode (spit ratio = 15:1). The oven temperature was set to 130 °C for 5 min, followed by a temperature ramp from 130 to 250 °C at 4 °C/min, and finally, held at 250 °C for 5 min, using helium as the carrier gas. Mass spectra data were obtained after electron impact ionization (EI, 70 eV, ion source temperature 200 °C) in full scan mode between 50 and 600 amu. The temperatures of the injection port and the detector were set at 250 °C. The data was processed using the GC/MS LabSolutions Postrun Analysis software (Shimadzu) for metabolite identification using the NIST 2014 spectral mass library (National Institute of Standards and Technology). Fatty acid yields were calculated as follows: First, the millimoles of each fatty acid were calculated using the formula mmol FA = (AFA/AIS)*CIS*Vchl, where (AFA/AIS) is the ratio of the areas of each fatty acid and the internal standard calculated from the gas chromatogram, CIS is the known concentration of the internal standard (M), and Vchl is the volume of chloroform:MeOH used to resuspend the concentrated fatty acids (mL). From here, the milligrams of each fatty acid per liter of culture were calculated as mg FA/L = (mmol FA*gcell*MW)/(gext*Vtot), where gcell is the total dry cell mass (g), gext is the dried cell mass used for the extraction (g), Vtot is the total culture volume (L), and MW is the molecular weight for each fatty acid (g/mol). Individual fatty acid yields were pooled in order to determine the amount of total fatty acids per liter of culture. Statistical analysis was performed using GraphPad Prism 6. Data are shown as means ± SEM and p -values were calculated using the Student’s t-test, p < 0.05.
References
Cronan, J. E. & Thomas, J. Bacterial fatty acid synthesis and its relationships with polyketide synthetic pathways. Methods Enzymol. 459, 395–433 (2009).
Byers, D. M. & Gong, H. Acyl carrier protein: structure-function relationships in a conserved multifunctional protein family. Biochem. Cell Biol. 85, 649–662 (2007).
Staunton, J. & Weissman, K. J. Polyketide biosynthesis: a millennium review. Nat. Prod. Rep. 18, 380–416 (2001).
Allen, E. E. & Bartlett, D. H. Structure and regulation of the omega-3 polyunsaturated fatty acid synthase genes from the deep-sea bacterium Photobacterium profundum strain SS9. Microbiology 148, 1903–1913 (2002).
Metz, J. G. et al. Production of polyunsaturated fatty acids by polyketide synthases in both prokaryotes and eukaryotes. Science 293, 290–293 (2001).
Santín, O. & Moncalián, G. Loading of malonyl-CoA onto tandem acyl carrier protein domains of polyunsaturated fatty acid synthases. J. Biol. Chem. 293, 12491–12501 (2018).
Gu, L. et al. Tandem acyl carrier proteins in the curacin biosynthetic pathway promote consecutive multienzyme reactions with a synergistic effect. Angew. Chem. Int. Ed. 50(12), 2795–8 (2011).
Okuyama, H., Orikasa, Y., Nishida, T., Watanabe, K. & Morita, N. Bacterial genes responsible for the biosynthesis of eicosapentaenoic and docosahexaenoic acids and their heterologous expression. Appl. Environ. Microbiol. 73, 665–670 (2007).
Trujillo, U. et al. Solution structure of the tandem acyl carrier protein domains from a polyunsaturated fatty acid synthase reveals beads-on-a-string configuration. PLoS One 8, e57859 (2013).
Metz, J. G. et al. Biochemical characterization of polyunsaturated fatty acid synthesis in Schizochytrium: release of the products as free fatty acids. Plant Physiol. Biochem. PPB 47, 472–478 (2009).
El-Sayed, A. K. et al. Characterization of the mupirocin biosynthesis gene cluster from Pseudomonas fluorescens NCIMB 10586. Chem. Biol. 10, 419–430 (2003).
Fujii, I., Watanabe, A., Sankawa, U. & Ebizuka, Y. Identification of Claisen cyclase domain in fungal polyketide synthase WA, a naphthopyrone synthase of Aspergillus nidulans. Chem. Biol. 8, 189–197 (2001).
Rahman, A. S., Hothersall, J., Crosby, J., Simpson, T. J. & Thomas, C. M. Tandemly duplicated acyl carrier proteins, which increase polyketide antibiotic production, can apparently function either in parallel or in series. J. Biol. Chem. 280, 6399–6408 (2005).
Jiang, H. et al. The role of tandem acyl carrier protein domains in polyunsaturated fatty acid biosynthesis. J. Am. Chem. Soc. 130, 6336–6337 (2008).
Hayashi, S., Satoh, Y., Ujihara, T., Takata, Y. & Dairi, T. Enhanced production of polyunsaturated fatty acids by enzyme engineering of tandem acyl carrier proteins. Sci. Rep. 6, (2016).
Keating, D. H., Carey, M. R. & Cronan, J. E. The unmodified (apo) form of Escherichia coli acyl carrier protein is a potent inhibitor of cell growth. J. Biol. Chem. 270, 22229–22235 (1995).
Kikhney, A. G. & Svergun, D. I. A practical guide to small angle X-ray scattering (SAXS) of flexible and intrinsically disordered proteins. FEBS Lett. 589(19A), 2570–2577 (2015).
Blanchet, C. E. & Svergun, D. I. Small-angle X-ray scattering on biological macromolecules and nanocomposites in solution. Annu. Rev. Phys. Chem. 64, 37–54 (2013).
Marr, A. G. & Ingraham, J. L. Effect of temperature on the composition of fatty acids in Escherichia coli. J. Bacteriol. 84, 1260–1267 (1962).
Tria, G., Mertens, H. D. T., Kachala, M. & Svergun, D. I. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2, 207–217 (2015).
Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J. & Svergun, D. I. PRIMUS - a Windows-PC based system for small-angle scattering data analysis. J Appl Cryst. 36, 1277–1282 (2003).
Franke, D. & Svergun, D. I. DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Cryst. 42, 342–346 (2009).
Svergun, D. I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 (1992).
Yang, J. et al. The I-TASSER Suite: Protein structure and function prediction. Nature Methods 12, 7–8 (2015).
Bligh, E. G. & Dyer, W. J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 (1959).
Acknowledgements
This work was supported by National Science Foundation (NSF) grant CHE0953254 to ABO and National Institutes of Health (NIH) grant R25GM061838 (MBRS-RISE Program) to CRL and MOR. Some of the shared instrumentation was purchased with NIH Grant G12RR03051 (RCMI Program). The Metabolomics Facility is funded through the PR-INBRE program (NIGMS P20 GM103475). The authors thank the UPR Material Characterization Center for their support through shared instrumentation. This research used resources of LiX beamline at the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No.DE-AC02- 98CH10886. The LiX beamline is part of the Life Science and Biomedical Technology Research Resource (LSBR) funded by NIH grants GM111244 and BER KP1605010.
Author information
Authors and Affiliations
Contributions
C.R.L. and A.B.O. designed the study. A.B.O. obtained the funding necessary for the study. C.R.L., A.G.G. and R.B.P. performed the experiments. C.R.L., R.B.P. and V.S. obtained and analyzed the X_ray scattering data. N.E.C. and M.O.R. performed chromatographic and mass spectrometric analyses. C.R.L. and R.B.P. performed data analyses and interpretation of the data. C.R.L., R.B.P., M.O.R. and A.B.O. prepared the figures. All authors contributed in the preparation of tables and figures, and in drafting the manuscript. Some of this work was published as part of the doctoral thesis of C.R.L.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rullán-Lind, C., Ortiz-Rosario, M., García-González, A. et al. Artificial covalent linkage of bacterial acyl carrier proteins for fatty acid production. Sci Rep 9, 16011 (2019). https://doi.org/10.1038/s41598-019-52344-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52344-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
