
Abstract
Quantitative trait loci (QTLs) mapped in different genetic populations are of great significance for marker-assisted breeding. In this study, an F2:3 population were developed from the crossing of two maize inbred lines SG-5 and SG-7 and applied to QTL mapping for seven yield-related traits. The seven traits included 100-kernel weight, ear length, ear diameter, cob diameter, kernel row number, ear weight, and grain weight per plant. Based on an ultra-high density linkage map, a total of thirty-three QTLs were detected for the seven studied traits with composite interval mapping (CIM) method, and fifty-four QTLs were indentified with genome-wide composite interval mapping (GCIM) methods. For these QTLs, Fourteen were both detected by CIM and GCIM methods. Besides, eight of the thirty QTLs detected by CIM were identical to those previously mapped using a F2 population (generating from the same cross as the mapping population in this study), and fifteen were identical to the reported QTLs in other recent studies. For the fifty-four QTLs detected by GCIM, five of them were consistent with the QTLs mapped in the F2 population of SG-5 × SG-7, and twenty one had been reported in other recent studies. The stable QTLs associated with grain weight were located on maize chromosomes 2, 5, 7, and 9. In addition, differentially expressed genes (DEGs) between SG-5 and SG-7 were obtained from the transcriptomic profiling of grain at different developmental stages and overlaid onto the stable QTLs intervals to predict candidate genes for grain weight in maize. In the physical intervals of confirmed QTLs qKW-7, qEW-9, qEW-10, qGWP-6, qGWP-8, qGWP-10, qGWP-11 and qGWP-12, there were 213 DEGs in total. Finally, eight genes were predicted as candidate genes for grain size/weight. In summary, the stable QTLs would be reliable and the candidate genes predicted would be benefit for maker assisted breeding or cloning.
Similar content being viewed by others


Introduction
Maize is a very important crop which plays an important role in food, animal feed and the raw materials of bioenergy worldwide1. Obtaining high grain yield is of great significance for maize breeders. Yield-related traits are all complex quantitative traits, controlled by multiple genes. It is difficult to explain yield formation mechanism just from maize phenotypies. To improve maize yield, it is important to study on the relations between yield and yield-related traits at molecular level. Since Helentjaris and Slocum et al. published the first piece of molecular marker linkage map of maize in 19862, large amounts of QTLs have been mapped for yield traits3,4,5,6. To date, QTL mapping methods have been successfully used in maize and many QTLs associated with maize yield-related traits were identified. The identified QTLs included 36, 45, 149, 46 and 23 associated with cob diameter, ear diameter, grain weight, ear length and kernel row number, respectively (https://archive.gramene.org/qtl/).
In earlier plant breeding, researchers directly utilized the markers linked with identified QTLs in marker assisted breeding to enhance breeding efficiency7. However, QTL mapping results are usually vary among different experimental materials and different experimental environments8. Thus, it is important for QTLs to be confirmed or to be fine mapped before used for marker assisted breeding9. For QTL confirmation, one scheme is to detect QTLs in early generations like F2 and F2:3, and then confirmed in advanced generations from the same cross. Such approach have been successfully conducted in rice9, sweet sorghum10, soybean11, cucumber12. And the early generations with beneficial effect would be served as new breeding materials for cultivating new varieties7. Another scheme for confirming QTLs is to analyze if the target QTLs are stable and common between or among different populations. For example, the trichome density trait was confirmed with four recombinant inbred lines (RIL) populations of A. thaliana13, the stay green trait was confirmed with two RILs populations of sorghum14, and the kernel length trait of barley was confirmed by different RILs population15.
Grain weight is one of the most important yield-related trait in crops. It is of great significance to clone the genes controlling grain weight and then to clarify their molecular genetic mechanism. Great achievements have been made in genes/QTLs identification and dissection for grain size and grain weight in many crops, such as rice16,17,18,19,20, soybean21,22, wheat23,24. Especially many genes related to grain weight or grain size in rice, including GS325, GS516, qGL326, GW218, GW827, GS228, qGW7/GL729, have been successfully cloned. The evidence showed that the grain size trait was regulated by multiple signaling pathways, and the main pathway included ubiquitin-proteasome degradation pathway30, phytohormone signaling pathway and G protein independent pathway. Earlier studies showed that crop’s yield was greatly influenced by grain size and grain weight. Although great achievements on the genes controlling grain size and grain weight have been made in maize in recent years5,31,32,33, it was relatively low compared to rice and Arabidopsis thaliana. It is necessary for maize yield-related traits to confirm stable QTLs, to decrease functional gene number in stable QTL intervals and to predict candidate genes. All these works would provide basis for cloning functional genes and marker-assisted breeding.
The purposes of this study were: (1) to identify QTLs for yield-related traits with an F2:3 population from SG5 × SG7; (2) to compared these QTLs detected in this study with the QTLs identified in other populations, including an F2 population from the same parents as the F2:3 used in this study and recent studies; (3) RNA-seq technology was applied to identify transcriptional variations between maize inbred lines SG5 and SG7 subjected to grain weight; (4) to identify the DEGs related to grain weight between SG5 and SG7 and to predict candidate genes.
Results
Phenotype evaluation of the mapping population
The phenotypic data of seven yield traits, i.e., ear length (EAL), ear diameter (EAD), cob diameter (CD), kernel row number (KRN), ear weight (EW), grain weight per ear (GWP) and 100-kernel weight (KW), were collected from the F2:3 mapping population in 2016. The mean values of the seven traits were shown in Table 1. The phenotypic values of the two parents SG5 and SG7 were different in all the seven traits. The seven yield traits all displayed in bell-shaped normal distribution (Supplemental Fig. 1). Table 2 listed the pearson’s correlation coefficients and the significance tests between every two of the seven observed traits. The closes relation occurs between GWP and EW (0.975).
QTL analysis using a high-density linkage map
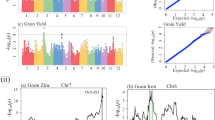
CIM and GCIM procedures were applied to identify QTLs associated with the seven yield traits based on a high-density linkage map. The map constructed in previous study34, had 3305 bin-markers. Using CIM, a total of thirty-three QTLs were detected for the seven yield traits. These QTLs were distributed on all of the 10 maize chromosomes. Among them, four QTLs controlled KW trait and were located on maize chromosomes 3, 7, 8, and 9; five related to EAL trait were located on chromosomes 1, 2, 3, and 5; four associated with EAD trait were located on chromosomes 1, 4, and 10; four controlling CD trait were located on chromosomes 1, 3, and 6; five controlled KRN trait and were located on chromosomes 2, 3, 4, and 9; four were associated with EW trait and located on 2, 5, 7, and 9; and seven controlling GWP trait were located on chromosomes 2, 3, 5, 7, and 9. QTL mapping result from CIM procedure is shown in Fig. 1 and Table 3. The physical intervals of 33 QTLs ranged from 0.15 to 23.75 Mb according to reference genome. The phenotypic variation explained by single QTL ranged from 4.5 to 25.6%, the means of KW, EAL, EAD, CD, KRN, EW, and GWP being 7.28, 12.2, 10.15, 12.2, 9.56, 10.4, and 9.6%, respectively. The logarithm of odds (LOD) scores ranged from 3.0 (qKW-12) to 7.4 (qEAD1-1).

Plots of test statistic −Log10(p) against genome location for seven traits of maize using the CIM method. The horizontal blue line of each panel is the critical value of the test statistic. The seven traits are: 100-kernel weight (KW), ear length (EAL), ear diameter (EAD), cob diameter (CD), kernel row number (KRN), ear weight (EW), and grain yield per plant (GWP). Dotted rectangle with green color indicate pleiotropism phenomenon might exist.
For GCIM, a total of fifty-four QTLs were detected. Among these QTLs, fourteen were also detected by CIM (Table 4), three KW QTLs were located on chromosomes 7, 8 and 9, two EAD QTLs on chromosomes 1, and 2, nine CD QTLs on chromosomes 1, 2, 4, 5, 6, and 7, thirty-four KRN QTLs on over all chromosomes except chromosomes 6 and 9, two EW QTLs on chromosomes 2 and 7, and four GWP QTLs on chromosomes 5, 7, and 9. Their LOD scores ranged from 3.1 (qKW-7) to 19.4 (qKRN-8). The GCIM approach seems more powerful in detecting small effects QTLs, especially for early generation population. The related information is summarized in Table 4
.
Confirmation of QTLs in different generation materials
It is essential to confirm QTLs before used in marker assisted breeding35. We compared the QTLs mapped with the F2:3 population with those detected using other populations, including an F2 generating the same parents as the mapping population in this study34. For the 33 QTLs mapped by CIM in this study (Table 3), eight of them located on chromosomes 1, 2, 3, and 7 were consistent with those detected in the F2 population (Fig. 2). There were fifteen major QTLs overlapped with the those identified in previous studies36,37,38 (Fig. 2), and they were located on chromosomes 1, 2, 3, 5, 7 and 9. For the fifty-four QTLs detected by GCIM, five of them(qKW-7, qEAD1-1, qCD-3, qKRN2-1, qKRN-1) were also mapped in the F2 population from the same parents, and twenty one were identical to the reported QTLs in other studies (Table 4). The QTL detected in more than one mapping populations is considered to be stable QTL.

QTL locations for seven yield-related traits studied in the SG5/SG7 F2:3 population. QTLs were represented in different colors for seven traits including blue for EAD (ear diameter, mm), pink for CD (cob diameter, mm), green for EAL (ear length, mm), brown for KRN (kernel row number), light blue for EW (ear weight, g), yellow for GWP (grain weight per ear) and red for KW (100-kernel weight, g) on chromosomes C1 to C10. QTLs represented by bars are shown on the right of the linkage groups, close to their corresponding markers. Supported intervals for each QTL are indicated by the length of vertical bars. The eight QTLs circled in red were stably detected in both F2 and F2:3 populations with CIM. The fifteen QTLs circled in blue were also detected in same or similar physical positions by previous studies with CIM.
To sum up, for all the QTLs detected by CIM and GCIM methods, thirty of them were stable QTLs, and fourteen and twenty-two were confirmed by CIM and GCIM, respectively. The detailed information about confirmed QTLs were listed in Tables 3 and 4. The confirmed QTLs for grain weight include one major QTL for KW on chromosome 7 (qKW-7, mapped by CIM and GCIM), two major QTLs for EW on chromosome 7 and 9 respectively (qEW-9, mapped by CIM and GCIM; and qEW-10, mapped by CIM only), two major QTLs for GWP on chromosome 2 and 7 respectively (qGWP-6, mapped by CIM only; qGWP-10, mapped by CIM and GCIM), two major QTLs for GWP on chromosome 5 (qGWP-8 and qGWP-13, detected by CIM and GCIM respectively), and three major QTLs for GWP on chromosome 9 (qGWP-11 and qGWP-12, mapped by CIM; and qGWP-14, mapped by GCIM).
Comparison of QTL regions
In this study, a total of thirty-three QTLs and fifty-four QTLs were respectively mapped by using CIM and GCIM methods for the seven yield-related traits. However, more than half of these QTLs were not stable, implying that these traits were controlled by multiple minor genes. The closely connected QTLs might be one locus, such asqEAD1 and qEAD1-1 on chromosome 1, qCD-3 and qCD3-1 on chromosome 1, qKRN-2 and qKRN-2-1 on chromosome 2. In addition, some of the detected QTLs in this study tended to have pleiotropism phenomenon. For example, qEAD-1 and qCD-3 were located on the same position on chromosome 1, but they were related to EAD and CD respectively. qEW-8 and qGWP-9 were located on the same position on chromosome 5, but they were related to EW and GWP respectively. qEW-10 and qGWP-11 located on the same position on chromosome 9 were related to EW and GWP respectively. And qKW-7, qEW-9 and qGWP-10 were located on the same position on chromosome 7, but they were related to KW, EW and GWP respectively.
Identification of candidate DEG for grain weight
RNA-seq procedure was carried out on an Illumina NovaSeq instrument. Totally, 18 RNA samples from SG5 and SG7 were collected form grains at three developmental stages (three biological replicates per stage) and used for RNA-seq analysis. The Pearson correlation analysis for RNA-seq data between samples was showed in Supplemental Table 2 and Fig. 2. A total of 932,738,890 with average 51, 818,827 clean reads on each chromosome were obtained after we filtering and performing quality control against the raw reads. (Supplemental Table 1). The clean reads were mapped onto the maize B73 genome by TopHat v. 2.0.1239 software. For each replicate, 40,277,620–62,763,408 reads were obtained. Of these reads, 81.97–86.23% were mapped to the reference genome as unique and multiple matches (Supplemental Table 1). Based on these mapped reads, DEGs, novel transcripts, alternative splicing and events were detected. The fragments per kilobase of exon per million fragments mapped (FPKM) model was used for calculating all genes expression values. The threshold of corrected P-value 0.05 and log2 (Fold change) of 0.5 (absolute value) were set as the thresholds for pair-wise comparison to detect DEGs between SG5 and SG7. To analyze the functions of DEGs, Gene Ontology function enrichment was conducted by Blast swiss prot database (website). The top significant biological process (BP), cellular component (CC), and molecular function (MF) terms were shown in Supplemental Fig. 3. In the BP group, DEGs are enriched in carbohydrate metabolic and lipid metabolic processes. In the MF group, DEGs are enriched in catalytic activity, hydrolase activity, oxidoreductase activity and so on. In the CC group, DEGs enrichment do not reach up to a significant level.
To decrease the candidate DEG number, we focused on those overlapped within the physical intervals of those confirmed QTLs related to grain weight. These QTLs were located on chromosomes 1, 2, 5, 7, and 9, including qKW-7, qEW-9, qEW-10, qGWP-6, qGWP-8, qGWP-10, qGWP-11, qGWP-12, qGWP-13 and qGWP-14. There were more than 1000 protein-coding genes in the mapped physical intervals in total, but after removing those genes that were not DEGs between parents, only 213 genes left and applied for further comparative genomics analysis and candidate gene prediction (Supplemental Table 3).
Candidate gene prediction
In ubiquitin-proteasome degradation pathway, grain size/weight was regulated by the interactions among three kinds of protein enzymes, i.e., ubiquitin protein ligase E3, ubiquitin-binding enzyme E2 and ubiquitin-activating enzyme E130.The functional mechanism of DA240 in Arabidopsis and GW218 in rice in regulating grain size were both involved in the ubiquitin-proteasome degradation pathways. Previous studies have proved that abscisic acid (ABA) plays functions during seed development, and ABA-deficient Arabidopsis mutants produce seeds with increased size and mass41. MADS-box transcription factor genes have also been proven to play critical role in regulating grain size/weight indirectly42. FEM111/AGL80 is such a kind of gene in Arabidopsis43. Among the candidate genes in the intervals of qGWP-6, there were two genes GRMZM2G097089 and GRMZM2G158191 encoding E3 ubiquitin protein ligase. At qGWP-8 interval, one gene GRMZM2G169994 encoding E3 ubiquitin protein ligase was located. At the physical interval of the three identical QTLs qKW-7, qEW-9 and qGWP-10, there were two genes, one was GRMZM2G113039 that encodes E3 ubiquitin protein ligase and the other was GRMZM2G134480 that encodes ubiquitin-activating enzyme E1. At the physical interval of the two consistent QTLs qEW-10 and qGWP-11, two genes GRMZM2G057959 and GRMZM2G128953 were located and encoded ABA receptor MADS-box transcription factor respectively. And at qGWP-12 interval, one gene GRMZM2G036697 that encodes probable E3 ubiquitin-protein ligase was located.
These eight genes were chosen as candidate genes for grain weight in maize for the future prospects (Table 5).
Discussion
High yield is a permanent objective for maize breeders. Studies on QTL mapping and gene dissection for yield-related traits have become a research focus in maize in recent years37,44,45,46. It is of great significance for maize breeder to map QTLs, to predict candidate genes, to clone functionlal genes and to clarify gene’s genetic mechanism for yield-related traits, especially for grain weight. Compared to rice and Arabidopsis thaliana, the studies on maize grain weight candidate genes were relatively slow. Stable QTLs are useful for marker-assisted breeding. QTLs mapped in one population might well be not detected in another population. Thus, it is critical important for QTLs to be confirmed to rule out statistical anomalies while used in marker assisted breeding7. To validate QTLs, the first scheme is to confirm them in other mapping populations, the second scheme is to confirm them in different generations from the same crossing, and the third is to confirm QTLs using the same population evaluated in multiple locations and in multiple years. Mapping QTLs in early generations is that the QTLs with beneficial effect for breeding lines in early generations can be transferred to the high generations directly. In addition, the QTLs detected in early generation are more than those in late generation, especially for those minor QTLs47. In this study, an F2:3 population developed from the cross between SG-5 and SG-7 was used for mapping QTLs for seven yield-related traits. In terms of phenotypic segregation issue in F2:3, Zhang et al. (2004) proposed to combine the F2 phenotypes with the F2:3 average phenotypes to further increase the power of QTL mapping48. In this study, the F2:3 phenotypic value were the mean value of eight F2:3 individuals of F3 progeny derived from F2 plant selfing. The confirmed QTLs for yield-related traits are considered as stable QTLs and could be applied in marker assisted breeding, gene cloning and function analyzing, etc.
Compared to low-density linkage map, Hori et al.49 indicated that the higher-density linkage maps were more beneficial for QTL mapping, that is, the markers tightly linked to target QTL are more effective in marker assisted breeding49. Furthermore, it is possible to separate two closely linked QTLs by a high-density map with high resolution49. In this study, a high -density linkage map developed from an earlier study result34 was applied in QTL mapping. However, the confidence intervals of identified QTLs always were not narrow enough to predict candidate genes directly. Gene density is within a wide range of 0.5–10.7 genes per 100 kb in maize genome50. It is of great significance to study how to decrease the number of candidate genes located on confirmed QTL intervals. For the purpose, a common approach is to develop advanced generation population such as NIL(near isogenic line) population, thus to narrow down the QTL confidence interval significantly, even to clone the QTL or gene based on primary mapping results51,52. However, the construction of NILs is time - consuming and tedious, which limit the usage of large amount of objective QTLs in marker assisted breeding to a certain extent. In this study, the DEGs between parents were obtained from transcriptomic profiling of grains at different developmental stages and overlaid onto the confirmed QTL intervals to predict candidate genes for grain weight. Based on these DEGs, the candidate genes in the physical intervals of the ten confirmed QTLs (qKW-7, qEW-9, qEW-10,qGWP-6, qGWP-8, qGWP-10,qGWP-11, qGWP-12, qGWP-13 and qGWP-14) were decreased from over 1000 to 213 (Supplemental Table 3). Comparative genomics analysis was carried out to further predict candidate genes. Finally, a total of eight genes that might be involved in ubiquitin-proteasome degradation30, phytohormone signaling and transcription factor pathways were chosen as candidate genes controlling grain size and grain weight in maize.
In this study, a total of ten QTLs associated with grain weight were confirmed as stable QTLs, and eight candidate genes were predicted. All these results would be basis for cloning related functional genes and marker-assisted breeding.
Methods
Development of the F2:3 mapping population and field trails’ investigation
The F1 hybrid seeds were obtained from an intraspecific cross between two inbred lines SG5 and SG7 in Liupanshui, Guizhou in 2013 summer. There are significant differences in yield-related traits for the two inbred lines (Table 1). The obtained F1 seeds were also planted in Liupanshui, Guizhou in 2014 summer. A total of 199 F2 plants grew up from F1 seeds In November 2014, field trials were conducted at the Panxian Maize Breeding Station in Sanya, Hainan, China. The segregating population of 199 F2:3 lines, P1, P2 and F1 were all planted in field with a randomized block design of three replications, single-row plot with 15 plants, row spacing being 50 cm, and plant spacing being 35 cm. Seven agronomic traits including 100-kernel weight (KW), ear length (EL), ear diameter (ED), cob diameter (CD), kernel row number (KRN), ear weight (EW), and grain weight per plant (GWP) were investigated for the F2:3 population. Eight plants located in the middle of each plot were sampled for investigation after harvest and natural drying.
High density linkage map, QTL analysis and validation
Methods of extracting genomic DNA, sequencing genotype, grouping sequence data, identifying single nucleotide polymorphisms (SNPs) and constructing high-density bin map were exhibited in our previous study34. QTL were detected by two methods: one is CIM method included in QTL Cartographer v. 2.5, and the other is genome-wide composite interval mapping (GCIM) method53. GCIM method was applied by using random model with maximum likelihood (ML) function. For CIM and GCIM mapping procedures, the walking speed was 1 cM and the logarithm of odds (LOD) threshold value was set up as 3.0. The position of a significant QTL was determined by its LOD peaks. The positive or negative additive effect of a QTL indicated that the increase or decrease in phenotypic value for a trait is provided by the alleles from SG5 or SG7. The graphic of QTLs positions on 10 maize chromosomes were drawn by MapChart 2.32 software54. The physical intervals between the QTLs identified in F2:3 population were compared with the intervals of QTLs mapped using a F2 population previously34, and Those QTLs both detected from F2 and from F2:3 populations show overlapped physical intervals will be regarded as confirmed QTLs.
RNA sample preparation
The two maize inbred lines SG5 and SG7 were grown at the Panxian Maize Breeding Station, in Hainan, China in November 2016. Ear shoot were covered before silking. Some parental plants were hand pollinated when the length of corn silk was about 5 cm. Kernels were sampled from SG5 and SG7 on the 5-, 10, and 15-the days after pollination. Each sample consisted of three biological replicates in parallel. All samples were collected and frozen immediately with liquid nitrogen and stored in refrigerator at −80 °C. The total RNA of kernels at different growing stages was extracted by using TRIzol reagent (Invitrogen). One percent of agarose gels were used for RNA degradation and contamination. The NanoPhotometer® spectrophotometer (IMPLEN, CA, USA) was used for checking RNA purity. Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, USA) was used for measuring RNA concentration. The RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, USA) was used for assessing RNA integrity. The Illumina NovaSeq platform was then applied to RNA-seq.
Illumina sequencing and data analysis
A total of 18 samples with three repeats were collected and sequenced at the Illumina NovaSeq platform. Raw reads with fastq format were firstly handled by in-house perl scripts. Clean reads were then obtained after deleting reads containing adapter and ploy-N and removing reads of low quality in raw data. The GC content, Q20 and Q30 of the clean reads were calculated. High quality clean data were then carried out for further downstream analyzing. Maize reference genome and correlated files of gene annotation were downloaded directly from website (https://www.maizegdb.org/). Bowtie v. 2.2.3 and TopHat v2.0.1239 were used for building reference genome index and aligning paired-end clean reads to the reference genome, respectively. The reads mapped to every gene were counted by HTSeq v. 0.6.1.
For each gene, the expected number of Fragments Per Kilobase of transcript sequence per Millions base pairs (FPKM) was calculated by analyzing the gene length and mapped reads. FPKM is a widely accepted methodfor evaluating gene expression levels based on sequencing depth effect and gene length of the read count simultaneously55. The DEGSeq R package (v. 1.20.0) was used for analyzing differential expression genes between two conditions. The P value was corrected by using the Benjamini & Hochberg method. The threshold of corrected P-value 0.05 and log2 (Fold change) of 0.5 (absolute value) were considered as significantly differential expression. The GOseq R package was used for analyzing Gene Ontology (GO) enrichment of DEGs. The GO term with corrected P-value less than 0.05 was considered as DEG.
Screening of candidate DEGs associated with QTLs for grain weight
In this study, the obtained RNA-seq data were used to explore DEGs between parental lines SG5 and SG7. Pair-wise comparison of transcriptomes between SG5 and SG7 was conducted for detecting DEGs. The DEGs were overlaid onto the physical intervals of confirmed QTLs to predict candidate genes for grain weight in maize.
References
Li, H. et al. Quantitative trait locus analysis of heterosis for plant height and ear height in an elite maize hybrid zhengdan 958 by design III. BMC Genet 18, 36 (2017).
Helentjaris, T., Slocum, M., Wright, S., Schaefer, A. & Nienhuis, J. Construction of genetic linkage maps in maize and tomato using restriction fragment length polymorphisms. Theoretical and applied genetics 72, 761–769 (1986).
Austin, D. & Lee, M. Comparative mapping in F 2∶ 3 and F 6∶ 7 generations of quantitative trait loci for grain yield and yield components in maize. Theoretical and Applied Genetics 92, 817–826 (1996).
Messmer, R. et al. Drought stress and tropical maize: QTL-by-environment interactions and stability of QTLs across environments for yield components and secondary traits. Theoretical and Applied Genetics 119, 913–930 (2009).
Peng, B. et al. QTL analysis for yield components and kernel-related traits in maize across multi-environments. Theoretical and applied genetics 122, 1305–1320 (2011).
Yan, J.-b., Tang, H., Huang, Y.-q., Zheng, Y.-l. & Li, J.-s. Quantitative trait loci mapping and epistatic analysis for grain yield and yield components using molecular markers with an elite maize hybrid. Euphytica 149, 121–131 (2006).
Gelli, M. et al. Validation of QTL mapping and transcriptome profiling for identification of candidate genes associated with nitrogen stress tolerance in sorghum. BMC Plant Biol 17, 123 (2017).
Holland, J. B. Genetic architecture of complex traits in plants. Curr Opin Plant Biol 10, 156–161 (2007).
Wickneswari, R. et al. Identification and validation of quantitative trait loci for agronomic traits in advanced backcross breeding lines derived from Oryza rufipogon× Oryza sativa cultivar MR219. Plant molecular biology reporter 30, 929–939 (2012).
Wang, H.-L. et al. Identification and validation of QTLs controlling multiple traits in sorghum. Crop and Pasture Science 67, 193–203 (2016).
Su, C., Lu, W., Zhao, T. & Gai, J. Verification and fine-mapping of QTLs conferring days to flowering in soybean using residual heterozygous lines. Chinese Science Bulletin 55, 499–508 (2010).
Weng, Y. et al. QTL mapping in multiple populations and development stages reveals dynamic quantitative trait loci for fruit size in cucumbers of different market classes. Theoretical and applied genetics 128, 1747–1763 (2015).
Symonds, V. V. et al. Mapping quantitative trait loci in multiple populations of Arabidopsis thaliana identifies natural allelic variation for trichome density. Genetics (2005).
Haussmann, B. et al. QTL mapping of stay-green in two sorghum recombinant inbred populations. Theoretical and Applied Genetics 106, 133–142 (2002).
Zhou, H. et al. Mapping and validation of major quantitative trait loci for kernel length in wild barley (Hordeum vulgare ssp. spontaneum). BMC Genet 17, 130 (2016).
Li, Y. et al. Natural variation in GS5 plays an important role in regulating grain size and yield in rice. Nat Genet 43, 1266 (2011).
Qiu, X., Gong, R., Tan, Y. & Yu, S. Mapping and characterization of the major quantitative trait locus qSS7 associated with increased length and decreased width of rice seeds. Theoretical and applied genetics 125, 1717–1726 (2012).
Song, X.-J., Huang, W., Shi, M., Zhu, M.-Z. & Lin, H.-X. A QTL for rice grain width and weight encodes a previously unknown RING-type E3 ubiquitin ligase. Nat Genet 39, 623 (2007).
Wan, X. et al. QTL analysis for rice grain length and fine mapping of an identified QTL with stable and major effects. Theoretical and Applied Genetics 112, 1258–1270 (2006).
Kang, Y.-J. et al. Fine mapping and candidate gene analysis of the quantitative trait locus gw8. 1 associated with grain length in rice. Genes & genomics 40, 389–397 (2018).
Xu, Y. et al. Mapping quantitative trait loci for seed size traits in soybean (Glycine max L. Merr.). Theoretical and Applied Genetics 122, 581–594 (2011).
Han, Y. et al. QTL analysis of soybean seed weight across multi-genetic backgrounds and environments. Theoretical and Applied Genetics 125, 671–683 (2012).
Sun, X.-Y. et al. QTL analysis of kernel shape and weight using recombinant inbred lines in wheat. Euphytica 165, 615 (2009).
Ramya, P. et al. QTL mapping of 1000-kernel weight, kernel length, and kernel width in bread wheat (Triticum aestivum L.). J Appl Genet 51, 421–429 (2010).
Fan, C. et al. GS3, a major QTL for grain length and weight and minor QTL for grain width and thickness in rice, encodes a putative transmembrane protein. Theoretical and Applied Genetics 112, 1164–1171 (2006).
Zhang, X. et al. Rare allele of OsPPKL1 associated with grain length causes extra-large grain and a significant yield increase in rice. Proceedings of the National Academy of Sciences 109, 21534–21539 (2012).
Wang, S. et al. Control of grain size, shape and quality by OsSPL16 in rice. Nat Genet 44, 950 (2012).
Hu, J. et al. A rare allele of GS2 enhances grain size and grain yield in rice. Mol Plant 8, 1455–1465 (2015).
Wang, S. et al. The OsSPL16-GW7 regulatory module determines grain shape and simultaneously improves rice yield and grain quality. Nat Genet 47, 949 (2015).
Verma, R., Oania, R., Graumann, J. & Deshaies, R. J. Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin-proteasome system. Cell 118, 99–110 (2004).
Gupta, P. K., Rustgi, S. & Kumar, N. Genetic and molecular basis of grain size and grain number and its relevance to grain productivity in higher plants. Genome 49, 565–571 (2006).
Li, Y. et al. Three main genetic regions for grain development revealed through QTL detection and meta-analysis in maize. Molecular breeding 30, 195–211 (2012).
Liu, J. et al. The conserved and unique genetic architecture of kernel size and weight in maize and rice. Plant Physiol, pp. 00708.02017 (2017).
Su, C. et al. High density linkage map construction and mapping of yield trait QTLs in maize (Zea mays) using the genotyping-by-sequencing (GBS) technology. Front Plant Sci 8, 706 (2017).
Langridge, P. et al. Trends in genetic and genome analyses in wheat: a review. Aust J Agric Res 52, 1043–1077 (2001).
Chen, L. et al. Candidate loci for yield-related traits in maize revealed by a combination of metaQTL analysis and regional association mapping. Front Plant Sci 8, 2190 (2017).
Xiao, Y. et al. Genome‐wide dissection of the maize ear genetic architecture using multiple populations. New Phytologist 210, 1095–1106 (2016).
Xue, Y. et al. Genome-wide association analysis for nine agronomic traits in maize under well-watered and water-stressed conditions. Theoretical and applied genetics 126, 2587–2596 (2013).
Trapnell, C. et al. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol 31, 46 (2013).
T, X. et al. The ubiquitin receptor DA1 interacts with the E3 ubiquitin ligase DA2 to regulate seed and organ size in Arabidopsis. Plant Cell 25, 3347–3359 (2013).
Cheng, Z. J. et al. Abscisic acid regulates early seed development in Arabidopsis by ABI5-mediated transcription of short hypocotyl under blue1. Plant Cell 26, 1053–1068 (2014).
Kapazoglou, A. et al. The study of two barley type I-like MADS-box genes as potential targets of epigenetic regulation during seed development. BMC Plant Biol 12, 166 (2012).
Portereiko, M. F. et al. AGL80 is required for central cell and endosperm development in Arabidopsis. Plant Cell 18, 1862–1872 (2006).
Jiménez-Galindo, J. C., Ordás, B., Butrón, A., Samayoa, L. F. & Malvar, R. A. QTL Mapping for Yield and Resistance against Mediterranean Corn Borer in Maize. Front Plant Sci 8, 698 (2017).
Yang, N. et al. Genome wide association studies using a new nonparametric model reveal the genetic architecture of 17 agronomic traits in an enlarged maize association panel. PLoS Genet 10, e1004573 (2014).
Chen, L. et al. Fine-mapping of qGW4. 05, a major QTL for kernel weight and size in maize. BMC Plant Biol 16, 81 (2016).
Vales, M. et al. Effect of population size on the estimation of QTL: a test using resistance to barley stripe rust. Theoretical and Applied Genetics 111, 1260–1270 (2005).
Zhang, Y.-M. & Xu, S. Mapping quantitative trait loci in F2 incorporating phenotypes of F3 progeny. Genetics 166, 1981–1993 (2004).
Hori, K. et al. Efficient construction of high-density linkage map and its application to QTL analysis in barley. Theoretical and Applied Genetics 107, 806–813 (2003).
Haberer, G. et al. Structure and architecture of the maize genome. Plant Physiol 139, 1612–1624 (2005).
Yano, M. et al. Hd1, a major photoperiod sensitivity quantitative trait locus in rice, is closely related to the Arabidopsis flowering time gene CONSTANS. Plant Cell 12, 2473–2483 (2000).
Kojima, S. et al. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant and Cell Physiology 43, 1096–1105 (2002).
Wen, Y.-J. et al. An efficient multi-locus mixed model framework for the detection of small and linked QTLs in F2. Brief Bioinform (2018).
Voorrips, R. MapChart: software for the graphical presentation of linkage maps and QTLs. Journal of heredity 93, 77–78 (2002).
Trapnell, C. et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28, 511 (2010).
Acknowledgements
This work was supported by the Natural Science Foundation of China (Grant #31460359), Research Foundation for Advanced Talents of Qingdao Agricultural University (Grant 6631119035), the Natural Science Foundation of Guizhou Province of China (Grant #Qian Kehe J word [2014]2155 and Grant #Qian Kehe LH word [2015]7605).
Author information
Authors and Affiliations
Contributions
C.S. have finished phenotyping and genotyping of F2:3 progeny, analyzing the data, and drafted the manuscript. Y.Z. have finished in developing the F2:3 population and phenotypes investigation. Both authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, Y., Su, C. Mapping quantitative trait loci for yield-related traits and predicting candidate genes for grain weight in maize. Sci Rep 9, 16112 (2019). https://doi.org/10.1038/s41598-019-52222-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-52222-5
This article is cited by
-
QTL mapping and candidate gene analysis for yield and grain weight/size in Tartary buckwheat
BMC Plant Biology (2023)
-
Genetic mapping of loci determining seed coat color and size in watermelon
Euphytica (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
