
Abstract
A transcriptome analysis of G. pallida juveniles collected from S. tuberosum or S. sisymbriifolium 24 h post infestation was performed to provide insights into the parasitic process of this nematode. A total of 41 G. pallida genes were found to be significantly differentially expressed when parasitizing the two plant species. Among this set, 12 were overexpressed when G. pallida was parasitizing S. tuberosum and 29 were overexpressed when parasitizing S. sisymbriifolium. Out of the 12 genes, three code for secretory proteins; one is homologous to effector gene Rbp-4, the second is an uncharacterized protein with a signal peptide sequence, and the third is an ortholog of a Globodera rostochiensis effector belonging to the 1106 effector family. Other overexpressed genes from G. pallida when parasitizing S. tuberosum were either unknown, associated with a stress or defense response, or associated with sex differentiation. Effector genes namely Eng-1, Cathepsin S-like cysteine protease, cellulase, and two unknown genes with secretory characteristics were over expressed when G. pallida was parasitizing S. sisymbriifolium relative to expression from S. tuberosum. Our findings provide insight into gene regulation of G. pallida while infecting either the trap crop S. sisymbriifolium or the susceptible host, S. tuberosum.
Similar content being viewed by others


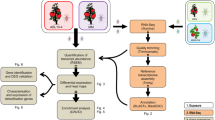
Introduction
Cyst nematodes have been identified as some of the greatest threat to agricultural crops worldwide1. The potato cyst nematodes (PCN) Globodera pallida Stone (1973) and G. rostochiensis Wollenweber, (1923) Skarbilovich, (1959) are found in potato production areas and are of regulatory significance throughout the world1,2. In the United States, Globodera pallida was first detected in 2006 in a potato processing facility in Idaho. Currently, there are no commercially acceptable potato varieties with G. pallida resistance suitable for production in the northwestern United States3.
Globodera pallida possesses highly sophisticated machinery to locate and parasitize its host plant4,5,6. Development and reproduction of potato cyst nematodes relies on the establishment and maintenance of a syncytium inside the host root. Once a feeding site is established, the nematode engages in a sustained biotrophic interaction with its host. Effectors, small proteins produced in the nematode’s esophageal glands, are delivered into plant cells through the stylet, and mediate this biotrophic interaction4,7. Effector proteins manipulate the host cell by modulating a variety of cellular processes such as suppression of host defense or stress responses and cause significant transcriptional re-programming in the host cell nucleus8. The early infection stages of the nematode life cycle are crucial in deciding the fate of the nematode. The ability of the nematode to overcome hostile conditions presented by the plant determines whether or not the nematode completes its life cycle. In a resistant host, the plant has acquired the capacity to recognize the parasite (often through the detection of specific effectors) to prevent the establishment of a feeding site. One response is the initiation of a hypersensitive response (HR) by the plant which triggers programmed cell death in infected cells9,10,11, but other processes may also be at work.
Solanum sisymbriifolium, a non-tuber bearing solanaceous plant, was identified as a promising trap crop for the control of G. pallida12,13,14. Solanum sisymbriifolium stimulates hatch of juveniles from the cysts, while preventing subsequent reproduction of G. pallida15,16,17. Microscopic observation revealed cell necrosis around the infective J2s in S. sisymbriifolium roots due to an HR as early as 2–4 days post infestation (dpi)15, and several dead nematodes were observed at this time point in S. sisymbriifolium roots. Due to unfavorable conditions in resistant plants, the nematode may experience stress and starvation. Nematodes are able to sense the physiological response of the plant and activate oxidative stress response pathways18. To evade the plant defense responses plant parasitic nematodes express an array of proteins such as antioxidants, catalases, peroxidases, heat shock proteins and body morphology and structural proteins19,20.
In this study, G. pallida juveniles that were parasitizing the roots of the susceptible host S. tuberosum and the resistant trap crop S. sisymbriifolium were extracted at 24 hours post infestation (hpi) and the transcriptome of these nematodes were analyzed using RNAseq methods. The transcriptome of G. pallida was studied at early stages of parasitism of because of the rapid defense response observed in S. sisymbriifolium15. This study has revealed several potentially important differences in the way the Globodera transcriptome changes according to the suitability of the plant as a host.
Results
Extraction of RNA from low numbers of nematodes
Globodera pallida juveniles were extracted from S. tuberosum or S. sisymbriifolium roots 24 h post infestation and a comparative transcriptome analysis was performed using RNAseq. Because an average of only 30 individual nematodes were isolated from S. sisymbriifolium roots 24 hours post infestation, the number of nematodes were normalized to 30 individuals from both the plant species for RNA isolation. Even though the samples were derived from just 30 nematodes collected from the roots of S. sisymbriifolium and S. tuberosum, sufficiently high-quality RNA (RNA quality number (RQN) > 8) was obtained for library preparation and sequencing.
Sequencing quality analysis and mapping
Sequencing of three biological replicates of G. pallida extracted from S. tuberosum and S. sisymbriifolium generated a total of 402,702,714 paired-end reads (2 × 100 bp) at an average of 67,117,119 per sample. The average % mapping of sequence reads to the G. pallida reference genome was 19.21% or 18.06%, when the nematodes were parasitizing potato or S. sisymbriifolium, respectively. All the mapped RNA seq reads have been submitted to the National Center for Biotechnology Information (NCBI) with Sequence read archive (SRA) accession no. SRP159274.
Differential gene expression of G. pallida parasitizing susceptible S. tuberosum or resistant S. sisymbriifolium
For analysis of differential gene expression, only the reads that mapped to the reference G. pallida genome were considered. A total of 29,551 transcripts were identified across all the samples. Expression analyses identified 41 differentially expressed genes (DEGs) (P ≤ 0.01) (Fig. 1). Out of these 41 DEGs, 22 were expressed only when G. pallida was parasitizing S. sisymbriifolium and 9 were expressed only when parasitizing S. tuberosum. The remaining 10 genes among the 41 were expressed in both the cases with significant difference in expression values. Blast2GO analysis performed on these 41 DEGs returned 39% with annotations (see Supplementary Fig. S1). Gene ontology terms assigned to these transcripts are presented in Supplementary Fig. S2.

Heatmap showing the significant differentially expressed Globodera pallida genes when parasitizing susceptible Solanum tuberosum and resistant Solanum sisymbriifolium at 24 h post infestation. Each row represents a gene and column represents different plant species. Labels on the left show the transcript ID, followed by gene ID and closest gene name according to BLAST results. The color key is given on the right side of the heatmap.
Differential regulation of genes involved in nematode development, metabolism and sex differentiation
Functional analysis of the 41 DEGs revealed the overexpression of a protease coding gene Clp-1 (Ca2+ dependent cysteine protease calpain) when G. pallida was parasitizing S. sisymbriifolium (log2 fold change = 6.74). Other genes that were overexpressed when G. pallida was parasitizing S. sisymbriifolium were cathepsin S-like cysteine proteinase (log2 fold change = 3.38), an essential gene for invasion and reproduction, and the stress related gene hsp-20 (heat shock protein) (log2 fold change = 3.40). Genes encoding sex related proteins Hsd2 (hydroxysteroid 17-beta dehydrogenase 2) (no expression in S. tuberosum) and Srp-8, (signal recognition particle-8) (no expression in S. tuberosum) and genes encoding epidermal proteins cornifin-A (no expression in S. tuberosum) and cytoskeleton related actin (no expression in S. tuberosum) were also over expressed when parasitizing S. sisymbriifolium. Additionally, uncharacterized genes (without a signal peptide) namely, GPLIN_000296600, NAIXLOC_002944, NAIXLOC_006444, NAIXLOC_018501 and NAIXLOC_019666 were uniquely expressed when G. pallida was parasitizing S. sisymbriifolium. When G. pallida was parasitizing S. tuberosum, overexpression of genes with functions in autophagy (Epg-5, Ectopic p granules protein-5), nematode immunity (Bath-38 BTB and MATH domain containing protein), a guanine nucleotide exchange factor (GEF, Ephexin-1), and genes encoding cuticle collagen Dpy-2 were found. None of these genes were expressed when G. pallida was parasitizing S. sisymbriifolium.
Differential regulation of effector genes
Among the 41 DEGs we found several characterized effectors as well as novel sequences with signal peptide which are putative effectors. Effector genes cellulase (log2 fold change = 3.55), cathepsin S-like cysteine protease (log2 fold change = 3.38), and Globodera rostochiensis 1106 effector ortholog (Gr1106, GPLIN_000812600) (log2 fold change = 4.26) were overexpressed when G. pallida was parasitizing S. sisymbriifolium. Other potential effector Eng-1 (Beta-1, 4-endoglucanase precursor) was only expressed when G. pallida was parasitizing S. sisymbriifolium but was not expressed when G. pallida was parasitizing S. tuberosum. Two other uncharacterized G. pallida gene sequences with signal peptide, GPLIN_000044400 and GPLIN_000028880 were also overexpressed when G. pallida was parasitizing S. sisymbriifolium but not when parasitizing S. tuberosum. Only, Rbp-4, and Gr1106 effectors (GPLIN_000235300) were overexpressed when G. pallida was parasitizing S. tuberosum. One G. pallida gene sequence of unknown function with a signal peptide, GPLIN_000925800, was expressed only when G. pallida was parasitizing S. tuberosum but not when parasitizing S. sisymbriifolium. The three above mentioned G. pallida genes (GPLIN_000044400, GPLIN_000028880 and GPLIN_000925800) with signal peptide did not show a dorsal gland promoter element motif (DOG Box) in their promoter region, which is a characteristic feature of dorsal gland cell effectors in Globodera species7. These genes need to be further studied for confirmation of their importance in parasitism.
Confirmation of gene expression by qRT-PCR
To verify the RNA seq data, the expression profiles of the three G. pallida genes Rbp4 (GPLIN_000311600), Gr1106 (GPLIN_000812600), and cellulase (GPLIN_000304900) were determined by qRT‐PCR. A significant difference (P ≤ 0.05) in G. pallida gene expression was obtained when parasitizing S. tuberosum or S. sisymbriifolium. Similar to the results of the RNAseq differential expression analysis, qRT-PCR results indicated Gr1106 (47 fold higher, P = 0.02) and cellulase (32 fold higher, P = 0.04) in G. pallida was overexpressed when parasitizing S. sisymbriifolium, but expression of Rbp4 (7 fold higher, P = 0.02) when G. pallida was parasitizing S. sisymbriifolium was not detected (Fig. 2).

Quantitative PCR (qRT‐PCR) validation of RNA sequencing (RNAseq) data for the Globodera pallida genes 1106 orthologue, Rbp4 and cellulase during resistance and susceptible responses. The y axes represent the relative fold change (calculated using the ΔΔCt method) in gene expression. The data are representative of three independent biological replicates. Bars indicate standard errors.
Discussion
In this study the differential expression of transcripts involved in early stages of parasitism for G. pallida was determined by analyzing the transcriptome of G. pallida from either the susceptible host S. tuberosum or the resistant host S. sisymbriifolium. The functional analysis of the DEGs revealed the expression of genes important for parasitism such as proteases, cell wall modifying, stress related protein coding, and sex related protein coding. Several effector genes were also differentially expressed when G. pallida was parasitizing the susceptible host S. tuberosum or the resistant host S. sisymbriifolium at the early stages of parasitism. Of the 41 DEGs, 14 DEGs (either identified or uncharacterized) as illustrated in Fig. 1 (either known or unknown) have not been studied in relation to nematode parasitism.
Proteases play a variety of roles in host-parasite interactions, such as participation during invasion of host tissues, nutrition of the parasite, and escape from host defense responses21. A cysteine protease family gene Clp-1 and cathepsin S was highly expressed when G. pallida was parasitizing S. sisymbriifolium. The higher expression of Clp-1 and cathepsin S may be due to the defense response in the S. sisymbriifolium roots. Proteases have been identified as a primary target for the control of plant parasitic nematodes22,23,24,25.
This toxic environment induced by the host plant causes a stress response in the pathogen18. The stress response gene, Epg-5, was found to be over expressed when G. pallida was parasitizing S. tuberosum. A recent study reported the role of autophagy genes in the pine wood nematode Bursaphelenchus xylophilus as a response to stressful environmental conditions26. The gene, Epg-5, needs to be studied further to determine the expression over time when infecting S. tuberosum. Gene silencing studies will also provide information about the role of autophagy genes in the parasitism of G. pallida. One reason for the over expression may be to protect G. pallida at the early stages of parasitism. Earlier studies in other pathogens and insect pests show that autophagy has an important role in the growth, development, reproduction and pathogenicity for these organisms27,28,29,30. Epg-5, in human systems, encodes a large coiled coil domain-containing protein that functions in autophagy during starvation conditions31. Brennand et al. (2011) reviewed the strategy of human parasitic protists to starvation or stress conditions where autophagy plays a major role in adaptation to changes in the nutritional status of the cells32.
Heat shock proteins are stress tolerance proteins which are highly conserved in a wide range of organism and are reported to be essential candidate genes which can be targeted for plant mediated resistance development33,34. Over expression of the heat shock protein gene hsp-20 was also observed when G. pallida was parasitizing S. sisymbriifolium. Hsp-20 is in the small heat shock protein family (sHSPs), which function as molecular chaperones. Under stress conditions, sHSPs bind to denatured proteins and maintain them in a folded state which prevents the irreversible aggregation of these proteins. When stressful conditions such as starvation, high temperature etc. are removed, sHSPs are released and re-naturation takes place35,36. This may be an attempted adaptive mechanism for G. pallida to overcome the stress encountered when invading S. sisymbriifolium roots. Metabolic pathway analysis revealed that heat shock protein (hsp) hsp-20 was classified under both stress related and sex related protein categories. However, detailed research would need to be conducted to determine the relationship between hsp and sex determination for plant parasitic nematodes. Two other genes encoding sex related proteins namely, Hsd-2 and Srp-8 were also overexpressed when G. pallida was isolated from S. sisymbriifolium. Although this may not be the case for G. pallida while infecting S. sisymbriifolium, in some resistant plants delayed HR known as male-based resistance is evident, and results in sexual differentiation to males37. The relationship of stress and sex differentiation is poorly understood and needs to be further studied.
Overexpression of G. pallida genes encoding epidermal proteins (cornifin-A), and cystoskeleton (actin) while infecting S. sisymbriifolium was observed, possibly due to changes in the cytoskeleton and body wall muscles under stress conditions. In a resistant plant such as S. sisymbriifolium a feeding cell is not formed which leads to starvation‐induced stress in nematodes and, subsequently, cessation of their development19.
Effector genes modulate host metabolism for a successful infection and life cycle completion. Effector molecules which protect nematodes from plant defenses called immune-modulators play decisive roles in a successful parasitism4,5,38,39. Plants have evolved defense proteins that can recognize pathogen effectors, and induce ETI40,41, often involving a type of cell death known as the hypersensitive response (HR). An HR to G. pallida was observed in S. sisymbriifolium15 (Supplementary Fig. S3). In the present study, genes with secretory characteristics were identified as Rbp-4, Globodera rostochiensis 1106 effector (Gr1106), and one uncharacterized gene with a signal peptide. These genes were found to be highly expressed when G. pallida was parasitizing S. tuberosum. Another member of the Gr1106 effector family GPLIN_000812600 was found to be highly expressed when G. pallida was isolated from S. sisymbriifolium. The G. rostochiensis effectors 1106 (Gr1106 effectors) suppress effector-triggered immunity (ETI) for development of the nematode in susceptible plants42. Although detailed studies are not available on the Rbp-4 gene, a closely related (protein sequence similarity is presented in Supplementary Fig. S4) G. pallida gene, Rbp-1 (Gp-Rbp-1), has been found to provoke an ETI response from potatoes when it interacts with Gpa2, a resistance protein containing coiled- coil, nucleotide-binding, and leucine-rich-repeat domains9,43.
During nematode invasion and migration, cell wall‐degrading or modifying enzymes including endoglucanases (glycosylhydrolase (GH) family) are expressed39. Higher expression of cell wall-modifying enzymes coding effector genes beta-1,4-endoglucanase (Eng-1 and cellulase) and hydroxyacyl-coenzyme A dehydrogenase was observed when G. pallida was parasitizing S. sisymbriifolium compared to when G. pallida was parasitizing S. tuberosum. Eng-1 was not expressed when G. pallida was parasitizing S. tuberosum. The over expression of GH family enzymes in G. pallida when infecting a resistant plant species compared to their expression in susceptible plant species could be attributed towards the structural complexity of plant cell wall, however a detailed study is required to better understand this phenomenon. Shukla et al. (2018) also reported the over expression of GH family genes in resistance response19.
In conclusion, this study provides insight into the molecular mechanisms of a susceptible and resistant interactions at early stages of G. pallida parasitism. Our results indicate that, at an early stage of infection, over expression of several effector genes, including cell wall modifying enzyme coding genes, may play a role to overcome plant defenses from S. sisymbriifolium in G. pallida. We have also observed over expression of several stress related genes when G. pallida was isolated from S. sisymbriifolium. The role of these genes in parasitism is poorly understood and need further investigation. Several novel and known nematode genes were identified, which are differentially expressed when G. pallida was infecting a resistant or susceptible plant species, which will help to advance research in plant-nematode interactions. Genetic engineering experiments aimed at creating nematode resistant potatoes may benefit by targeting DEGs produced during the earliest stages of parasitism.
Materials and Methods
Rearing of G. pallida
Globodera pallida cysts collected from infested fields in Shelly, ID, USA were reared on a susceptible potato cultivar ‘Desiree’ in clay pots filled with sterilized sandy loam soil and sand (2:1) under greenhouse conditions of 18 °C ± 2 °C (day time), 14 ± 2 °C (night time), and 16:8-h light: dark period12,15,44. After 16 weeks, cysts for experimental use were recovered by extraction from soil using the Fenwick Can method45. Prior to experimental use, all cysts were incubated at 4 °C for a minimum of 16 weeks. For the current experiment the cysts were put inside sterile nylon 250 μm mesh bags (McMaster Carr, Elmhurst, IL). The nylon mesh was sealed along the edges with a hand sealer (Sealer 8″ F-200, Sealer sales Inc., Northridge, CA), surface sterilized in a solution of 0.5% NaOCl for 5 min and rinsed thoroughly with sterile distilled water. The cyst bags were then amended with potato cultivar ‘Desiree’ root diffusate (collected from 4 weeks grown plants) containing gentamicin (1.5 mg/ml) and nystatin (0.05 mg/ml) and incubated at 20 °C in a sterile 6-well plate. After 2 weeks the hatched juveniles (J2s) were collected and surface sterilized in 100 μg/ml of ampicillin and streptomycin (w/v) and in 0.125% w/v benzethonium chloride15,46.
Plant materials
Solanum tuberosum L. cv. ‘Desiree’ were propagated in standard tissue culture media47. After 4 weeks the tissue culture plants were transplanted into root trainers (Haxnicks™, TDI Brands, USA) and kept under greenhouse conditions 18 °C ± 2 °C (day time), 14 ± 2 °C (night time), 16:8-h light: dark period. Solanum sisymbriifolium seeds (Accession PI 381291), were obtained from Chuck Brown, USDA‐ARS, Prosser, WA, USA and were grown in plastic seed trays for 4 weeks before being transplanted into root trainers and kept under greenhouse conditions, as above. Both the plant species were grown in root trainers for 4 weeks prior to inoculation.
Infecting plants with G. pallida
Four weeks post planting, root trainers were opened and single root from each plant was inserted into a glass tube (10 cm L × a.5 cm W, Supplementary Fig. S5). The tube was then filled with sterilized sandy loam soil and sand (2:1) mix. Surface sterilized G. pallida J2s suspension (1500 J2s/ml) in 0.2% agarose was inoculated onto the single root inserted in to the glass tube. There were 3 replicates for each plant species.
Extraction of infected G. pallida from the roots of S. tuberosum and S. sisymbriifolium
The infected roots were removed from the tube, and gently washed 24 h post- infestation. The washed roots were cut into 1 mm length and blended for 20 seconds in a blender (WARING Commercial®, WARING Products Division, CT, USA) on a low speed setting with ice cold water. The suspension was passed through three sieves stacked together (no. 60 on top, 200 in the middle and 500 at the bottom). The nematodes were collected from the no. 500 sieve and transferred to a Petri dish (modified from Coolen and D’Herde 197248). Under a microscope (Leica, M 80, Germany) the nematodes were pipetted individually using a Pasteur pipette into an Eppendorf tube (kept on ice). Approximately 30 nematodes were isolated from S. sisymbriifolium root samples, therefore, only 30 nematodes from each plant species were used for RNA extraction. The extra water was pipetted out from each tube and the nematodes were immediately frozen in liquid nitrogen and stored at −80 °C.
RNA isolation, sequencing and differential expression analysis
A previously published RNA extraction protocol was modified by supplementing frozen nematode samples with 200 µl Proteinase K solution (100 µg/ml Proteinase K in 1% sodium dodecyl sulfate, 50 mM Tris, pH 7.5, and 10 mM Ethylenediaminetetraacetic acid (EDTA) - pH 8, 5% β-mercaptoethanol)49. The suspension was then incubated at 50 °C for 15 minutes, and then mixed with 500 µl RNAzol (RNAzol® RT, Sigma-Aldrich, USA), and incubated at room temperature for 15 min. The suspension was next centrifuged at 13,000 g for 12 min and the supernatant was transferred to a new tube. The volume of supernatant was measured and precipitated by mixing freshly made 70% ethanol to bring the final solution to 40% ethanol. The tubes were incubated at room temperature for 45 min and centrifuged at 21,000 g for 8 min49. After removal of the supernatant, the pelleted RNA was purified by adding 400 µl magnetic bead buffer provided in the Agencourt RNAdvance Tissue total RNA extraction kit (Beckman Coulter, USA). RNA was further purified as per manufacturer protocol (Agencourt RNAdvance Tissue total RNA extraction kit, Beckman Coulter, USA). The final elution was done in 20 µl RNAse free water. RNA quality and quantity were estimated on a FRAGMENT ANALYZER™ (Advanced Analytical Technologies Inc., USA) and Qubit™ 3 fluorometer (Life Technologies, USA). Only samples with good RNA quality (RQN > 8) were kept for further analyses.
For each sample an average of 25 ng total RNA was used for library preparation using TruSeq® Stranded mRNA Library Prep Kit for NeoPrep™ (Illumina, San Diego, USA). The libraries were sequenced on Illunima HiSeq 4000 (100 bp paired end (PE)) at the QB3 Vincent J. Coates Genomics Sequencing Laboratory, University of California, Berkely, CA, USA. The 100 bp PE reads were assessed for quality using FastQC program50. Trimmomatic was used for trimming the adapters and primers from the reads51. The quality trimmed reads were processed through the Tuxedo suite52 for differential gene expression analysis by aligning the reads onto the G. pallida genome, G.pal.v153 downloaded from Welcome Trust Sanger Institute data base using tophat v2.0.1454 on default parameter settings. The aligned reads were further processed through cufflinks, cuffcompare, cuffmerge and cuffdiff of Tuxedo suite. The cuffdiff output data was visualized on an R program cummeRbund55. Pairwise similarities between conditions and replicates were visualized using the csDistHeat function on the cummeRbund program (Supplementary Fig. S6). A confidence level of 99% (P = 0.01) was set to evaluate the significant difference in G. pallida gene expression extracted from two different plant species 24 hours-post-infestation.
Functional characterization of transcripts
To investigate functions, the transcripts were processed using Blast2GO PRO (version 4.1.9) software56. The data was processed through Cloud BLAST (bastx-fast, e-Value = 1.0E-5) of Blast2GO. In addition, significant differentially expressed gene (DEG) sequences were individually extracted and blasted (blastn) to the NCBI database. Those transcripts without a gene annotation name from the differential expression data set were blasted to Wormbase (http://parasite.wormbase.org/Multi/Tools/Blast) to compare with G. pallida genome data. Transcripts that did not overlap with annotated genes (during the cufflinks step) are given an ID, that is XLOC, instead of GPLIN. Transcripts were also translated using ExPASy (http://web.expasy.org/translate/) and blasted to ExPASy BLAST (http://web.expasy.org/blast/) to confirm their identity. To predict possible effector genes among the differential expression data, the protein sequences were assessed for signal peptides using SignalP 4.157.
Gene expression analysis using quantitative PCR (qRT-PCR)
To validate DEGs on qRT-PCR, three potential candidate effector genes were selected namely, Rbp4, 1106 ortholog and cellulase from the DEGs list. The G. pallida infected roots of S. tuberosum and S. sisymbriifolium were extracted 24 h post infestation, and RNA was isolated as per the methods described above. An average of 25 ng total RNA was used for cDNA synthesis using SUPERSCRIPT® II reverse transcriptase (Invitrogen) on a Thermal Cycler C1000™, (BIO-RAD, USA). The qRT-PCR (ViiA™ 7 Real-Time PCR System, Applied Biosystems, USA) reaction was performed in 20 µL total volume containing 10 µL master mix (FAST SYBR™ Green Master Mix, Thermo Fisher Scientific Inc, MA, USA), 1 µL template (10 ng), 200 nM (primer efficiency = 104.11, R2 = 0.98) each of forward and reverse primers of elongation factor (reference gene), 150 nM (primer efficiency = 99.65, R2 = 0.98) each of forward and reverse primers of RBP4, 200 nM (primer efficiency = 100.51, R2 = 0.98) each of forward and reverse primers of 1106 ortholog, 200 nM (primer efficiency = 101.88, R2 = 0.97) each of forward and reverse primers of cellulase, and nuclease free water was added. The cycling conditions included 95 °C for 2 min, 40 cycles of 95 °C for 15 sec, and an annealing step for 30 sec at 55 °C. The relative expression level was calculated in comparison to elongation factor by means of 2(control sample Ct of target−infected sample Ct of target) ÷ 2(control sample Ct of reference−infected sample Ct of reference)58. The results presented are the average of three biological replicates. T-test was performed to determine significant differences between the relative expression of G. pallida genes when isolated from S. tuberosum and S. sisymbriifolium.
Data Availability
The aligned RNAseq reads are available at National Center for Biotechnology Information (NCBI) with Sequence read archive (SRA) accession no. SRP159274.
References
Nicol, J. M. et al. Current nematode threats to world agriculture. In: Genomics and Molecular Genetics of Plant-Nematode Interactions (Eds Jones, J. T., Gheysen, G. & Fenoll, C.) 21–43 (Springer, 2011).
Yu, Q., Ye, W., Sun, F. & Miller, S. Characterization of Globodera rostochiensis (Tylenchida: Heteroderidae) associated with potato in Quebec, Canada. Can. J. Plant Pathol. 32, 264–271 (2010).
Whitworth, J. L. et al. Resistance of potato breeding clones and cultivars to three species of potato cyst nematode. Plant Dis. 102, 2120–2128 (2018).
Hewezi, T. & Baum, T. J. Manipulation of plant cells by cyst and root-knot nematode effectors. Mol. Plant-Microbe Interact. 26, 9–16 (2013).
Quentin, M., Abad, P. & Favery, B. Plant parasitic nematode effectors target host defense and nuclear functions to establish feeding cells. Front. Plant Sci. 13, 53, https://doi.org/10.3389/fpls.2013.00053 (2013).
Sobczak, M. & Golinowski, W. Structure of cyst nematode feeding site. In: Cell biology of plant nematode parasitism (eds Berg, R. H. & Taylor, C. G.) 153–187 (Springer, 2009).
Eves-van den Akker, S. & Birch, P. R. J. Opening the effector protein toolbox for plant– parasitic cyst nematode interactions. Mol. Plant 9, 1451–1453 (2016).
Gheysen, G. & Mitchum, M. G. How nematodes manipulate plant development pathways for infection. Curr. Opin. Plant Biol. 14, 415–21 (2011).
Sacco, M. A. et al. The cyst nematode SPRYSEC protein RBP-1 elicits Gpa2-and RanGAP2-dependent plant cell death. PLoS Pathog. 5, p. e1000564 (2009).
Sobczak, M. et al. Characterization of susceptibility and resistance responses to potato cyst nematode (Globodera spp.) infection of tomato lines in the absence and presence of the broad-spectrum nematode resistance hero gene. Mol. Plant-Microbe Interact. 18, 158–168 (2005).
Ernst, K. et al. The broad-spectrum potato cyst nematode resistance gene (Hero) from tomato is the only member of a large gene family of NBS-LRR genes with an unusual amino acid repeat in the LRR region. Plant J. 31, 127–136 (2002).
Dandurand, L. M. & Knudsen, G. R. Effect of the trap crop Solanum sisymbriifolium and two biocontrol fungi on reproduction of the potato cyst nematode, Globodera pallida. Ann. Appl. Biol. 169, 180–189 (2016).
Timmermans, B. G. H., Vos, J., Stomph, T. J., Van Nieuwburg, J. & Van Der Putten, P. E. L. Growth duration and root length density of Solanum sisymbriifolium (Lam.) as determinants of hatching of Globodera pallida (Stone). Ann. of Appl. Biol. 148, 213–222 (2006).
Scholte, K. Screening of non-tuber bearing solanceae for resistance to and induction of juvenile hatch of potato cyst nematodes and their potential for trap cropping. Ann. Appl. Biol. 136, 239–246 (2000).
Kooliyottil, R., Dandurand, L. M., Govindan, B. N. & Knudsen, G. R. Microscopy method to compare cyst nematode infection of different plant species. Adv. Biosci. Biotechnol. 7, 311–8 (2016).
Sasaki-Crawley, A. et al. Signaling and behavior of potato cyst nematode in the rhizosphere of the trap crop, Solanum sisymbriifolium. Aspects Appl. Biol. 103, 45–51 (2010).
Sasaki-Crawley, A. et al. Quantitative difference in gene expression of defence genes in Solanum tuberosum and S. sisymbriifolium infected with Globodera pallida. Indian J. Nematol. 44, 62–72 (2014).
Gillet, F. X., Bournaud, C., de Souza Júnior, J. D. A. & Grossi-de-Sa, M. F. Plant-parasitic nematodes: towards understanding molecular players in stress responses. Ann. Bot. 119, 775–789 (2017).
Shukla, N. et al. Transcriptome analysis of root‐knot nematode (Meloidogyne incognita) ‐infected tomato (Solanum lycopersicum) roots reveals complex gene expression profiles and metabolic networks of both host and nematode during susceptible and resistance responses. Mol. Plant Pathol. 19, 615–633 (2018).
Palomares-Rius, J. E. et al. Comparison of transcript profiles in different life stages of the nematode Globodera pallida under different host potato genotypes. Mol. Plant Pathol. 13, 1120–1134 (2012).
Tort, J., Brindley, P. J., Knox, D., Wolfe, K. H. & Dalton, J. P. Proteinases and associated genes of parasitic helminths. Adv. Parasitol. 43, 161–266 (1999).
Wang, K. et al. The cathepsin S cysteine proteinase of the burrowing nematode Radopholus similis is essential for the reproduction and invasion. Cell Biosci. 6, 1–15 (2016).
Thakur, P. K., Kumar, M., Kumar, J., Gantasala, N. P. & Rao, U. Structural and functional analysis of cathepsin S of Heterodera spp: a promising candidate for its control. Indian J. Exp. Biol. 52, 223–231 (2014).
Antonino de Souza Júnior, J. D. et al. Knocking down Meloidogyne incognita proteases by plant-delivered dsRNA has negative pleiotropic effect on nematode vigor. PLoS One 8, e85364 (2013).
Urwin, P. E., Lilley, C. J., McPherson, M. J. & Atkinson, H. J. Characterization of two cDNAs encoding cysteine proteinases from the soybean cyst nematode Heterodera glycines. Parasitol. 114, 605–613 (1997).
Deng, L. N., Wu, X. Q., Ye, J. R. & Xue, Q. Identification of autophagy in the pine wood nematode Bursaphelenchus xylophilus and the molecular characterization and functional analysis of two novel autophagy-related genes, BxATG1 and BxATG8. Int. J. Mol. Sci. 17, 279, https://doi.org/10.3390/ijms17030279 (2016).
Tindwa, H. et al. Molecular cloning and characterization of autophagy-related gene TmATG8 in Listeria-invaded hemocytes of Tenebrio molitor. Dev. Comp. Immunol. 51, 88–98 (2015).
Fernández, J. M. F. et al. Molecular cloning and characterization of two novel autophagy-related genes belonging to the ATG8 family from the cattle tick Rhipicephalus (Boophilus) microplus (Acari: Ixodidae). Exp. Appl. Acarol. 64, 533–542 (2014).
Liu, X. H. et al. Involvement of a Magnaporthe grisea serine/threonine kinase gene, MgATG1, in appressorium turgor and pathogenesis. Eukaryot Cell. 6, 997–1005 (2007).
Valent, B., Farrall, L. & Chumley, F. G. Magnaporthe grisea genes for pathogenicity and virulence identified through a series of backcrosses. Genetics 127, 87–101 (1991).
Wang, Z. et al. The vici syndrome protein epg5 is a rab7 effector that determines the fusion specificity of autophagosomes with late endosomes/lysosomes. Mol. Cell 63, 781–795 (2016).
Brennand, A. et al. Autophagy in parasitic protists: unique features and drug targets. Mol. Biochem. Parasitol. 177, 83–99 (2011).
Lourenço-Tessutti, I. T. et al. Knock-down of heat-shock protein 90 and isocitrate lyase gene expression reduced root-knot nematode reproduction. Phytopathol. 105, 628–637 (2015).
De Luca, F. et al. Characterization of the heat shock protein 90 gene in the plant parasitic nematode Meloidogyne artiellia and its expression as related to different developmental stages and temperature. Gene 440, 16–22 (2009).
Pérez-Morales, D. & Espinoza, B. The role of small heat shock proteins in parasites. Cell Stress Chaperones. 20, 767–780 (2015).
Sun, Y. & MacRae, T. H. Small heat shock proteins: molecular structure and chaperone function. Cell. Mol. Life Sci. 62, 2460–76 (2005).
Sobczak, M. & Golinowski, W. Cyst nematodes and syncytia. In: Genomics and Molecular Genetics of Plant-Nematode Interactions (eds Jones, J. T., Gheysen, G. & Fenoll, C.) 61–82 (Springer, 2011).
Niu, J. et al. Msp40 effector of root-knot nematode manipulates plant immunity to facilitate parasitism. Sci. Rep. 6, 19443, https://doi.org/10.1038/srep19443 (2016).
Mitchum, M. G. et al. Nematode effector proteins: an emerging paradigm of parasitism. New Phytol. 199, 879–894 (2013).
Chisholm, S. T., Coaker, G., Day, B. & Staskawicz, B. J. Host-microbe interactions: shaping the evolution of the plant immune response. Cell 124, 803–814 (2006).
Jones, J. D. G. & Dangl, J. L. The plant immune system. Nature 444, 323–329 (2006).
Makunde, P. T. Functional analysis of potato cyst nematode effectors. PhD Thesis, Wageningen University, The Netherlands (2012).
Bendahmane, A., Kanyuka, K. & Baulcombe, D. C. The Rx gene from potato controls separate virus resistance and cell death responses. Plant Cell 11, 781–792 (1999).
Contina, J. B., Dandurand, L. M. & Knudsen, G. R. Use of GFP-tagged Trichoderma harzianum as a tool to study the biological control of the potato cyst nematode Globodera pallida. Appl. Soil Ecol. 115, 31–37 (2017).
Fenwick, D. W. Methods for the recovery and counting of cysts of Heterodera schachtii from soil. J. Helminth. 18, 155–155 (1940).
Kooliyottil, R., Dandurand, L. M., Kuhl, J. C., Caplan, A. & Xiao, F. Microaspiration of Solanum tuberosum root cells at early stages of infection by Globodera pallida. Plant Methods 13, 68, https://doi.org/10.1186/s13007-017-0219-x (2017).
Murashige, T. & Skoog, F. A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol. Plantarum 15, 473–497 (1962).
Coolen, W. A. & D’Herde, C. J. A Method for the Quantitative Extraction of Nematodes from Plant Tissue. p. 77 (State Agriculture Research Centre, Ghent, Belgium, 1972).
Casavant, N. C., Kuhl, J. C., Xiao, F., Caplan, A. B. & Dandurand, L. M. Assessment of Globodera pallida RNA extracted from Solanum roots. J. Nematol. 49, 12–20 (2017).
Andrews, S. FastQC: a quality control tool for high throughput sequence data, http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120, https://doi.org/10.1093/bioinformatics/btu170 (2014).
Trapnell, C. et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578 (2012).
Cotton, J. A. et al. The genome and life-stage specific transcriptomes of Globodera pallida elucidate key aspects of plant parasitism by a cyst nematode. Genome Biol. 15, R43, https://doi.org/10.1186/gb-2014-15-3-r43 (2014).
Kim, D. et al. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36, https://doi.org/10.1186/gb-2013-14-4-r36 (2013).
Goff, L., Trapnell, C. & Kelley, D. CummeRbund: Analysis, exploration, manipulation, and visualization of Cufflinks high-throughput sequencing data. R package version 2.18.0 (2013).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Petersen, T. N., Brunak, S., Heijne, G. & Nielsen, H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8, 785–786 (2011).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−∆∆CT method. Methods 25, 402–408 (2001).
Acknowledgements
The authors are thankful to USDA-APHIS, Idaho Potato Commission and North West Potato Research Consortium for financial support and The Institute for Bioinformatics and Evolutionary Studies (IBEST), University of Idaho for analytical support.
Author information
Authors and Affiliations
Contributions
R.K. performed experiments. R.K. and J.L.L. conducted the data analysis. R.K. and L.M.D. designed the research and wrote the paper. J.C.K., A.C., F.X. and B.M. helped to design experiments and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kooliyottil, R., Dandurand, LM., Kuhl, J.C. et al. Transcriptome analysis of Globodera pallida from the susceptible host Solanum tuberosum or the resistant plant Solanum sisymbriifolium. Sci Rep 9, 13256 (2019). https://doi.org/10.1038/s41598-019-49725-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49725-6
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
