
Abstract
Environmentally induced epigenetic modifications have been proposed as one mechanism underlying rapid adaptive evolution of invasive species. Didemnum vexillum is an invasive colonial ascidian that has established in many coastal waters worldwide. Phylogenetic analyses have revealed that D. vexillum populations consist of two distinct clades; clade B appears to be restricted to the native range (Japan), whereas clade A is found in many regions throughout the world, including New Zealand. The spread of D. vexillum clade A suggests that it might be intrinsically more invasive than clade B, despite low levels of genetic diversity compared to populations from the native region. This study investigated whether D. vexillum clade A exhibits epigenetic signatures (specifically differences in DNA methylation) associated with invasiveness. Global DNA methylation patterns were significantly different between introduced clade A colonies, and both clades A and B in the native range. Introduced colonies also showed a significant reduction in DNA methylation levels, which could be a mechanism for increasing phenotypic plasticity. High levels of DNA methylation diversity were maintained in the introduced population, despite reduced levels of genetic diversity, which may allow invasive populations to respond quickly to changes in new environments. Epigenetic changes induced during the invasion process could provide a means for rapid adaptation despite low levels of genetic variation in introduced populations.
Similar content being viewed by others


Introduction
A major threat to biodiversity worldwide is the introduction, establishment and spread of invasive species beyond their native range1,2. Due to the considerable ecological and economic impacts of biological invasions3, research has focused on the prevention, control and eradication of invasive species4. Molecular-based approaches are often utilized to characterize the ecological and evolutionary factors involved in the invasion process5,6,7, and are effective for estimations of propagule pressure8, early detection of introduced populations9, identifying the source of introductions10, and identifying genetic characteristics associated with successful invaders11. A major goal of invasion biology is to understand why certain species are successful invaders in order to facilitate assessment and management of risks7.
Phenotypic plasticity, a wide tolerance to environmental stress, rapid growth and reproduction, and a high level of competitive ability have all been proposed to facilitate invasiveness12. Higher genetic diversity within populations is also thought to aid in the successful colonization of new environments13,14. Specifically, low levels of genetic variation are known to increase the risk of inbreeding depression and limit the adaptive and evolutionary potential of a species12. Yet, invasive populations with little or no genetic variation can be extremely successful in new environments15, sometimes outcompeting native species16. Reduced competition from conspecifics17,18, the maintenance of a high level of quantitative trait diversity19, and the release from natural enemies20 have been proposed as key factors determining invasion success when genetic diversity is low. More recently, epigenetic mechanisms (e.g., DNA methylation), which can increase both phenotypic plasticity and heritable variation, have been suggested to play a key role in invasion success21,22,23. Epigenetic mechanisms can alter a given genotype’s influence on an organism’s phenotype, without changes in the underlying DNA nucleotide sequences. By increasing, decreasing, or silencing the activity of genes, epigenetic changes can lead to novel phenotypic variation in the absence of additional genetic diversity24. Despite this, few studies have investigated epigenetic patterns in invasive populations, which is an important step in understanding the role of epigenetic modifications in successful biological invasions.
A valuable approach for identifying genotypes and phenotypes associated with successful invasive species is to compare populations in their native and non-native habitats25,26. Molecular analyses have revealed that invasive species are often comprised of distinct sibling species or clades across their expanded and native ranges27,28,29, and in some cases, only certain clades become invasive18,25,30. Didemnum vexillum is an invasive marine colonial ascidian that has become established in temperate coastal waters worldwide, including several regions in New Zealand31,32, North America33, and Europe34,35,36. D. vexillum is believed to have been translocated throughout the world as fouling on aquaculture species and equipment, such as cultured Pacific oysters (Crassostrea gigas), and on ship hulls37,38,39. Additional to this well-defined history of spread, morphological37 and molecular comparisons18,40,41 have concluded that the native range of D. vexillum is likely to be the Northwest Pacific Ocean, including Japan. Phylogenetic analyses have revealed that D. vexillum populations consist of two distinct clades18. One clade (referred to as clade B) appears to be geographically restricted to D. vexillum’s native region (Northwest Pacific Ocean), whereas colonies belonging to clade A have become established in temperate coastal areas around the world, including New Zealand. Clades A and B are morphologically identical37, and the level of the mitochondrial cytochrome c oxidase subunit I (COI) gene sequence divergence between the two clades is far below the comparable divergence values for other ascidian species, such as the recently classified cryptic species Ciona robusta and Ciona intestinalis (formally C. intestinalis type A and type B, respectively)26,42,43,44. Therefore, evidence to date suggests that D. vexillum clades A and B represent an ongoing speciation event18,26. Diversification of clades A and B within the Northwest Pacific following a human-mediated introduction from another region is not consistent with known evolutionary rates at COI, as divergence of the A and B COI clade lineages is estimated to have occurred 1.5–2.5 million years ago, which pre-dates anthropogenic vectors18. The persistence of clade B’s restricted distribution may indicate that it is intrinsically less invasive than clade A18. Introduced populations of clade A in New Zealand have low levels of genetic diversity compared to populations within the native region of Japan, indicating a founder effect18. Despite this, introduced D. vexillum populations have been extremely successful in their new environments, forming large colonies that can extend over significant areas, including commercially important aquaculture farms39.
The most well-studied epigenetic mechanism related to invasion success is DNA methylation, which generally refers to the addition of a methyl group to cytosine nucleotides to form 5-methylcytosine. DNA methylation can respond to environmental cues45,46,47, and has been associated with adaptation to different environmental conditions in invaded habitats48,49. Additionally, some introduced populations have higher DNA methylation diversity than populations in their native range despite reductions in genetic diversity due to founder effects50, and lower levels of global or genome-wide DNA methylation51, both of which have been associated with phenotypic variation and phenotypic plasticity51,52,53,54. Given the tendency of D. vexillum clade A to successfully invade multiple regions throughout the world, we hypothesized that (1) introduced populations of D. vexillum clade A would have high levels of DNA methylation diversity, despite low genetic diversity, (2) introduced populations of D. vexillum clade A would have lower levels of global DNA methylation compared to populations within the native region, and (3) populations would be differentially methylated when collected from sites with different environmental conditions. To test these hypotheses, we compared whole genome DNA methylation in D. vexillum colonies collected from New Zealand (clade A) with those from Japan (clade A and clade B), and between sites with different environmental conditions (differences in temperature and salinity). Samples used in the present study were a subset of samples previously collected and analysed by Smith, et al.18 using mitochondrial cytochrome c oxidase subunit I (COI) partial coding sequences.
Materials and Methods
Tissue sample collection
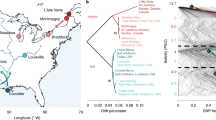
Didemnum vexillum tissue samples were collected between April 2008 and July 2009 from four sites in Japan (n = 39) and three sites in New Zealand (n = 35) by Smith, et al.18. Samples were collected in the summer months of the respective countries. Tissue samples were preserved in approximately 2.0 ml of 96% (v/v) ethanol and stored at -20 °C before being assigned to clade A or clade B by mitochondrial COI coding region analysis18. All colonies from New Zealand were clade A (n = 35), whereas samples collected from Japan were comprised of clade A (n = 16) and clade B (n = 23). In Japan, the distributions of clades A and B overlap at several sites (Fig. 1). The earliest worldwide record of D. vexillum was in Japan in 1926, but it was only identified as Didemnum sp. at the time37. In New Zealand, D. vexillum was first reported in 2001 in the North Island, in Whangamata Harbour31 and was described initially as a native species to New Zealand. Two months later, D. vexillum was discovered in the South Island, in Queen Charlotte Sound55. It is thought to have been transported to the South Island as fouling the hull of a large barge that had earlier been anchored in Whangamata Harbour55. Due to the continued movement of vessels, D. vexillum subsequently spread throughout the Marlborough Sounds, including to Pelorus Sound39.

Locations and details of Didemnum vexillum colonies used for analyses. Invasive populations in New Zealand (left) consist of clade A colonies only, whereas populations from within the native range of Japan (right) are comprised of both clade A and clade B colonies. Pie charts represent sampling sites used for mitochondrial cytochrome c oxidase subunit I (COI) gene analysis in Smith et al.18, and the relative proportions of each clade. Labelled sites represent the location of samples selected for DNA methylation analysis in this study. Figure adapted from Smith et al.18. The date D. vexillum populations were first recorded in each location in New Zealand is indicated below each site and the number of colonies sampled is indicated in brackets.
Environmental conditions
Site-specific environmental data (temperature and salinity) were collated from publicly available reports and websites (Table 1). Based on experiments identifying D. vexillum tolerance limits and the optimal temperature and salinity range for growth46, we allocated a temperature and salinity ‘stress score’ to each site, with 1 being the lowest stress score and 3 being the highest (Table 2). Temperature and salinity stress scores related to tolerance ranges were as follows; Temperature stress: D. vexillum grows well in temperatures less than 23 °C, so a stress score of 1 was allocated to sites with maximum temperatures of <23 °C, D. vexillum growth is usually reduced at temperatures higher than 24 °C, so sites with a maximum temperature of 23–25 °C were given a stress score of 2, growth is further reduced above 25 °C, and 28 °C is close to the upper tolerance limit for D. vexillum46,56, so sites with a maximum summer temperature of 25–28 °C were given a stress score of 3; Salinity stress: D. vexillum can grow well in sanities above 26 PSU46, so sites with minimum salinity values ≥26 were given a stress score of 1, growth is generally reduced below 26 PSU, and ascidians are rarely found in salinities lower than 25 PSU57, so a stress score of 2 was given to sites with minimum salinity values of 23–<26 PSU, a stress score of 3 was allocated to sites that drop below 23 PSU, as 20 PSU is thought to be close to the low salinity limit for D. vexillum, with extended periods of exposure resulting in mortality58,59. We then combined the temperature and salinity stress score at each site to obtain a ‘summative stress score’ which was used for further analysis (see Table 2). This is a similar approach to the method described in Ardura et al.60.
The sampling sites represent different environments within both New Zealand and Japan (Table 1). New Zealand seawater temperatures are similar between sites and within the optimal range for D. vexillum, with winter minima around 11–12 °C, and maxima between 19 °C and 23 °C. Therefore, we allocated all New Zealand sites a temperature stress score of 1 (Table 2). While the mean and maximum salinity values are similar between sites in New Zealand (mean approx. 33–35 PSU), the minimum salinity is more variable, with salinity above 30 PSU in Queen Charlotte Sound but dropping below 25 PSU in Pelorus Sound and Whangamata Harbour. The lowest salinity values were reported in Whangamata (13 PSU). Minimum and maximum temperatures and salinities are most likely to influence global DNA methylation differences (see Hawes et al.46). Therefore, each site in New Zealand was allocated a different salinity stress score based on minimum salinity values (1, 2 and 3 respectively; Table 2).
For Japan, minimum water temperatures were similar, or slightly cooler, than minimum water temperatures at sites in New Zealand. Minimum temperatures ranged from 9–12.5 °C at all sites, except for Shizugawa Bay which can be <7 °C in winter. The maximum temperature in Shizugawa Bay was slightly warmer than the maximum water temperatures at New Zealand sites (19–23.5 °C), while Sagami Bay, Ise Bay, and Izu Peninsular all reach maximum water temperatures of 26–28 °C (Table 1). Therefore, all sites in Japan were allocated stress scores of 2 or 3 (Table 2). For salinity, Shizugawa Bay salinity values were most similar to Queen Charlotte Sound, while Ise Bay salinity values were lower, and more similar to Pelorus Sound and Whangamata Harbour. Major rivers empty into Pelorus Sound (Pelorus River), Whangamata Harbour (Wentworth River) and Ise Bay (Kiso Three Rivers) which can result in low minimum salinity values following precipitation events, so these sites were given stress scores of 2 or 3 while all other sites obtained a salinity stress score of 1 (Table 2).
DNA extraction and methylation-sensitive amplified polymorphism
Tissue samples were finely chopped up using a sterile scalpel blade and genomic DNA was obtained using G-spin Total DNA extraction kits (animal tissue protocol; Intron, Gyeonggi-do, South Korea). Methylation-sensitive amplified polymorphism (MSAP) was then used to determine changes in global methylation patterns as previously described in detail in Hawes et al.46.
Data analysis
The Peak Scanner software (Applied Biosystems, Foster City, CA, USA) was used to assign the MSAP fragments peak height and size. To establish the peak height with the lowest error rate (the lowest number of fragment presence/absence mismatches between triplicate samples), and the error rate for msap (v. 1.1.8) analysis61 described below, we completed the MSAP procedure described above in triplicate for one individual sample. The following settings were associated with the lowest error rate between triplicates: error rate per primer, 0.07; analysis range, 50–500 base pairs (bp); minimum peak height, 50 relative fluorescent units (RFU). Peak presence/absence data corresponding to HpaII and MspI fragments was then converted to a binary matrix (presence = 1, absence = 0), so that the methylation state of each restriction site could be identified. The MSAP profiles were then assessed in R62 using the msap package (v. 1.1.8)61.
The msap package determines whether individual fragments (loci) are methylation-susceptible loci (MSL) or non-methylated loci (NML) by analyzing the contents of the binary matrix. Subsequent analyses are performed independently, with MSL used to assess DNA methylation variation and NML used to assess genetic variation61. While NML has been used to assess genetic variation in several studies51,60,63, it is only a proxy for genetic variation and should be interpreted with some caution64. Methylation state is categorized as either: Type I = unmethylated state, Type II = methylation of internal cytosine, Type III = hemimethylation, and Type IV, which is considered uninformative as this type could be due to either hypermethylation or the absence of the target restriction site61,65. Within msap, epigenetic (MSL) and genetic (NML) differentiation between populations were assessed by principal coordinates analyses (PCoA) followed by analyses of molecular variance AMOVA66 with 10000 permutations61. A Mantel test was used to establish the correlation between MSL and NML, an indication of how much epigenetic variation might be influenced by the genetic background61. The amount of genetic and epigenetic variation was estimated using the Shannon diversity index (S). Differences between NML and MSL diversity were tested using the Wilcoxon Rank Sum test with continuity correction.
Following analysis with msap, the overall MSL and NML variation estimated by the Shannon diversity index was compared between populations using the number of samples (N), mean diversity (I) and the standard deviation (obtained from msap) to perform and one-way analysis of variance (ANOVA) and post-hoc pairwise comparisons using Tukey’s honest significance difference test (Tukey’s HSD). The global methylation level was calculated as the proportion of methylated loci (the number of methylated loci (Types II and III) divided by the scorable loci (Types I, II and III)) following51,67. Using R (v. 3.3.0)62, global methylation levels were compared among populations using a generalised linear model (GLM) with a quasibinomial distribution to correct for overdispersion. Residuals were checked and assumptions validated. We then compared global methylation differentiation in colonies collected from sites with the same summative stress scores using msap (v. 1.1.8)61 (PCoA followed by AMOVA) to determine whether site-specific MSL differences were associated with differences in environmental conditions (temperature and salinity). Only summative stress scores ‘3’ and ‘4’ could be analysed with msap because scores ‘2’ and ‘6’ were only associated with one site respectively. To assess the relationship between global methylation levels and environmental conditions, we used a GLM with a quasibinomial distribution to compare the proportion of methylated loci in colonies collected from sites with different summative stress scores, followed by post-hoc pairwise comparisons using Tukey’s HSD tests.
Results
Environmental differences between sites
DNA methylation patterns in colonies from sites with the same summative stress scores were significantly differentiated (Supplementary Table S1), suggesting that global DNA methylation differences between sites in this study are not clearly associated with summative temperature and salinity scores. There were also no significant differences between the proportion of methylated loci (Types II and III/Types I, II and III) associated with different summative stress scores (Supplementary Table S2).
DNA methylation and genetic differentiation
In New Zealand colonies (clade A), 1784 loci were identified, 1309 of these were MSL (73.4%, 94% polymorphic) and 475 were NML (26.6%, 97% polymorphic). In Japan clade A, 1780 loci were identified, 1496 of these were MSL (84%, 92% polymorphic), and 284 were NML (16.6%, 97% polymorphic). Finally, in Japan clade B, 1785 loci were identified, 1658 were MSL (92.9%, 93% polymorphic) and 127 were NML (7.1%, 96% polymorphic). Combined, 1799 loci were identified. 1617 were MSL (89%, 99% polymorphic) and 182 were NML (10%, 99% polymorphic). We found no significant differences in MSL or NML patterns between Japan clade A (JAA) and Japan clade B (JAB). Instead, significant differences were due to colonies collected in New Zealand being different to colonies collected in Japan, regardless of clade affiliation (Table 3 and Fig. 2). When clades collected in Japan (JAA and JAB) were compared, the methylation patterns almost fully overlapped (Fig. 2a). However, when colonies collected in New Zealand (NZA) and Japan (JAA and JAB) were compared, they were partially differentiated, regardless of clade (Fig. 2b,c). MSL differences between NZA and JAA were due to colonies collected from Pelorus (PelA) and Whangamata (WhaA) (New Zealand) being different to colonies collected from Japan (Shizugawa (ShiA) and Ise (IseA)). In contrast, there were no significant MSL differences between colonies collected from Queen Charlotte, New Zealand (QueA) and clade A colonies collected from Japan (Supplementary Table S3, MSL). For NML, there were significant differences between NZA and JAA at all sites, except for QueA and ShiA (Supplementary Table S3, NML). MSL differences between NZA and JAB were due to colonies collected from Izu (IzuB) and Sagami (SagB) being different to colonies from all sites in New Zealand (PelA, WhaA, and QueA) (Supplementary Table S4, MSL). For NML, there were significant differences between JAB and NZA colonies from all sites, with the exception of WhaA and QueA (NZ) which were not significantly different from ShiB (JA) (Supplementary Table S4, NML).

Principal coordinates analyses (PCoA) showing: (a) non-significant methylation (MSL) differences between Didemnum vexillum colonies from Japan clade A (JAA) and Japan clade B (JAB), (b) significant MSL differences between colonies from Japan clade A (JAA) and New Zealand clade A (NZA) and (c) significant MSL differences between colonies from Japan clade B (JAB) and New Zealand clade A (NZA). The first two coordinates (C1 and C2) are shown with the percentage of variance explained by them. Points in each group cloud represent individuals from different groups. Country and clade labels show the centroid for the points cloud in each group. Ellipses represent average dispersion of those points around their centre61. AMOVA tests for MSL differences are shown in Table 3. NZA (n = 35), JAA (n = 16) and JAB (n = 23).
There were no significant MSL or NML differences between colonies collected from different sites within Japan (MSL ɸST = 0.01559, p = 0.1072, Supplementary Fig. S5; NML ɸST = −0.01433, p = 0.7505). In contrast, there were significant differences between all sites in New Zealand (MSL, ɸST = 0.05517, p = < 0.001, Supplementary Fig. S5; NML ɸST = 0.07178, p = 0.0033, Supplementary Table S6). NML and MSL were significantly positively correlated (Mantel test; JAA, r = 0.725, p = <0.001; JAB, r = 0.368, p = <0.001; NZA, r = 0.746, p = <0.001).
DNA methylation and genetic diversity
MSL diversity was similar in all three populations (JAA, JAB, and NZA), and DNA methylation diversity (MSL) was significantly higher than genetic diversity (NML) (Fig. 3a and Supplementary Table S7). In contrast, there were significant differences in NML diversity between populations (Supplementary Table S8). NML diversity was significantly higher in JAA compared to both JAB and NZA (Supplementary Table S9).

Bar graphs showing MSL and NML diversity and global DNA methylation levels in Didemnum vexillum colonies collected from Japan clade A (JAA, n = 16), Japan clade B (JAB, n = 23) and New Zealand clade A (NZA, n = 35): (a) DNA methylation (MSL) and genetic (NML) diversity; (b) methylation-susceptible loci classified as: unmethylated (Type I), internal methylation (Type II), hemimethylation (Type III), and hypermethylation or absence of restriction site (Type IV); and (c) global methylation levels measured as the proportion of methylated loci (Type II + III/scorable loci (Types I + II + III)) following Nicotra et al.67 and Ardura et al.51. Significant differences between treatments are denoted by different letters (p < 0.05).
DNA methylation levels
The proportion of MSL classified as unmethylated, internally methylated, hemimethylated or hypermethylated for JAA, JAB, and NZA is shown in Fig. 3b. In all three groups, a greater proportion of Type IV methylation (hypermethylation) was detected compared to unmethylated or methylated loci. However, as Type IV methylation can also be due to absence of the restriction target (mutation) it is considered uninformative and is not used to calculate the proportion of methylated loci51,60. For the proportion of methylated loci, recently introduced New Zealand colonies had significantly lower methylation levels than colonies collected from within the native region (Japan) (p < 0.05, Supplementary Table S10). For JAA, JAB and NZA, the methylated loci (Types II and III) were 49%, 53% and 41% of the scoreable loci respectively (Fig. 3c). There were no significant differences in the proportion of methylated loci between JAA and JAB.
Discussion
Comparing closely related clades, only one of which has become invasive, provides a unique opportunity to identify genotypes and phenotypes that might be associated with invasion success26. Significant global DNA methylation (MSL) and genetic (NML) differences were detected between Didemnum vexillum colonies introduced to New Zealand, and both clades in the native range (Japan). These differences were evident when looking at both differentiation and global DNA methylation levels, which will be discussed in detail below. In contrast, we found no significant differences in MSL or NML patterns between D. vexillum clades A and B collected from within the native range, despite the predicted divergence of D. vexillum COI clade lineages being between 12,000 and 2.6 million years ago18. A recent study of genome-wide DNA methylation variation within and between five house sparrow subspecies in their native range (Middle East, Iranian plateau) also found the majority of subspecies were not significantly differentiated, despite also inhabiting different environmental conditions (e.g., arid desert, humid forest, tropical coast). Most of the observed DNA methylation variation could be attributed to differences within subspecies rather than among subspecies, and appeared to follow a similar pattern to genetic differentiation among populations, which was also very low68. In contrast, a recent study of global DNA methylation in co-occurring subspecies of the facultatively clonal plant, P. australis, found significant differentiation of DNA methylation patterns between subspecies that inhabit the same environment69. However, these subspecies have very different life histories; one subspecies is native and the other introduced. Therefore, epigenetic differences between these subspecies might be associated with changes induced by invasion history69. In D. vexillum, the level of COI sequence variation between clades A and B (as estimated by percentage of polymorphic sites) is low compared to values reported for other ascidian species18, and colonies from the two clades are morphologically identical and occur in sympatry at several sites in Japan37. The lack of global DNA methylation differentiation between clades A and B in Japan might, therefore, represent similar genetic and life history backgrounds.
Clade A colonies collected from Japan had significantly higher levels of genetic (NML) diversity compared to the two other populations studied (NZA and JAB). While differences in genetic variation do not necessarily reflect differences in quantitative traits, it has been theorized that a high level of standing genetic variation will increase adaptive and evolutionary potential, which may facilitate the colonisation of new environments70. High levels of genetic variation within the native range (as found in JAA) may facilitate the spread of invasive species like D. vexillum. In the copepod, Eurytemora affinis, two clades have overlapping distributions in several estuaries and salt marshes, but one clade has invaded freshwater habitats while the other remains restricted to the native range25. The invasive clade was found to have higher DNA nucleotide diversity than the non-invasive clade within the native range25, and has evolved increased haemolymph osmolality following the invasion of freshwater habitats71,72. Comparisons of geographically restricted and widespread species also suggest that higher levels of genetic variation in ecologically important traits contribute to larger niche breadth73,74. While it is possible that future sampling might reveal that clade B has also invaded other regions, the rate of discovery suggests that if it is present, it is rare. Clade A has been identified in five broad sampling areas (New Zealand, Japan, West Coast North America, East Coast North America and Europe), whereas clade B haplotypes have been exclusively amplified from Japan18. It is also possible that clade A is only invasive ‘by chance’. Clade A and clade B haplotypes are not equally abundant nor equally distributed among sites in Japan41, and the probability of a haplotype being transported during an invasion event is proportional to the frequency of that haplotype in the native population where the transport vector originated75. This effect would be amplified if introductions to new regions were secondary invasion events rather than multiple primary introductions originating from the native range41.
Genetic variation based on NML should be interpreted with caution64, but lower genetic diversity in NZA is consistent with previous studies that have concluded that introduced populations of D. vexillum have experienced a reduction in genetic diversity, indicating a founder effect18. If lower levels of genetic diversity are in fact limiting adaptive potential, alternative mechanisms for increasing phenotypic variation, such as epigenetic variation, might be especially important during the introduction and establishment phases. Despite differences in genetic diversity, there were no significant differences in DNA methylation diversity between NZA, JAA, and JAB. A similar result was found when DNA methylation diversity was compared across house sparrow populations with varying invasion histories53. Combined, these results support the idea that epigenetic variation could preserve the ability of invasive populations to adapt following reductions in genetic diversity that can be associated with colonisation53,54, although the functional significance of epigenetic diversity remains unknown.
DNA methylation patterns were significantly differentiated between sites in the introduced range of D. vexillum (New Zealand), but not in the native range (Japan). Recent studies of invasive populations have found DNA methylation differences between environments that may play a role in adaptation to different habitats49,76,77. In the present study, comparisons of DNA methylation patterns between sites with similar or different environmental stress (temperature and salinity) scores indicate that DNA methylation patterns are not driven by environmental variations alone. However, it remains possible that environmental stress is an important driver of epigenetic structure in the invasive New Zealand population. Random DNA methylation changes have been shown to be more likely following exposure to environmental stressors78. Invasive populations may accumulate novel epigenetic marks more quickly than populations in the native range if the invasion process can be considered a stress51, especially if combined with additional environmental challenges60. When unpredictable environmental conditions are encountered, epigenetically variable offspring will be favored, serving as a ‘bet-hedging’ strategy if some individuals are suited to the invaded habitat79,80,81,82. Additionally, it is possible that the founding genotypes that successfully established in New Zealand exhibit a higher propensity for methylation plasticity due to environmental filtering. Site-specific differentiation in the introduced populations could be due to the introduction and spread of epigenetic variants to different sites which, like genetic drift, is likely to have a large effect in small populations. However, these processes rely on a level of stability and heritability of methylation. It is not yet clear how much DNA methylation variation underlies selectable phenotypic variation, or how it responds to evolutionary forces83.
While we found DNA methylation differences between colonies from New Zealand and Japan, and between sites within New Zealand, these were associated with significant genetic (NML) differences. Invasive populations can include a different admixture of genes or genotypes compared to those in the native range, due to founder effects or multiple introductions from different source populations. Therefore, DNA methylation differences might reflect genetic differences between the native and introduced populations and between sites within the introduced range. Invasive populations can also be exposed to variable selection pressures across the newly colonised range, and have a faster evolutionary response to selection79. Models have shown that during early adaptation to new environments, epigenetic shifts could provide a “stepping stone”, facilitating the persistence of populations and allowing for the accumulation of neutral genetic variation84. Some epigenetic variation might become assimilated, meaning plastic responses or phenotypes are epigenetically encoded first, followed by genetic evolution85,86. Stress-induced reductions in DNA methylation can also affect genetic variation by increasing transposition events87. The results from this study cannot establish whether DNA methylation differences between sites are pure, obligatory or facilitated, but all three types are likely to play a role in range expansions23,69. Selection on epigenetic variation, in addition to genetic variation, can allow populations to adapt faster than selection on genetic variation alone84.
The introduced New Zealand population had lower methylation levels compared to populations in the native range. Reduced levels of DNA methylation have been suggested to be a common ‘signature of invasion’, increasing phenotypic plasticity during the expansive phase of invasion when it is most required51,60. However, the mechanistic link between levels of methylation, environmental stress, biological invasions and adaptation to new environments is not well understood. Few studies have reported methylation levels in aquatic invertebrates and, of those that have, it appears that increased genome-wide methylation is the most common response to environmental stress47,88,89,90,91. Evidence suggests that high levels of gene body methylation can reduce spurious transcription and transcriptional noise (variability of gene expression levels) and may be a defense mechanism to combat the damaging effects of environmental challenges88,92. In contrast, genes with reduced levels of germline DNA methylation have been linked with increased phenotypic plasticity via associations with alternative transcription start sites, exon skipping, alternative splicing and transient methylation52,92,93. Hypomethylation has also been associated with increased activity of transposable elements in several species, which can introduce phenotypic variation and may facilitate adaptation to new environments87,94. While stable and accurate gene expression could be beneficial within the native range of a species, where genetic diversity is generally high and environmental conditions are predictable from one generation to the next, following invasion, noisy or unstable genomes might be especially beneficial. If environmental stress is associated with higher levels of DNA methylation, it should be considered that lower levels of methylation might be associated with a reduction in stress. However, there is little evidence for this at present. One study has compared invasive populations of pygmy mussels (Xenostrobus secures) from two sites with apparently varying levels of environmental stress (an international port and a protected lagoon)51. It was expected that the population from the less stressful environment (the lagoon) would be less methylated, however it was found that methylation levels between sites were similar. In this case, it was suggested that the ‘invasive signature’ might be overriding any differences associated with the environment. It should be noted that while Type IV methylation (hypermethylation) is considered uninformative because it could be due to target absence, a large amount could also be due to methylation polymorphisms, so could also be important when comparing methylation levels in invasive and native populations.
Conclusion
This is the first study investigating DNA methylation differences between closely related invertebrate clades, one of which has become invasive and one of which has not. We found further evidence for a ‘signature of invasion’ associated with a recent population expansion51, with introduced populations having distinct DNA methylation patterns and levels compared to populations in the native range. These results suggest that genetic diversity might facilitate the spread of invasive populations outside the native range, while DNA methylation might be especially important in introduced populations when genetic diversity is reduced. The MSAP method used in this study only allows for analysis of global genomic patterns, so there may be important functional methylation differences between the invasive and non-invasive clades that were not detected here. Our future research will focus on examining the functional properties and stability of DNA methylation changes in response to different environmental conditions, which will aid our understanding of ecological adaptation and evolutionary dynamics, including the epigenetic basis of invasiveness.
Data Availability
The datasets analysed during the current study are available from the corresponding author on reasonable request.
Change history
13 December 2019
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Mooney, H. A. & Cleland, E. E. The evolutionary impact of invasive species. Proc. Natl. Acad. Sci. USA 98, 5446–5451 (2001).
Bax, N., Williamson, A., Aguero, M., Gonzalez, E. & Geeves, W. Marine invasive alien species: a threat to global biodiversity. Mar. Policy 27, 313–323 (2003).
Pimentel, D., Zuniga, R. & Morrison, D. Update on the environmental and economic costs associated with alien-invasive species in the United States. Ecol. Econ. 52, 273–288 (2005).
Mack, R. N. et al. Biotic invasions: causes, epidemiology, global consequences, and control. Ecol. Appl. 10, 689–710 (2000).
Estoup, A. & Guillemaud, T. Reconstructing routes of invasion using genetic data: why, how and so what? Mol. Ecol. 19, 4113–4130 (2010).
Lee, C. E. Evolutionary genetics of invasive species. Trends Ecol. Evol. 17, 386–391 (2002).
Blanchet, S. The use of molecular tools in invasion biology: an emphasis on freshwater ecosystems. Fish. Manag. Ecol. 19, 120–132 (2012).
Ficetola, G. F., Bonin, A. & Miaud, C. Population genetics reveals origin and number of founders in a biological invasion. Mol. Ecol. 17, 773–782 (2008).
Ficetola, G. F., Miaud, C., Pompanon, F. & Taberlet, P. Species detection using environmental DNA from water samples. Biol. Lett. 4, 423–425 (2008).
Goldstien, S. J. et al. Global phylogeography of the widely introduced North West Pacific ascidian Styela clava. PLoS One 6, e16755 (2011).
Smith, K. F., Thia, J., Gemmill, C. E., Cary, S. C. & Fidler, A. E. Barcoding of the cytochrome oxidase I (COI) indicates a recent introduction of Ciona savignyi into New Zealand and provides a rapid method for Ciona species discrimination. Aquat. Invasions 7, 305–313 (2012).
Sakai, A. K. et al. The population biology of invasive species. Annu. Rev. Ecol. Syst. 32, 305–332 (2001).
Lavergne, S. & Molofsky, J. Increased genetic variation and evolutionary potential drive the success of an invasive grass. Proc. Natl. Acad. Sci. USA 104, 3883–3888 (2007).
Crawford, K. & Whitney, K. Population genetic diversity influences colonization success. Mol. Ecol. 19, 1253–1263 (2010).
Geng, Y. P. et al. Phenotypic plasticity rather than locally adapted ecotypes allows the invasive alligator weed to colonize a wide range of habitats. Biol. Invasions 9, 245–256 (2007).
Mergeay, J., Verschuren, D. & De Meester, L. Invasion of an asexual American water flea clone throughout Africa and rapid displacement of a native sibling species. Proc. R. Soc. Lond. B Biol. Sci. 273, 2839–2844 (2006).
Tsutsui, N. D., Suarez, A. V., Holway, D. A. & Case, T. J. Reduced genetic variation and the success of an invasive species. Proc. Natl. Acad. Sci. USA 97, 5948–5953 (2000).
Smith, K. F. et al. Increased inter-colony fusion rates are associated with reduced COI haplotype diversity in an invasive colonial ascidian Didemnum vexillum. PloS one 7, e30473 (2012).
Dlugosch, K. M. & Parker, I. M. Founding events in species invasions: genetic variation, adaptive evolution, and the role of multiple introductions. Mol. Ecol. 17, 431–449 (2008).
Roy, H., Handley, L.-J. L., Schönrogge, K., Poland, R. & Purse, B. Can the enemy release hypothesis explain the success of invasive alien predators and parasitoids? BioControl 56, 451–468 (2011).
Pérez, J. E., Nirchio, M., Alfonsi, C. & Muñoz, C. The biology of invasions: the genetic adaptation paradox. Biol. Invasions 8, 1115–1121 (2006).
Prentis, P. J., Wilson, J. R. U., Dormontt, E. E., Richardson, D. M. & Lowe, A. J. Adaptive evolution in invasive species. Trends Plant Sci. 13, 288–294 (2008).
Hawes, N. A. et al. Understanding the role of DNA methylation in successful biological invasions: a review. Biol. Invasions, 1–16 (2018).
Bossdorf, O., Richards, C. L. & Pigliucci, M. Epigenetics for ecologists. Ecol. Lett. 11, 106–115 (2008).
Winkler, G., Dodson, J. J. & Lee, C. E. Heterogeneity within the native range: population genetic analyses of sympatric invasive and noninvasive clades of the freshwater invading copepod Eurytemora affinis. Mol. Ecol. 17, 415–430 (2008).
Smith, K. F., Abbott, C. L., Saito, Y. & Fidler, A. E. Comparison of whole mitochondrial genome sequences from two clades of the invasive ascidian, Didemnum vexillum. Mar. Genomics 19, 75–83 (2015).
Zhan, A. et al. Complex genetic patterns in closely related colonizing invasive species. Ecol. Evol. 2, 1331–1346 (2012).
Bock, D. G., MacIsaac, H. J. & Cristescu, M. E. Multilocus genetic analyses differentiate between widespread and spatially restricted cryptic species in a model ascidian. Proc. R. Soc. Lond. B Biol. Sci. 279, 2377–2385 (2012).
Eunmi Lee, C. Global phylogeography of a cryptic copepod species complex and reproductive isolation between genetically proximate “populations”. Evolution 54, 2014–2027 (2000).
Tsutsui, N. D., Suarez, A. V., Holway, D. A. & Case, T. J. Relationships among native and introduced populations of the Argentine ant (Linepithema humile) and the source of introduced populations. Mol. Ecol. 10, 2151–2161 (2001).
Coffey, B. Potentially invasive compound ascidian, Whangamata Harbour. Whangamata: Brian T. Coffey and Associates Limited (2001).
Kott, P. A complex didemnid ascidian from Whangamata. New Zealand. J. Mar. Biol. Assoc. U. K. 82, 625–628 (2002).
Bullard, S. G. et al. The colonial ascidian Didemnum sp. A: current distribution, basic biology and potential threat to marine communities of the northeast and west coasts of North America. J. Exp. Mar. Biol. Ecol. 342, 99–108 (2007).
Gittenberger, A. Recent population expansions of non-native ascidians in The Netherlands. J. Exp. Mar. Biol. Ecol. 342, 122–126 (2007).
Minchin, D. & Sides, E. Appearance of a cryptogenic tunicate, a Didemnum sp. fouling marina pontoons and leisure craft in Ireland. Aquat. Invasions 1, 143–147 (2006).
Griffith, K. et al. First records in Great Britain of the invasive colonial ascidian Didemnum vexillum Kott, 2002. Aquat. Invasions 4, 581–590 (2009).
Lambert, G., Locke, A. & Carman, M. Adventures of a sea squirt sleuth: unraveling the identity of Didemnum vexillum, a global ascidian invader. Aquat. Invasions 4, 5–28 (2009).
Herborg, L. M., O’Hara, P. & Therriault, T. W. Forecasting the potential distribution of the invasive tunicate Didemnum vexillum. J. Appl. Ecol. 46, 64–72 (2009).
Coutts, A. D. & Forrest, B. M. Development and application of tools for incursion response: lessons learned from the management of the fouling pest Didemnum vexillum. J. Exp. Mar. Biol. Ecol. 342, 154–162 (2007).
Stefaniak, L. et al. Genetic conspecificity of the worldwide populations of Didemnum vexillum Kott, 2002. Aquat. Invasions 4, 29–44 (2009).
Stefaniak, L. et al. Determining the native region of the putatively invasive ascidian Didemnum vexillum Kott, 2002. J. Exp. Mar. Biol. Ecol. 422, 64–71 (2012).
Nydam, M. L. & Harrison, R. G. Polymorphism and divergence within the ascidian genus Ciona. Mol. Phylogenet. Evol. 56, 718–726 (2010).
Nydam, M. L. & Harrison, R. G. Introgression despite substantial divergence in a broadcast spawning marine invertebrate. Evolution 65, 429–442 (2011).
Caputi, L. et al. Cryptic speciation in a model invertebrate chordate. Proc. Natl. Acad. Sci. USA 104, 9364–9369 (2007).
Rondon, R. et al. Effects of a parental exposure to diuron on Pacific oyster spat methylome. Environ. Epigenet. 3 (2017).
Hawes, N. A. et al. Effects of temperature and salinity stress on DNA methylation in a highly invasive marine invertebrate, the colonial ascidian Didemnum vexillum. PeerJ. 6, e5003 (2018).
Putnam, H. M., Davidson, J. M. & Gates, R. D. Ocean acidification influences host DNA methylation and phenotypic plasticity in environmentally susceptible corals. Evol. Appl. 9, 1165–1178 (2016).
Gao, L., Geng, Y., Li, B. O., Chen, J. & Yang, J. I. Genome‐wide DNA methylation alterations of Alternanthera philoxeroides in natural and manipulated habitats: implications for epigenetic regulation of rapid responses to environmental fluctuation and phenotypic variation. Plant Cell Environ. 33, 1820–1827 (2010).
Pu, C. & Zhan, A. Epigenetic divergence of key genes associated with water temperature and salinity in a highly invasive model ascidian. Biol. Invasions, 1–14 (2017).
Chwedorzewska, K. & Bednarek, P. Genetic and epigenetic variation in a cosmopolitan grass Poa annua from Antarctic and Polish populations. Pol. Polar Res. 33, 63–80 (2012).
Ardura, A., Zaiko, A., Morán, P., Planes, S. & Garcia-Vazquez, E. Epigenetic signatures of invasive status in populations of marine invertebrates. Sci. Rep. 7, 42193 (2017).
Roberts, S. B. & Gavery, M. R. Is there a relationship between DNA methylation and phenotypic plasticity in invertebrates? Front. Physiol. 2, 116 (2011).
Schrey, A. W. et al. Epigenetic variation may compensate for decreased genetic variation with introductions: a case study using house sparrows (Passer domesticus) on two continents. Genet. Res. Int. 2012, 979751 (2012).
Liebl, A. L., Schrey, A. W., Richards, C. L. & Martin, L. B. Patterns of DNA methylation throughout a range expansion of an introduced songbird. Integr. Comp. Biol. 53, 351–358 (2013).
Coutts, A. D. A biosecurity investigation of a barge in the Marlborough Sounds. Cawthron Report 744, 86 (2002).
Zerebecki, R. A. & Sorte, C. J. B. Temperature tolerance and stress proteins as mechanisms of invasive species success. PLoS One 6, e14806 (2011).
Lambert, G. Ecology and natural history of the protochordates. Can. J. Zool. 83, 34–50 (2005).
Gröner, F., Lenz, M., Wahl, M. & Jenkins, S. R. Stress resistance in two colonial ascidians from the Irish Sea: The recent invader Didemnum vexillum is more tolerant to low salinity than the cosmopolitan Diplosoma listerianum. J. Exp. Mar. Biol. Ecol. 409, 48–52 (2011).
Bullard, S. G. & Whitlatch, R. B. In situ growth of the colonial ascidian Didemnum vexillum under different environmental conditions. Aquat. Invasions 4, 275–278 (2009).
Ardura, A., Clusa, L., Zaiko, A., Garcia-Vazquez, E. & Miralles, L. Stress related epigenetic changes may explain opportunistic success in biological invasions in Antipode mussels. Sci. Rep. 8, 10793 (2018).
Pérez‐Figueroa, A. msap: a tool for the statistical analysis of methylation‐sensitive amplified polymorphism data. Mol. Ecol. Resour. 13, 522–527 (2013).
R Core Team. R: A Language and Environment for Statistical Computing. (R Foundation for Statistical Computing, Vienna, Austria) (2016).
Herrera, C. M. & Bazaga, P. Epigenetic differentiation and relationship to adaptive genetic divergence in discrete populations of the violet Viola cazorlensis. New Phytol. 187, 867–876 (2010).
Watson, R., Baldanzi, S., Pérez-Figueroa, A., Gouws, G. & Porri, F. J. M. B. Morphological and epigenetic variation in mussels from contrasting environments. 165, 50 (2018).
Schulz, B., Eckstein, R. L. & Durka, W. Scoring and analysis of methylation‐sensitive amplification polymorphisms for epigenetic population studies. Mol. Ecol. Resour. 13, 642–653 (2013).
Excoffier, L., Smouse, P. E. & Quattro, J. M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491 (1992).
Nicotra, A. B. et al. Adaptive plasticity and epigenetic variation in response to warming in an Alpine plant. Ecol. Evol. 5, 634–647 (2015).
Riyahi, S. et al. Natural epigenetic variation within and among six subspecies of the house sparrow. Passer domesticus. 220, 4016–4023 (2017).
Spens, A. E. & Douhovnikoff, V. Epigenetic variation within Phragmites australis among lineages, genotypes, and ramets. Biol. Invasions 18, 2457–2462 (2016).
Barrett, R. D. & Schluter, D. Adaptation from standing genetic variation. Trends Ecol. Evol. 23, 38–44 (2008).
Lee, C. E., Kiergaard, M., Gelembiuk, G. W., Eads, B. D. & Posavi, M. Pumping ions: rapid parallel evolution of ionic regulation following habitat invasions. Evolution 65, 2229–2244 (2011).
Lee, C. E., Posavi, M. & Charmantier, G. Rapid evolution of body fluid regulation following independent invasions into freshwater habitats. J. Evol. Biol. 25, 625–633 (2012).
Sheth, S. N. & Angert, A. L. The evolution of environmental tolerance and range size: a comparison of geographically restricted and widespread Mimulus. Evolution 68, 2917–2931 (2014).
Kellermann, V., van Heerwaarden, B., Sgrò, C. M. & Hoffmann, A. A. Fundamental evolutionary limits in ecological traits drive Drosophila species distributions. Science 325, 1244–1246 (2009).
Nei, M., Maruyama, T. & Chakraborty, R. The bottleneck effect and genetic variability in populations. Evolution 29, 1–10 (1975).
Thorson, J. L. M. et al. Epigenetics and adaptive phenotypic variation between habitats in an asexual snail. Sci. Rep. 7, 14139 (2017).
Ni, P. et al. Methylation divergence of invasive Ciona ascidians: Significant population structure and local environmental influence. Ecol. Evol. 8, 10272–10287 (2018).
Verhoeven, K. J. F., Jansen, J. J., van Dijk, P. J., Biere, A. & Stress‐induced DNA methylation changes and their heritability in asexual dandelions. New Phytol. 185, 1108–1118 (2010).
Colautti, R. I. & Lau, J. A. Contemporary evolution during invasion: evidence for differentiation, natural selection, and local adaptation. Mol. Ecol. 24, 1999–2017 (2015).
Herman, J. J., Spencer, H. G., Donohue, K. & Sultan, S. E. How stable ‘should’epigenetic modifications be? Insights from adaptive plasticity and bet hedging. Evolution 68, 632–643 (2014).
Jablonka, E. et al. The adaptive advantage of phenotypic memory in changing environments. Philos. Trans. R. Soc. Lond. B Biol. Sci. 350, 133–141 (1995).
O’Dea, R. E., Noble, D. W., Johnson, S. L., Hesselson, D. & Nakagawa, S. The role of non-genetic inheritance in evolutionary rescue: epigenetic buffering, heritable bet hedging and epigenetic traps. Environ. Epigenet. 2, dvv014 (2016).
Kronholm, I. & Collins, S. Epigenetic mutations can both help and hinder adaptive evolution. Mol. Ecol. 25, 1856–1868 (2016).
Klironomos, F. D., Berg, J. & Collins, S. How epigenetic mutations can affect genetic evolution: model and mechanism. BioEssays 35, 571–578 (2013).
Pigliucci, M., Murren, C. J. & Schlichting, C. D. Phenotypic plasticity and evolution by genetic assimilation. J. Exp. Biol. 209, 2362–2367 (2006).
Smith, G. & Ritchie, M. G. How might epigenetics contribute to ecological speciation? Curr. Zool. 59, 686–696 (2013).
Stapley, J., Santure, A. W. & Dennis, S. R. Transposable elements as agents of rapid adaptation may explain the genetic paradox of invasive species. Mol. Ecol. 24, 2241–2252 (2015).
Liew, Y. J. et al. Epigenome-associated phenotypic acclimatization to ocean acidification in a reef-building coral. Sci. Adv. 4, eaar8028 (2018).
Marsh, A. G. & Pasqualone, A. A. DNA methylation and temperature stress in an Antarctic polychaete, Spiophanes tcherniai. Front. Physiol. 5, 173–181 (2014).
Lee, S.-W. et al. Integrated approach of eco-epigenetics and eco-metabolomics on the stress response of bisphenol-A exposure in the aquatic midge Chironomus riparius. Ecotoxicol. Environ. Saf. 163, 111–116 (2018).
Cribiu, P. et al. Natural variability and modulation by environmental stressors of global genomic cytosine methylation levels in a freshwater crustacean, Gammarus fossarum. Aquat. Toxicol. (Amst.) 205, 11–18 (2018).
Dixon, G. B., Bay, L. K. & Matz, M. V. Bimodal signatures of germline methylation are linked with gene expression plasticity in the coral Acropora millepora. BMC Genomics 15, 1109 (2014).
Dimond, J. L. & Roberts, S. B. Germline DNA methylation in reef corals: patterns and potential roles in response to environmental change. Mol. Ecol. 25, 1895–1904 (2015).
Gavery, M. R. & Roberts, S. B. A context dependent role for DNA methylation in bivalves. Brief. Funct. Genomics 13, 217–222 (2014).
Takahashi, D., Endo, H., Minegishi, Y., Gomi, Y. & Kaneko, K. Wind-and density-driven circulation in a bay on the Sanriku ria coast, Japan: study of Shizugawa Bay facing the Pacific Ocean. J. Oceanogr. 74, 81–100 (2018).
Acknowledgements
This work was supported by the Marsden Fund of the Royal Society of New Zealand (contract number CAW1401). The authors would like to thank staff at Shimoda Marine Research Center (University of Tsukuba), the Shizugawa Nature Center, Misaki Marine Biological Station (University of Tokyo), Sugashima Marine Biological Laboratory (Nagoya University), and the Asamushi Research Center for Marine Biology (Tohoku University) for assistance with sample collection in Japan, and Aaron Pannell (Marlborough Mussel Company), Brian Coffey (Brian T. Coffey and Associates Limited), and Drs Barrie Forrest and Lauren Fletcher (Cawthron Institute) for assistance with sample collection in New Zealand. Thank you to Eric Goodwin (Cawthron Institute) for help with R coding, Jacqui Stuart for assistance with Figure 2 (Cawthron Institute), and Dr. Andrés Pérez-Figueroa (University of Vigo) and Prof. Carlos M. Herrera (Estacion Biologica de Doñana) for helpful discussions about NML scoring.
Author information
Authors and Affiliations
Contributions
K.S. and A.F. collected the samples; N.H. and A.A. performed the laboratory work; N.H., analysed the data, N.H. wrote the first drafts of the manuscript; N.H., K.S., L.T., X.P., B.D., contributed substantially to revisions; A.F. and K.S. secured the funding; all authors approved the final draft.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hawes, N.A., Amadoru, A., Tremblay, L.A. et al. Epigenetic patterns associated with an ascidian invasion: a comparison of closely related clades in their native and introduced ranges. Sci Rep 9, 14275 (2019). https://doi.org/10.1038/s41598-019-49813-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49813-7
This article is cited by
-
Prokaryotic symbiont communities in three ascidian species introduced in both Ireland and New Zealand
Environmental Science and Pollution Research (2023)
-
Studying phenotypic variation and DNA methylation across development, ecology and evolution in the clonal marbled crayfish: a paradigm for investigating epigenotype-phenotype relationships in macro-invertebrates
The Science of Nature (2022)
-
Genetic diversity and relatedness in aquaculture and marina populations of the invasive tunicate Didemnum vexillum in the British Isles
Biological Invasions (2021)
-
Epigenetic variation in animal populations: Sources, extent, phenotypic implications, and ecological and evolutionary relevance
Journal of Biosciences (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
