
Abstract
The loss of functional beta cell mass characterises all forms of diabetes. Beta cells are highly susceptible to stress, including cytokine, endoplasmic reticulum (ER) and oxidative stress. This study examined the role of pleckstrin homology-like, domain family A, member 3 (Phlda3) in beta cell survival under stress conditions and the regulatory basis. We found that the mRNA levels of Phlda3 were markedly upregulated in vivo in the islets of diabetic humans and mice. In vitro, exposure of MIN6 cells or islets to cytokines, palmitate, thapsigargin or ribose upregulated Phlda3 mRNA and protein levels, concurrent with the induction of ER stress (Ddit3 and Trb3) and antioxidant (Hmox1) genes. Furthermore, H2O2 treatment markedly increased PHLDA3 immunostaining in human islets. Phlda3 expression was differentially regulated by adaptive (Xbp1) and apoptotic (Ddit3) unfolded protein response (UPR) mediators. siRNA-mediated knockdown of Xbp1 inhibited the induction of Phlda3 by cytokines and palmitate, whereas knockdown of Ddit3 upregulated Phlda3. Moreover, knockdown of Phlda3 potentiated cytokine-induced apoptosis in association with upregulation of inflammatory genes (iNos, IL1β and IκBα) and NFκB phosphorylation and downregulation of antioxidant (Gpx1 and Srxn1) and adaptive UPR (Xbp1, Hspa5 and Fkbp11) genes. Knockdown of Phlda3 also potentiated apoptosis under oxidative stress conditions induced by ribose treatment. These findings suggest that Phlda3 is crucial for beta cell survival under stress conditions. Phlda3 regulates the cytokine, oxidative and ER stress responses in beta cells via the repression of inflammatory gene expression and the maintenance of antioxidant and adaptive UPR gene expression. Phlda3 may promote beta cell survival in diabetes.
Similar content being viewed by others


Introduction
The loss of functional beta cell mass plays a crucial role in the pathogenesis of both type 1 and type 2 diabetes1. Compelling evidence has revealed that prolonged exposure to the (pre)diabetic milieu triggers beta cell stress and death. Indeed, inflammatory, endoplasmic reticulum (ER) and oxidative stress are potential mechanisms through which cytokines and elevated levels of glucose and free fatty acids induce beta cell apoptosis2,3,4,5,6. This has been associated with the activation of numerous proapoptotic effectors such as Ddit3, Trb3, Txnip and c-Jun N-terminal kinase7,8,9,10,11,12,13,14. However, stress stimuli also activate defense and adaptive responses, including the unfolded protein response (UPR) and the antioxidant response, that maintain homeostasis and promote beta cell survival5,15,16. The balance between protective and deleterious responses to stress determines beta cell fate, but the pathways involved are not fully characterized5. A better understanding of the beta cell stress response signaling pathways is needed to identify novel targets to preserve the functional beta cell mass in (pre)diabetic individuals.
Phlda3 encodes for a member of the pleckstrin homology-like, domain family of proteins. The pleckstrin homology domain is an amino acid sequence of about 100 residues with a specific three-dimensional structure allowing binding to phosphoinositides and protein-protein interaction. It is present in a variety of proteins involved in signal transduction, phospholipid processing, membrane trafficking and organization of cytoskeleton17. The first member of this family, Phlda1, has previously been implicated in the modulation of energy metabolism and obesity18. On the other hand, Phlda3 has been identified as a tumor suppressor in pancreatic neuroendocrine tumors19, and its expression is induced by ER stress in hepatocytes20. However, whether Phlda3 expression is altered in diabetes or plays a role in beta cell pathophysiology are unclear.
In the present study, we report for the first time that Phlda3 expression is upregulated in the islets of diabetic rodents and humans. Our findings in isolated islets and MIN6 beta cells suggest that Phlda3 is induced in response to inflammatory, ER and oxidative stress and that it plays an important adaptive role during these stresses. Indeed, Phlda3 knockdown potentiates inflammatory- and oxidative stress-induced apoptosis. Mechanistically, we demonstrate that the adaptive UPR effector Xbp1 is required for Phlda3 induction, whereas the pro-apoptotic effector Ddit3 inhibits its expression. Moreover, we show that the Phlda3-mediated protection against stress involves the modulation of proinflammatory, adaptive UPR and antioxidant gene expression. Our results therefore suggest that Phlda3 is a novel adaptive gene induced under conditions of stress that promotes beta cell survival.
Material and Methods
Reagents
Cytokines IL1β, IFNγ and TNFα were obtained from R&D Systems (Minneapolis, MN, USA). Ribose, thapsigargin and 4-hydroxytamoxifen were from Sigma (St. Louis, MI, USA). Control Non-Targeting and ON-TARGETplus SMARTpool siRNAs and transfection reagent DharmaFECT3 were from Thermo Fisher Scientific (Lafayette, CO, USA).
Human islets
Human islets were obtained from 8 non-diabetic and 5 diabetic subjects at the Tom Mandel Islet Transplant Program in Melbourne21. Human islets were purified from heart-beating, brain-dead donors, with written informed consent from next of kin. All human studies were approved by the St Vincent’s Hospital Human Research Ethics Committee (approval number HREC011/04) and all methods were carried out in accordance with guidelines and regulations. Characteristics of organ donors and islet preparations are indicated in Supplementary Table 1. To evaluate the impact of oxidative stress on Phlda3 protein expression ex vivo, human islets were obtained from 3 non-diabetic subjects through the JDRF award 31-2008-416 (ECIT Islet for Basic Research program) with written informed consent from next of kin and approved for use under the ethics reference B403/2017/05JUL/355 (Comité d'éthique hospitalo-facultaire Saint-Luc, UCLouvain). Characteristics of these donors and islet preparations are listed in Supplementary Table 2. All experiments were performed in accordance with relevant guidelines and regulations.
Mice
14–16 weeks old C57BL/KsJ db/db mice and age-matched lean control mice (C57BL/KsJ), and 11–13 weeks old female nonobese diabetic (NOD) mice and age-matched control Balb/c mice were obtained from the Garvan Institute breeding colonies (Australian BioResources, Moss Vale, NSW, Australia). Xbp1flox/flox mice were kindly provided by L.H. Glimcher and A.H. Lee (Weill Cornell Medical College, New York, NY, USA). They were crossed with Pdx1-CreER mice to generate Xbp1+/+-Pdx1-CreER (controls) and Xbp1flox/flox-Pdx1-CreER mice. For Xbp1 deletion, control and Xbp1flox/flox-Pdx1-CreER islets were treated with 100 nmol/l 4-hydroxytamoxifen as previously described14. For ex vivo islet experiments, 8–10 week-old wild-type C57BL/6 J mice were used. All experiments were approved by the Garvan Institute/St. Vincent’s Hospital Animal Experimentation Ethics Committee and by the Institutional Committee on Animal Experimentation of the Health Sciences Sector at UCLouvain (Project 2017/UCL/MD/014). All experiments were performed in accordance with relevant guidelines and regulations.
Islet isolation and culture
Islets were isolated by liberase digestion, separated by a density gradient and handpicked under a stereomicroscope. Islets were cultured in RPMI medium (Invitrogen, Carlsbad, CA, USA) containing 11.1 mmol/l glucose, 2 mmol/l glutamine, 10% heat-inactivated FBS, 50 units/ml penicillin and 50 µg/ml streptomycin.
Cell culture
MIN6 beta cells (P26–43)22 were grown in Dulbecco’s modified Eagle’s medium (Invitrogen) containing 25 mmol/l glucose, 10 mmol/l HEPES, 10% FCS, 50 units/ml penicillin and 50 µg/ml streptomycin.
Islet and cell treatment
Isolated islets and cells were treated with 100 U/ml IL1β, 250 U/ml IFNγ and 100 U/ml TNFα (15 min-24h). To assess the effects of lipotoxicity, islets and cells were treated with 0.92 g/100 ml BSA or 400 µmol/l palmitate coupled to 0.92 g/100 ml BSA (48 h). Thapsigargin (300 nmol/l, 1 µmol/l; 24 h) was used to induce ER stress. Ribose (50 mmol/l, 48 h) was used to induce oxidative stress. Cells were transfected with 100 nmol/l control, Phlda3, Xbp1 or Ddit3 siRNA using DharmaFECT3 transfection reagent following manufacturer’s instruction. Human islets were cultured in RPMI medium containing 5.5 mmol/l glucose in the absence or presence of 50 µmol/l H2O2 for 24 h.
Apoptosis assay
Cell death was determined by quantification of cytoplasmic histone-associated DNA fragments using the Cell Death Detection ELISA (Roche Diagnostics, Castle Hill, NSW, Australia). Absorbance values were normalized to total DNA content measured by SYBR Green I (Roche Diagnostics, Castle Hill, NSW, Australia).
RNA analysis
Total RNA was extracted using RNAeasy kit (Qiagen, Victoria, Australia) and cDNA synthesized using the QuantiTect reverse transcription kit (Qiagen, Victoria, Australia). Real-time RT-PCR was performed using power SYBR Green PCR Master Mix and a 7900HT Real-Time PCR system (Applied Biosystems, Foster City, CA, USA). Primer sequences are listed in Supplementary Table 3. The value obtained for a specific gene product was normalized to the control gene cyclophilin A and expressed as a fold-change of the value in control condition. For human samples, RNA was extracted using Trizol and cDNA was synthesized using a ‘High Capacity cDNA Reverse Transcription Kit’ as previously described23. Taqman gene expression assays were used for Phlda3 (Hs00385313_ml) and the control gene Gapdh (Hs02758991_g1) (Applied Biosystems, Foster City, CA) using a TaqMan Fast Universal PCR Master Mix on a ViiA7 PCR machine.
Protein analysis
Western blotting and band quantification were performed as previously described24,25. Phospho-v-akt murine thymoma viral oncogene homolog (AKT) (S473) and total AKT antibodies (9271 and 9272) were from Cell Signaling (Danvers, MA USA) and actin antibody (A2066) from Sigma. Activation of Nuclear factor κB (NFκB) was assessed by quantification of subunit p65 phosphorylation (pS536) using the NFκB p65 (pS536) SimpleStep ELISA kit (ab176647, Abcam, Cambridge, UK). Absorbance values were normalized to total protein content measured with the Pierce BCA protein assay kit (Thermo Fisher Scientific, Lafayette, CO, USA).
Immunodetection of PHLDA3
After culture, cells on coverslips were washed with ice cold PBS and fixed in 4% paraformaldehyde for 15 min, permeabilized with PBS-Triton 0.05%, blocked with 5% BSA and incubated overnight at 4 °C with goat polyclonal anti-PHLDA3 antibody (ab22822, Abcam, Cambridge, UK) diluted 1:70 in 1% BSA. The next day, cells were washed and endogenous peroxidases inactivated with 3% H2O2 (vol/vol) before incubation for 1 h at room temperature with HRP-conjugated donkey polyclonal anti-goat secondary antibody (705-035-003, Jackson ImmunoResearch) diluted 1:500 in 1% BSA. Human islets were washed and fixed in 4% paraformaldehyde for 4 h and embedded in paraffin. Antigen retrieval was performed on 5 µm islet sections using a microwave in the presence of citrate buffer (pH 5,7). Islet sections were then treated with 3% H2O2 (vol/vol), blocked with 5% BSA and incubated overnight at 4 °C with goat polyclonal anti-PHLDA3 antibody diluted 1:100 in 1% BSA. The next day, islet sections were incubated for 1 h at room temperature with HRP-conjugated secondary antibody diluted 1:500 in 1% BSA. For cells and islet sections, the signal was revealed by 3,3’-diaminobenzidine (K3468, DAKO, Carpintera, USA). Islet sections were counterstained with hematoxylin (S3301, DAKO). Insulin and glucagon immunostainings were performed on adjacent islet sections as previously reported24,25. Anti-insulin antibody (3014, Cell Signaling Technology, Danvers, MA, USA) was diluted 1:500 and anti-glucagon antibody (G2654, Sigma) was diluted 1:2000. Secondary Alexa fluor antibodies (Thermo Fisher Scientific) were diluted 1:1000.
Statistical analysis
Results are means ± SEM for the indicated number of experiments. Statistical significance was assessed by unpaired two-tailed student t-test, one-way ANOVA and a post-test of Newman-Keuls or two-way ANOVA and a post-test of Bonferroni.
Results
Phlda3 mRNA levels are upregulated in the islets of diabetic mice and humans
We first assessed whether the Phlda3 expression is altered in diabetes. The mRNA levels of Phlda3 were markedly upregulated in the islets of db/db mice, a model of type 2 diabetes, in comparison to age-matched lean control C57BL/KsJ mice (Fig. 1a). In NOD mice, a model of type 1 diabetes, we found a significant upregulation of Phlda3 mRNA levels in comparison to control Balb/c mice (Fig. 1b). In humans, the mRNA levels of Phlda3 were markedly upregulated in the islets of type 2 diabetes donors in comparison to control subjects (Fig. 1c). These findings show for the first time that the expression of Phlda3 is upregulated in islets from diabetic rodents and humans. The findings raise two important questions: (1) what is (are) the mechanism(s) of Phlda3 induction in beta cells? and (2) what is the role(s) of Phlda3 in beta cell pathophysiology?

Phlda3 mRNA levels are upregulated in the islets of diabetic animal models and human subjects. Changes in the mRNA levels of Phlda3 in the islets of (a) control (C, white bar) and db/db mice (black bar), (b) control (C, white bar) and NOD mice (black bar) and (c) human non-diabetic (N, white bar) and type 2 diabetic (T2D, black bar) subjects. n = 6 animals per group for (a), n = 5–6 animals per group for (b) and n = 5–8 human subjects per group for (c). *p < 0.05, **p < 0.01, ***p < 0.001 vs control animals or non-diabetic subjects.
Phlda3 expression is induced by cytokine and palmitate treatment
To explore the mechanism(s) underlying Phlda3 induction in type 1 and type 2 diabetes, we exposed MIN6 cells and isolated mouse islets to factors that characterize the diabetic milieu, including proinflammatory cytokines and saturated free fatty acids3,6. We found that exposure of MIN6 cells to the cytokines IL1β, IFNγ and TNFα or the saturated fatty acid palmitate markedly upregulated the mRNA levels of Phlda3 (Fig. 2a,f) with parallel induction of ER stress (Ddit3, Trb3) and antioxidant (Hmox1) genes (Fig. 2b,c,g,h). In agreement, PHLDA3 protein immunostaining was also increased by cytokine (Fig. 2e) and palmitate (Fig. 2j) treatments. These treatments have previously been demonstrated to be associated with increased beta cell death14. The upregulation of Phlda3 mRNA levels by cytokine and palmitate treatments was confirmed in primary mouse islets (Fig. 2d,i). These results suggest that Phlda3 is induced by common features of the diabetic environment in beta cells.

Phlda3 mRNA and protein levels are upregulated by cytokine and palmitate treatment in parallel with the induction of ER stress and antioxidant genes. MIN6 cells (n = 4–9 experiments) or primary mouse islets (n = 5–10 experiments) were cultured in the absence (white bars) or presence (black bars) of cytokines (24 h) or palmitate (48 h). Changes in the mRNA levels of Phlda3, Ddit3, Trb3 and Hmox1 and PHLDA3 immunostaning in cytokine- (a–c,e) or palmitate-treated (f–h,j) MIN6 cells. Changes in the mRNA levels of Phlda3 in cytokine- (d) or palmitate-high glucose-treated (i) islets. **p < 0.01, ***p < 0.001 vs control. Scale bars, 50 µm.
Phlda3 expression is induced by ER stress
ER stress is a key mechanism through which proinflammatory cytokines and palmitate have been shown to affect rodent and human beta cells11,13,14,26,27. We therefore tested whether exposure of beta cells to the pharmacological ER stress inducer thapsigargin affected Phlda3 expression. Interestingly, Phlda3 mRNA levels were markedly upregulated by thapsigargin treatment (Fig. 3a) in parallel with increased mRNA levels of adaptive (Hspa5, Hsp90b1 and Fkbp11) and proapoptotic (Ddit3 and Trb3) UPR genes (Fig. 3b–f). This finding was also confirmed at the protein level. Indeed, PHLDA3 protein immunostaining was markedly increased by thapsigargin treatment (Fig. 3g). These results demonstrate that Phlda3 is a novel ER stress-responsive gene in beta cells.

Phlda3 mRNA and protein levels are upregulated by thapsigargin treatment in parallel with the induction of ER stress genes. MIN6 cells were cultured for 24 h in the absence (white bars) or presence of 300 nmol/l (grey bars) or 1000 nmol/l (black bars) thapsigargin (Tg). Changes in the mRNA levels of Phlda3, Hmox1, Trb3, Hspa5, Hsp90b1 and Fkbp11 (a–f) and PHLDA3 immunostaining (g). *p < 0.05, ***p < 0.001 vs control. ###p < 0.001 vs thapsigargin 300 nM. Scale bar, 50 µm.
Phlda3 expression is induced by oxidative stress
Besides ER stress, cytokines and palmitate affect beta cells via the induction of oxidative stress. To determine the effects of oxidative stress on Phlda3 in beta cells, we exposed MIN6 cells to ribose. Ribose is a sugar that produces ROS more potently than glucose and is an established model of beta cell glucotoxicity and oxidative stress28,29. Ribose treatment strongly upregulated Phlda3 mRNA levels (Fig. 4a) with parallel upregulation of antioxidant genes (Hmox1, Gpx1 and Srxn1) (Fig. 4b). We also confirmed upregulation of Phlda3 mRNA levels by ribose treatment in primary mouse islets (Fig. 4c). In agreement, ribose treatment strongly increased PHLDA3 protein immunostaining in MIN6 cells (Fig. 4d). We next verified whether prolonged exposure to elevated glucose levels, thereby mimicking the diabetic milieu, may have an impact on Phlda3 expression. Interestingly, culture of mouse islets for 3 weeks in the presence of 30 mmol/l glucose instead of 10 mmol/l markedly upregulated Phlda3 mRNA levels (Fig. 4e) in parallel with the upregulation of the antioxidant gene Srxn1 (Fig. 4f). Confirming these findings in humans, we found that exposure of human islets to H2O2 strongly upregulated PHLDA3 immunostaining (Fig. 4g and Supplementary Fig. 1). Interestingly, insulin and glucagon immunostaining on adjacent islet sections revealed that PHLDA3 protein expression was induced throughout the islets in beta cells as well as in islet non-beta cells including alpha cells (Fig. 4g).

Phlda3 mRNA and protein levels are upregulated by oxidative stress-inducing agents in parallel with the induction of antioxidant genes. MIN6 cells or primary mouse islets were cultured in the absence (white bars) or presence of 50 mmol/l ribose for 48 h or high glucose (30 mmol/l) for 3 weeks (black bars). Human islets were cultured in the absence or presence of 50 µmol/l H2O2. Changes in the mRNA levels of Phlda3, Hmox1, Gpx1 and Srxn1 in ribose-treated MIN6 cells (a,b). Changes in the mRNA levels of Phlda3 in ribose-treated islets (c). Changes in PHLDA3 immunostaining in ribose-treated MIN6 cells (d). Changes in the mRNA levels of Phlda3 and Srxn1 in high glucose-treated islets (e,f). Changes in PHLDA3 immunostaining in H2O2-treated human islets and immunostaining for insulin and glucagon on adjacent islet sections (g). n = 3–5 experiments. **p < 0.01, ***p < 0.001 vs control. Scale bars, 50 µm.
Altogether, these results demonstrate that, in addition to ER stress, Phlda3 is an oxidative stress responsive gene in beta cells.
Phlda3 is induced downstream of XBP1
ER stress triggers several signaling cascades in beta cells to restore homeostasis or otherwise induce apoptosis. X-box Binding Protein 1 (XBP1) is a key ER stress-inducible transcription factor that modulates the expression of several adaptive UPR genes including chaperones, foldases and components of the ER-associated degradation machinery14. It has previously been reported that Phlda3 expression is regulated by XBP1 in hepatocytes20. To determine whether Phlda3 is regulated by XBP1 in beta cells, we used siRNA-mediated inhibition of Xbp1 expression in MIN6 cells14. In cytokine-treated MIN6 cells, Xbp1 inhibition partially prevented Phlda3 mRNA induction (Fig. 5a). We also used islets from Xbp1flox/flox-Pdx1-CreER mice14. Phlda3 induction by cytokines was partially prevented in islets with beta cell specific XBP1 deficiency in comparison to control islets (Fig. 5b). Moreover, in the MIN6 cell model of lipotoxicity, Xbp1 inhibition using siRNA almost completely abolished the palmitate-mediated upregulation of Phlda3 mRNA (Fig. 5c). We have previously demonstrated that inhibition of Xbp1 is associated with increased cytokine and palmitate-induced cell death14. Taken together, the studies demonstrate a novel association between Xbp1-Phlda3 and protection against cytokine toxicity and lipotoxicity in beta cells. Conversely, siRNA-mediated inhibition of the proapoptotic UPR gene Ddit3 significantly upregulated the mRNA levels of Phlda3 under basal conditions and they tended to be increased after palmitate treatment (Fig. 5c).

Phlda3 induction under ER stress is downstream of XBP1. 4-Hydroxytamoxifen-treated control (C) and Xbp1flox/flox-Pdx1-CreER (Xbp1f/f) mouse islets (n = 7 experiments) and MIN6 cells transfected with either control siRNA (si-C), siRNA against Xbp1 (siXbp1) or siRNA against Ddit3 (siDdit3) (n = 9–12 experiments) were cultured in the absence (white bars) or presence (black bars) of cytokines (24 h) or palmitate (48 h). Changes in the mRNA levels of Phlda3 in cytokine-treated MIN6 cells (a) and primary islets (b). Changes in the mRNA levels of Phlda3 in palmitate-treated MIN6 cells (c). **p < 0.01, ***p < 0.001, ****p < 0.0001 vs untreated. #p < 0.05, ##p < 0.01 vs control siRNA or control islets.
Altogether, these results demonstrate that Phlda3 induction in beta cells is differentially regulated by adaptive (Xbp1) and proapoptotic UPR (Ddit3) effectors under conditions of cytokine and lipotoxic stress.
Phlda3 induction protects against cell death during stress
We next explored the role of Phlda3 in stressed beta cells. To this end, we evaluated cell death under stress conditions after siRNA-mediated inhibition of Phlda3 in MIN6 cells. Interestingly, we found that Phlda3 knockdown (Fig. 6a) potentiated cytokine-induced apoptosis (Fig. 6b). Similarly, under oxidative stress conditions induced by ribose treatment, Phlda3 knockdown (Fig. 6c) potentiated apoptosis (Fig. 6d). These results strongly suggest that Phlda3 induction in beta cells under stress is adaptive and may play an important role to promote survival.

Phlda3 knockdown potentiated cytokine- and ribose-induced apoptosis. MIN6 cells transfected with either control siRNA (si-C) or siRNA against Phlda3 (si-Ph3) were cultured in the absence (white bars) or presence (black bars) of cytokines (24 h, a,b) or ribose (48 h, c,d). Changes in (a,c) mRNA levels and (b,d) apoptosis. n = 3–4 experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs untreated. #p < 0.05, ###p < 0.001 vs control siRNA.
Phlda3 regulates several stress response pathways in beta cells
The NFκB pathway
iNos is an important effector implicated in cytokine-mediated beta cell death26,30,31,32. Interestingly, we found that Phlda3 knockdown further increased iNos mRNA levels in control and cytokine-treated MIN6 cells (Fig. 7a). Since iNos is a downstream target of NFκB transcription factor33,34,35, we assessed the expression of Il1β and IκBα, two other known NFκB target genes. Remarkably, the mRNA levels of both genes were further upregulated in cytokine-treated MIN6 cells after Phlda3 knockdown (Fig. 7b,c). In agreement with these findings, cytokine-induced phosphorylation of NFκB subunit p65 on serine 536 (activation) was further increased after Phlda3 knockdown (Fig. 7d). Taken together, these results strongly suggest that NFκB activation is potentiated upon Phlda3 inhibition thereby leading to further upregulation of iNos, Il1β and IκBα. Therefore, the protective effect of Phlda3 induction under cytokines may stem, at least in part, from the repression of this pathway.

Phlda3 inhibition potentiated cytokine-induced activation of the NFκB pathway independently of AKT. MIN6 cells transfected with either control siRNA (si-C) or siRNA against Phlda3 (si-Ph3) were cultured in the absence (white bars) or presence (grey and black bars) of cytokines (15min-24h). Changes in the mRNA levels of iNos, Il1β and Ikbα (a–c). Changes in the phosphorylation of NFκB subunit p65 on serine 536 (d). Changes in phospho- and total AKT protein levels (e,f). n = 3–6 experiments. *p < 0.05, **p < 0.01, ***p < 0.001 vs untreated. #p < 0.05, ##p < 0.01, ###p < 0.001 vs control siRNA.
Evidence has suggested a role of the AKT pathway in NFκB activation in cancer cells36,37,38. Moreover, Phlda3 has been proposed as a repressor of AKT39. Therefore, we assessed whether Phlda3 inhibition affected AKT phosphorylation (activation) after cytokine treatment. Under basal conditions, differences in the phosphorylated AKT/AKT ratio were not detected in cells transfected with control or Phlda3 siRNA (Fig. 7e,f). After cytokine treatment, AKT phosphorylation was rapidly and strongly reduced to a similar extent in cells transfected with control or Phlda3 siRNA (Fig. 7e,f). These results argue against a potential contribution of AKT to the enhanced activation of the NFκB pathway observed after Phlda3 inhibition.
The antioxidant response
Since cytokine treatment induces oxidative stress in beta cells, we assessed whether the inhibition of Phlda3 affected the antioxidant response. Interestingly, we found that cytokine-mediated upregulation of Gpx1 mRNA levels was prevented after Phlda3 inhibition (Fig. 8a). In addition, Srxn1 expression was reduced in cytokine-treated cells after Phlda3 inhibition (Fig. 8b). On the other hand, the mRNA levels of Hmox1 were not affected (Fig. 8c). These results suggest that Phlda3 is required for maintaining an adequate expression of specific antioxidant genes under stress conditions.

Phlda3 inhibition negatively impacts antioxidant and adaptive UPR gene expression without affecting cytokine-induced alterations of beta cell differentiation. MIN6 cells transfected with either control siRNA (si-C) or siRNA against Phlda3 (si-Ph3) were cultured in the absence (white bars) or presence (black bars) of cytokines (24 h). Changes in the mRNA levels of (a–c) antioxidant genes, (d–g) adaptive UPR gene, (h) Ddit3, (i,j) beta cell enriched genes and (k,l) genes involved in beta cell dedifferentiation. n = 4 experiments. *p < 0.05, ***p < 0.001 vs untreated. #p < 0.05, ##p < 0.01, ###p < 0.001 vs control siRNA.
The adaptive UPR
We next assessed the influence of Phlda3 inhibition on the ability of cytokines to downregulate the adaptive UPR. We found that the mRNA levels of adaptive UPR genes were significantly lower (Xbp1, Hspa5, Fkbp11) or tended to be lower (Pdia4) following cytokine treatment in cells transfected with Phlda3 siRNA compared to control siRNA (Fig. 8d–g). On the other hand, the cytokine-mediated upregulation of the proapoptotic UPR gene Ddit3 was not different between control siRNA- and Phlda3 siRNA-treated cells (Fig. 8h). These results suggest that Phlda3 partially protects against the loss of adaptive UPR gene expression during inflammatory stress.
Finally, we tested whether the inhibition of Phlda3 influences cytokine-mediated beta cell dedifferentiation40. We found that the cytokine-mediated downregulation of beta cell genes Mafa and Slc2a2 (also known as Glut2) was not affected by Phlda3 knockdown (Fig. 8i,j). Furthermore, the cytokine-induced upregulation of genes associated with beta cell dedifferentiation, Id1 and Cdkn1a (also known as p21) were not affected by Phlda3 inhibition (Fig. 8k,l).
All together, these results demonstrate that Phlda3 plays an important adaptive role under stress conditions via the modulation of several stress responses including the NFκB pathway, the antioxidant response and the UPR. Our studies suggest that Phlda3 expression may be beneficial for preserving beta cell mass during the pathogenesis of diabetes.
Discussion
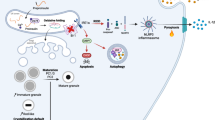
The elevated levels of fatty acids, glucose and proinflammatory cytokines of the (pre)diabetic environment play a major role in triggering beta cell stress and demise3,4,5,41. The complex interaction of stress-activated adaptive and proapoptotic responses determines the fate of beta cells. However, knowledge of the stress response pathways in beta cells is incomplete, which has hampered the development of strategies to preserve beta cell mass in diabetic subjects. In the present study, we have identified Phlda3 as a novel stress-responsive gene in beta cells. We report for the first time that: (1) Phlda3 is upregulated in islets of diabetic rodents and humans; (2) Phlda3 expression is induced in response to the major stress conditions associated with beta cell failure in diabetes, namely inflammatory, ER and oxidative stress; and (3) Phlda3 plays an important adaptive role to protect against beta cell death during stress (Fig. 9).

The proposed model. The elevated levels of proinflammatory cytokines, fatty acids and glucose play a key role in triggering beta cell stress and demise. Phlda3 is a novel adaptive beta cell stress response gene induced downstream of XBP1 that promotes beta cell survival via the repression of the NFκB pathway and the maintenance of adequate antioxidant and adaptive UPR gene expression. FFA; free fatty acids, NO; nitric oxide, ROS; reactive oxygen species, T1D; type 1 diabetes, T2D; type 2 diabetes.
Beta cells are highly vulnerable to ER stress because of their heavy engagement in proinsulin biosynthesis. Therefore, an intact and fine-tuned adaptive UPR is vital for beta cell viability. Indeed, previous evidence linked the failure of this response with beta cell decompensation and progression to diabetes42,43,44. Interestingly, our data suggest that Phlda3 plays a complex role both downstream and upstream of the XBP1 arm of the adaptive UPR (Fig. 9). Thus, we found that Xbp1 was required for stress-induced upregulation of Phlda3 in MIN6 cells and primary mouse islets. The Xbp1-dependent upregulation of Phlda3 is in agreement with previous findings in hepatocytes20. However, in the latter study, Phlda3 contributed to ER stress-mediated hepatocyte death20, in opposition to the protective role of Phlda3, and Xbp114, in stressed beta cells. These findings are suggestive of distinct tissue-specific roles of Xbp1-Phlda3 signalling during stress. Interestingly, inhibition of the proapoptotic ER stress gene Ddit3 was associated with marked upregulation of Phlda3 mRNA levels. This further supports the notion that Phlda3 is protective in beta cells since Ddit3 inhibition is associated with improved beta cell survival in vitro14,25 and in vivo12.
Moreover, our findings identify a previously unrecognized role of Phlda3 as an upstream regulator of adaptive UPR gene expression. Thus, Phlda3 inhibition reduced adaptive UPR gene mRNA levels after cytokine treatment (Fig. 8d–g). Interestingly, under these conditions Phlda3 inhibition potentiated the upregulation of iNos (Figs 7a and 9). Notably, nitric oxide is a known repressor of the adaptive UPR32. Thus, changes in iNos activation may provide a mechanism whereby Phlda3 regulates the adaptive UPR following cytokine stimulation.
In addition to iNos, Phlda3 inhibition resulted in the upregulation of other NFκB target genes (Il1β and Ikbα) in parallel with increased NFκB phosphorylation. This raises the possibility that Phlda3 acts as a brake on NFκB activation. Since the NFκB pathway is considered proapoptotic in beta cells45, the findings further support a protective role for Phlda3. How this repression may operate is unclear. We investigated AKT as a candidate because previous studies have linked it both with NFκB activation36,37,38 and repression by Phlda339. However, in our model, the phosphorylation of AKT (activation) was unaffected by Phlda3 inhibition under both basal conditions and following cytokine treatment (Fig. 7e,f). This suggests that the regulation of NFκB by Phlda3 occurs independently of AKT. Alternatively, cytokine treatment triggers oxidative stress with subsequent upregulation of antioxidant genes. Oxidative stress can also upregulate the expression of inflammatory genes in beta cells46. Therefore, dysregulation of the antioxidant response such as occurs with Phlda3 inhibition (reduced Gpx1 and Srxn1 mRNA levels, Fig. 8a,b) may lead to more severe oxidative stress. Accordingly, one could postulate that enhanced expression of NFκB target genes may result from increased oxidative stress under these conditions.
Our findings contrast with a recent report showing that islets from mice with Phlda3 deletion are more resistant to hypoxic stress in the context of islet transplantation47. In our model, Phlda3 knockdown had no significant effect on hypoxia-induced apoptosis (Supplementary Fig. 2). The reasons for these discrepant results are not clear. The studies of Sakata et al. adopted mice with constitutive whole body knockout of Phlda3. This raises the possibility of developmental effects in the knockout islets that are independent of Phlda3 expression in adult beta cells. Indeed, the Phlda3 knockout mice display a complex phenotype with the potential for metabolic changes in other tissues secondarily influencing islets. Phlda3 knockout mice develop islet hyperplasia only later in life with an altered distribution of small and large islets. Perhaps Phlda3 affects the stress response during beta cell ageing48,49, which may influence islet transplantation outcomes in the longer term47. Together with the knowledge that Phlda1 regulates insulin sensitivity and energy expenditure18, it is clear that experiments that employ inducible beta cell specific modulation of Phlda3 are needed. Moreover, hypoxic stress may involve unique regulatory networks compared with other stressors in beta cells, as exemplified by the strong suppression of XBP1 protein levels and downstream target genes25.
Interestingly, in mouse islets, Phlda3 mRNA levels were markedly upregulated in the type 2 diabetes model (Fig. 1a), but displayed comparatively modest induction in the type 1 diabetes model (Fig. 1b). This may be related to differences in the nature or duration of the diabetes stress conditions between these models. Finally, the observation that oxidative stress upregulated PHLDA3 expression in both beta and non-beta cells in human islets suggests that PHLDA3 may have a potential role in islet non-beta cells, including alpha cells, which are also crucial for glucose homeostasis. Therefore, upregulated mRNA levels of Phlda3 in human T2D islets may involve an effect in both beta and non-beta cells.
In conclusion, we have unveiled a novel role of Phlda3 in beta cell pathophysiology. Our studies show for the first time that: (1) Phlda3 is upregulated in the islets of diabetic mice and humans; (2) Phlda3 is induced by inflammatory, ER and oxidative stress; (3) Phlda3 is regulated positively by the adaptive UPR mediator, Xbp1 and negatively by the proapoptotic UPR mediator, Ddit3; and (4) Phlda3 contributes to the adaptive response to stress through modulation of the NFκB pathway, specific antioxidant genes and the adaptive UPR. These observations reveal a novel molecular mechanism regulating beta cell survival during stress and suggest the targeting of the Xbp1-Phlda3 axis as a potential therapeutic strategy in diabetes.
References
Matveyenko, A. V. & Butler, P. C. Relationship between beta-cell mass and diabetes onset. Diabetes Obes Metab 10(Suppl 4), 23–31, https://doi.org/10.1111/j.1463-1326.2008.00939.x (2008).
Cnop, M. et al. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 54(Suppl 2), S97–107 (2005).
Eizirik, D. L., Colli, M. L. & Ortis, F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol 5, 219–226, https://doi.org/10.1038/nrendo.2009.21 (2009).
Poitout, V. et al. Glucolipotoxicity of the pancreatic beta cell. Biochim Biophys Acta 1801, 289–298, https://doi.org/10.1016/j.bbalip.2009.08.006 (2010).
Bensellam, M., Laybutt, D. R. & Jonas, J. C. The molecular mechanisms of pancreatic beta-cell glucotoxicity: recent findings and future research directions. Mol Cell Endocrinol 364, 1–27, https://doi.org/10.1016/j.mce.2012.08.003 (2012).
Biden, T. J., Boslem, E., Chu, K. Y. & Sue, N. Lipotoxic endoplasmic reticulum stress, beta cell failure, and type 2 diabetes mellitus. Trends Endocrinol Metab 25, 389–398, https://doi.org/10.1016/j.tem.2014.02.003 (2014).
Oyadomari, S. et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest 109, 525–532, https://doi.org/10.1172/JCI14550 (2002).
Shalev, A. et al. Oligonucleotide microarray analysis of intact human pancreatic islets: identification of glucose-responsive genes and a highly regulated TGFbeta signaling pathway. Endocrinology 143, 3695–3698, https://doi.org/10.1210/en.2002-220564 (2002).
Elouil, H. et al. Acute nutrient regulation of the unfolded protein response and integrated stress response in cultured rat pancreatic islets. Diabetologia 50, 1442–1452, https://doi.org/10.1007/s00125-007-0674-4 (2007).
Qian, B. et al. TRIB3 [corrected] is implicated in glucotoxicity- and endoplasmic reticulum-stress-induced [corrected] beta-cell apoptosis. J Endocrinol 199, 407–416, https://doi.org/10.1677/JOE-08-0331 (2008).
Laybutt, D. R. et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia 50, 752–763, https://doi.org/10.1007/s00125-006-0590-z (2007).
Song, B., Scheuner, D., Ron, D., Pennathur, S. & Kaufman, R. J. Chop deletion reduces oxidative stress, improves beta cell function, and promotes cell survival in multiple mouse models of diabetes. J Clin Invest 118, 3378–3389, https://doi.org/10.1172/JCI34587 (2008).
Cnop, M. et al. RNA sequencing identifies dysregulation of the human pancreatic islet transcriptome by the saturated fatty acid palmitate. Diabetes 63, 1978–1993, https://doi.org/10.2337/db13-1383 (2014).
Chan, J. Y. et al. The balance between adaptive and apoptotic unfolded protein responses regulates beta-cell death under ER stress conditions through XBP1, CHOP and JNK. Mol. Cell Endocrinol 413, 189–201, https://doi.org/10.1016/j.mce.2015.06.025 (2015).
Scheuner, D. & Kaufman, R. J. The unfolded protein response: a pathway that links insulin demand with beta-cell failure and diabetes. Endocr Rev 29, 317–333, https://doi.org/10.1210/er.2007-0039 (2008).
Fonseca, S. G., Gromada, J. & Urano, F. Endoplasmic reticulum stress and pancreatic beta-cell death. Trends Endocrinol Metab 22, 266–274, https://doi.org/10.1016/j.tem.2011.02.008 (2011).
Scheffzek, K. & Welti, S. Pleckstrin homology (PH) like domains - versatile modules in protein-protein interaction platforms. FEBS Lett 586, 2662–2673, https://doi.org/10.1016/j.febslet.2012.06.006 (2012).
Basseri, S. et al. Loss of TDAG51 results in mature-onset obesity, hepatic steatosis, and insulin resistance by regulating lipogenesis. Diabetes 62, 158–169, https://doi.org/10.2337/db12-0256 (2013).
Ohki, R. et al. PHLDA3 is a novel tumor suppressor of pancreatic neuroendocrine tumors. Proc Natl Acad Sci USA 111, E2404–2413, https://doi.org/10.1073/pnas.1319962111 (2014).
Han, C. Y., Lim, S. W., Koo, J. H., Kim, W. & Kim, S. G. PHLDA3 overexpression in hepatocytes by endoplasmic reticulum stress via IRE1-Xbp1s pathway expedites liver injury. Gut 65, 1377–1388, https://doi.org/10.1136/gutjnl-2014-308506 (2016).
O’Connell, P. J. et al. Multicenter Australian trial of islet transplantation: improving accessibility and outcomes. Am J Transplant 13, 1850–1858, https://doi.org/10.1111/ajt.12250 (2013).
Miyazaki, J. et al. Establishment of a pancreatic beta cell line that retains glucose-inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 127, 126–132, https://doi.org/10.1210/endo-127-1-126 (1990).
Joglekar, M. V. & Hardikar, A. A. Isolation, expansion, and characterization of human islet-derived progenitor cells. Methods Mol Biol 879, 351–366, https://doi.org/10.1007/978-1-61779-815-3_21 (2012).
Bensellam, M. et al. Glucose-induced O(2) consumption activates hypoxia inducible factors 1 and 2 in rat insulin-secreting pancreatic beta-cells. PLoS One 7, e29807, https://doi.org/10.1371/journal.pone.0029807 (2012).
Bensellam, M. et al. Hypoxia reduces ER-to-Golgi protein trafficking and increases cell death by inhibiting the adaptive unfolded protein response in mouse beta cells. Diabetologia 59, 1492–1502, https://doi.org/10.1007/s00125-016-3947-y (2016).
Cardozo, A. K. et al. Cytokines downregulate the sarcoendoplasmic reticulum pump Ca2+ ATPase 2b and deplete endoplasmic reticulum Ca2+, leading to induction of endoplasmic reticulum stress in pancreatic beta-cells. Diabetes 54, 452–461 (2005).
Brozzi, F. et al. Cytokines induce endoplasmic reticulum stress in human, rat and mouse beta cells via different mechanisms. Diabetologia 58, 2307–2316, https://doi.org/10.1007/s00125-015-3669-6 (2015).
Tanaka, Y., Tran, P. O., Harmon, J. & Robertson, R. P. A role for glutathione peroxidase in protecting pancreatic beta cells against oxidative stress in a model of glucose toxicity. Proc Natl Acad Sci USA 99, 12363–12368, https://doi.org/10.1073/pnas.192445199 (2002).
Bensellam, M., Montgomery, M. K., Luzuriaga, J., Chan, J. Y. & Laybutt, D. R. Inhibitor of differentiation proteins protect against oxidative stress by regulating the antioxidant-mitochondrial response in mouse beta cells. Diabetologia 58, 758–770, https://doi.org/10.1007/s00125-015-3503-1 (2015).
Oyadomari, S. et al. Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci USA 98, 10845–10850, https://doi.org/10.1073/pnas.191207498 (2001).
Thomas, H. E., Darwiche, R., Corbett, J. A. & Kay, T. W. Interleukin-1 plus gamma-interferon-induced pancreatic beta-cell dysfunction is mediated by beta-cell nitric oxide production. Diabetes 51, 311–316 (2002).
Chan, J. Y., Cooney, G. J., Biden, T. J. & Laybutt, D. R. Differential regulation of adaptive and apoptotic unfolded protein response signalling by cytokine-induced nitric oxide production in mouse pancreatic beta cells. Diabetologia 54, 1766–1776, https://doi.org/10.1007/s00125-011-2139-z (2011).
Flodstrom, M., Welsh, N. & Eizirik, D. L. Cytokines activate the nuclear factor kappa B (NF-kappa B) and induce nitric oxide production in human pancreatic islets. FEBS Lett 385, 4–6 (1996).
Darville, M. I. & Eizirik, D. L. Regulation by cytokines of the inducible nitric oxide synthase promoter in insulin-producing cells. Diabetologia 41, 1101–1108, https://doi.org/10.1007/s001250051036 (1998).
Naamane, N., van Helden, J. & Eizirik, D. L. In silico identification of NF-kappaB-regulated genes in pancreatic beta-cells. BMC Bioinformatics 8, 55, https://doi.org/10.1186/1471-2105-8-55 (2007).
Li, B. et al. Id-1 activation of PI3K/Akt/NFkappaB signaling pathway and its significance in promoting survival of esophageal cancer cells. Carcinogenesis 28, 2313–2320, https://doi.org/10.1093/carcin/bgm152 (2007).
Bai, D., Ueno, L. & Vogt, P. K. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer 125, 2863–2870, https://doi.org/10.1002/ijc.24748 (2009).
Akca, H., Demiray, A., Tokgun, O. & Yokota, J. Invasiveness and anchorage independent growth ability augmented by PTEN inactivation through the PI3K/AKT/NFkB pathway in lung cancer cells. Lung Cancer 73, 302–309, https://doi.org/10.1016/j.lungcan.2011.01.012 (2011).
Kawase, T. et al. PH domain-only protein PHLDA3 is a p53-regulated repressor of Akt. Cell 136, 535–550, https://doi.org/10.1016/j.cell.2008.12.002 (2009).
Bensellam, M., Jonas, J. C. & Laybutt, D. R. Mechanisms of beta-cell dedifferentiation in diabetes: recent findings and future research directions. J Endocrinol 236, R109–R143, https://doi.org/10.1530/JOE-17-0516 (2018).
Kolb, H. & Mandrup-Poulsen, T. An immune origin of type 2 diabetes? Diabetologia 48, 1038–1050, https://doi.org/10.1007/s00125-005-1764-9 (2005).
Chan, J. Y., Luzuriaga, J., Bensellam, M., Biden, T. J. & Laybutt, D. R. Failure of the adaptive unfolded protein response in islets of obese mice is linked with abnormalities in beta-cell gene expression and progression to diabetes. Diabetes 62, 1557–1568, https://doi.org/10.2337/db12-0701 (2013).
Omikorede, O. et al. ER stress in rodent islets of Langerhans is concomitant with obesity and beta-cell compensation but not with beta-cell dysfunction and diabetes. Nutr Diabetes 3, e93, https://doi.org/10.1038/nutd.2013.35 (2013).
Herbert, T. P. & Laybutt, D. R. A Reevaluation of the Role of the Unfolded Protein Response in Islet Dysfunction: Maladaptation or a Failure to Adapt? Diabetes 65, 1472–1480, https://doi.org/10.2337/db15-1633 (2016).
Eizirik, D. L., Cardozo, A. K. & Cnop, M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev 29, 42–61, https://doi.org/10.1210/er.2007-0015 (2008).
Jonas, J. C. et al. Glucose regulation of islet stress responses and beta-cell failure in type 2 diabetes. Diabetes Obes Metab 11(Suppl 4), 65–81, https://doi.org/10.1111/j.1463-1326.2009.01112.x (2009).
Sakata, N. et al. Pleckstrin homology-like domain family A, member 3 (PHLDA3) deficiency improves islets engraftment through the suppression of hypoxic damage. PLoS One 12, e0187927, https://doi.org/10.1371/journal.pone.0187927 (2017).
Aguayo-Mazzucato, C. et al. Beta Cell Aging Markers Have Heterogeneous Distribution and Are Induced by Insulin Resistance. Cell Metab 25, 898–910 e895, https://doi.org/10.1016/j.cmet.2017.03.015 (2017).
Aguayo-Mazzucato, C. et al. Acceleration of beta Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab 30, 129–142 e124, https://doi.org/10.1016/j.cmet.2019.05.006 (2019).
Acknowledgements
We sincerely thank Michèle De Beukelaer (platform 2IP, UCLouvain) for her help with the setup of the PHLDA3 immunostainings; Fatma Belhaj Aissa (Pôle d’endocrinology, diabète et nutrition, UCLouvain) for her help with the preparation of human islet sections and immunostainings; and Cassandra Liang (Garvan Institute of Medical Research) for experimental help. Some of the data were presented as an abstract at the 52nd EASD meeting in Munich, Germany, 12–16 September 2016. This work was supported by a grant from the National Health and Medical Research Council (NHMRC) of Australia (APP1144206) and the Diabetes Australia Research Program (to DRL) and a grant from the Société Francophone du Diabète, Paris, France (no. SFD/MSD 2016) (to JCJ). JYC is supported by an NHMRC Early Career Fellowship. JCJ is Research Director of the Fonds de la Recherche Scientifique-FNRS, Belgium. St Vincent’s Institute receives support from the Operational Infrastructure Support Scheme of the Government of Victoria. MB was supported by a MOVE-in Louvain/EC Marie-Curie incoming postdoctoral fellowship and is actually supported by a fellowship from the Clinical Research Fund, Cliniques Universitaires Saint-Luc and UCLouvain.
Author information
Authors and Affiliations
Contributions
M.B. and D.R.L. conceived and designed experiments, acquired and analysed data and wrote the manuscript. K.L., J.Y.C., M.V.J., A.A.H., T.L., H.E.T. & J.C.J. designed experiments, acquired and analysed data and critically reviewed the manuscript. All authors approved the final version of the manuscript. D.R.L. is the guarantor of this work.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bensellam, M., Chan, J.Y., Lee, K. et al. Phlda3 regulates beta cell survival during stress. Sci Rep 9, 12827 (2019). https://doi.org/10.1038/s41598-019-49289-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-49289-5
This article is cited by
-
Identification of unique cell type responses in pancreatic islets to stress
Nature Communications (2024)
-
Characterizing the role of Phlda3 in the development of acute toxicity and malignant transformation of hematopoietic cells induced by total-body irradiation in mice
Scientific Reports (2023)
-
PHLDA3 promotes lung adenocarcinoma cell proliferation and invasion via activation of the Wnt signaling pathway
Laboratory Investigation (2021)
-
Low-intensity ultrasound inhibits melanoma cell proliferation in vitro and tumor growth in vivo
Journal of Medical Ultrasonics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
