
Abstract
Sulfate aerosol (SO42−) preserved in Antarctic ice cores is discussed in the light of interactions between marine biological activity and climate since it is mainly sourced from biogenic emissions from the surface ocean and scatters solar radiation during traveling in the atmosphere. However, there has been a paradox between the ice core record and the marine sediment record; the former shows constant non-sea-salt (nss-) SO42− flux throughout the glacial-interglacial changes, and the latter shows a decrease in biogenic productivity during glacial periods compared to interglacial periods. Here, by ensuring the homogeneity of sulfur isotopic compositions of atmospheric nss-SO42− (δ34Snss) over East Antarctica, we established the applicability of the signature as a robust tool for distinguishing marine biogenic and nonmarine biogenic SO42−. Our findings, in conjunction with existing records of nss-SO42− flux and δ34Snss in Antarctic ice cores, provide an estimate of the relative importance of marine biogenic SO42− during the last glacial period to be 48 ± 10% of nss-SO42−, slightly lower than 59 ± 11% during the interglacial periods. Thus, our results tend to reconcile the ice core and sediment records, with both suggesting the decrease in marine productivity around Southern Ocean under the cold climate.
Similar content being viewed by others


Introduction
Secondary sulfate plays an important role in aerosol and cloud interactions and influences solar radiation1. In Antarctica, because of isolation from major anthropogenic SO2 emissions over the continents, the main source of non-sea-salt (nss-) SO42− is dimethyl sulfide (DMS) produced by marine phytoplankton living in the Southern Ocean2. Therefore, the nss-SO42− preserved in Antarctic ice cores is used as a record of past marine biogenic activity, and its response and feedback to climate change are debated3,4,5.
It has been shown that nss-SO42− flux recorded in Antarctic ice cores has not significantly changed throughout the last eight glacial cycles, which is concluded to indicate a nonsignificant change in marine biogenic activity3. However, this conclusion is inconsistent with the implication derived from marine sediment cores that shows lower productivity at latitudes higher than 50°S during the last glacial period than during the current warm period6. To unravel the cause of this paradox, identification of the sulfur sources of those nss-SO42− preserved in ice cores potentially provides helpful insights. The stable sulfur isotopic composition of nss-SO42− (δ34Snss) is a potential tool for quantitative estimates of the relative importance of marine biogenic (mb-) and nonmarine biogenic (nmb-) SO42−. Indeed, the δ34Snss values in snow and ice in East Antarctica7,8,9,10 have provided estimates of 80–90% dominance of mb-SO42− in nss-SO42− for the last several hundred years.
In addition to the dependence on its sulfur sources, the δ34S signature could be modified by isotopic fractionation via oxidation of SO2 to SO42−. Although Alexander et al.11 observed the δ34Snss values in deep ice cores that showed ca. 4‰ lower values during the last glacial period than during the current warm period, they interpreted those δ34Snss values to be the result of significant isotopic fractionation through SO2 oxidation, which caused progressive washing out of isotopically heavier SO42− 12 and transport of the remaining SO2 with low δ34Snss values to inland. On the other hand, Uemura et al.7 examined remarkably uniform δ34Snss (14–17‰) in surface snow over the transect between Syowa Station (69°00′S, 39°35′E) and Dome F (77°19′S, 39°42′E), concluding the absence of considerable isotopic fractionation during transport under present-day climate. However, surface snow observations are not sufficient to draw firm conclusions on the reasons for this homogeneity, as inhomogeneous snow deposition throughout the year and remobilization of snow by wind mask real atmospheric spatial and temporal variations. It is thus necessary to observe the differences in this signature between inland and coastal sites before the deposition of SO42−, i.e., for SO42− in atmospheric aerosol samples, to clarify the mechanisms that cause constant flux throughout the ice ages.
To address the above discussion, we performed a year-round observation of δ34Snss values of atmospheric SO42− at Dome C (75°06′S, 123°12′E; 3233 m a.s.l.) and Dumont d’Urville Station (DDU) (66°40′S, 140°01′E; 40 m a.s.l.), inland and coastal sites in East Antarctica, respectively, by utilizing continuous aerosol samples at those sites. Furthermore, we utilized δ34Snss values to estimate changes in sulfur sources in both the present and the past Antarctic atmosphere to infer the major nonmarine sulfur sources that remain to be identified.
Results
Figure 1 shows the time series of concentrations and δ34S values of SO42− at Dome C and DDU throughout 2011. Both were corrected into nss-SO42− values as described in the Methods. At both sites, nss-SO42− concentrations ([SO42−]nss) show well-marked seasonality with maxima of up to ca. 300 ng m−3 in late austral summer (February), with minima less than 20 ng m−3 during winter (August) (Fig. 1b). These trends are consistent with continuous observations at Dome C13,14 and DDU15,16, which are known as a result of enhanced production of biogenic DMS over the Southern Ocean during the austral summer and its subsequent oxidation into SO42−.

Observed concentrations and δ34S values of non-sea-salt (nss-) SO42−. (a) Map of the aerosol sampling sites, with seasonal variations in (b) concentrations (solid line: total suspended particle, dashed line: fine mode particle) and (c) δ34S values of nss-SO42− at Dome C (red) and Dumont d’Urville (blue) with residual values between the two sites. Error bars represent the standard errors propagated from the analytical error of concentrations, δ34S values, and the uncertainty in SO42−/Na+ ratios in sea salt (see Methods).
Along with the seasonal cycle in concentrations, the δ34Snss values also show strong seasonality with the summer maxima and winter minima (Fig. 1b and Supplementary Table S2). During January–March, the δ34Snss values stay within the narrow range of 17.7 to 18.6‰ at both sites (n = 21). In April, the δ34Snss values decrease gradually and reach their lowest values during June–August with mean values of 8.2 ± 2.4‰ (n = 12) and 9.4 ± 2.6‰ (n = 11) at Dome C and DDU, respectively. The δ34Snss values then increase during August–December, with a considerable decrease in November at Dome C, in contrast to the gradual change observed at DDU. This specific November trend at Dome C is discussed below. Consequently, the seasonal δ34Snss cycles and their amplitudes are quite consistent between the two sites.
Discussion
Homogeneity in sulfur isotopic compositions of sulfate in the atmosphere
Figure 1b shows the difference in δ34Snss values between the two sites by subtracting the δ34Snss value of each Dome C sample from that of each DDU sample collected in the closest time period. It is obvious that most of the values do not statistically deviate from 0‰ within the propagated analytical error. Excluding the specifically low δ34Snss values at Dome C in November, the residual values exhibit no systematic trend and averaged 0.5 ± 2.6‰. The δ34Snss values were thus surprisingly homogeneous between Dome C and DDU throughout the year, ensuring that the isotopic fractionation for δ34Snss during transport towards inland is far smaller than the observed variability ranging from 8.2 to 18.6‰. Moreover, the annual weighted averages of the δ34Snss values were 15.8 ± 0.5‰ and 17.0 ± 0.4‰ for Dome C and DDU (Table S2), respectively, which are consistent with the δ34Snss values of 13.7–16.6‰ observed for surface snow over the transect between Syowa Station and Dome Fuji by Uemura et al.7. This result indicates that the δ34Snss signature is preserved in snow SO42− without any significant postdepositional effect.
This result suggests that the δ34Snss values in the Antarctic atmosphere are not significantly influenced by isotopic fractionation during transport and are therefore controlled by the relative importance of sulfur sources. Fifteen years after advocating the possible change in the signature by isotopic fractionation11, these results bring back the applicability of the δ34Snss signature for reconstruction of sulfur sources, i.e., the relative importance of mb-SO42− and nmb-SO42−, through the past climate changes.
Seasonal variation in marine biogenic SO4 2− and nonmarine SO4 2− derived from δ34Snss
Based on the above result, we utilized the signature to estimate the relative contribution of mb- and nmb-SO42− for the present atmospheric samples collected in this study (see Methods for the calculation process). Note that, for DDU samples, the estimate was applied to fine mode particle only due to the absence of δ34Snss data for coarse mode particle (Methods), although the estimate was applied to total suspended particle for Dome C samples. As a result, the [SO42−]mb clearly shows strong seasonality with summer maxima and winter minima at both Dome C and DDU (Fig. 2), which correspond to 79–84% and 89–92% for annual total nss-SO42− at Dome C and DDU, respectively, and consequently controls the seasonality in [SO42−]nss. By contrast, the [SO42−]nmb varies in a small range of 0–39 ng m−3 during most of the period throughout the year, except for November when the [SO42−]nmb increased significantly at both sites. Average values of [SO42−]nmb excluding November were 9.5 ± 7.7 ng m−3 and 5.9 ± 4.0 ng m−3 at Dome C and DDU, respectively.

Estimated concentrations of mb-SO42− and nmb-SO42−. (a) Dome C and (b) Dumont d’Urville with assuming δ34Snmb = 2.5‰. Dotted lines represent the ranges of the uncertainty (standard error) for [SO42−]nmb propagated from the analytical error of concentrations and δ34S values.
In addition to such small variations during January–October, a marked increase in [SO42−]nmb up to 93–127 ng m−3 was observed in November at Dome C. Such an anomalous event was obscured by the increasing trends of mb-SO42− towards summer. The sum of nmb-SO42− during November accounts for ca. 50% of monthly nss-SO42− and ca. 10% of annual nss-SO42− at Dome C (Supplementary Fig. S1). Furthermore, the slight increase in [SO42−]nmb (38–52 ng m−3) was observed at DDU in exactly the same time period as that at Dome C, in the second week of November. Given the same timing at both sites and its higher amplitude at the inland site than at the coastal site, we first explored the possibility that this [SO42−]nmb increase is caused by nmb-SO42− emission located in inland Antarctica. However, this possibility is unlikely given that there has not been observed specific increases in volcanic activity of Mt. Erebus (77°53′S, 167°17′E), an active volcano on the Antarctic continent, during November 2011 based on satellite imagery data17. SO2 emissions from anthropogenic activities over Antarctica related to scientific activities are unlikely since they are generally highest during January–February18 and not in November. One attributable process is long-range transport of nmb-SO42− from other continents, which is supported by the significant correlation between our estimated [SO42−]nmb and previously observed 210Pb14, a commonly used tracer of continental submicron aerosols19, for the period from October to December with a p-value of less than 0.01 (Fig. 3 and Supplementary Table S3). Note that plausible sulfur sources of this specific [SO42−]nmb increase remained uncertain (detailed in Supplementary Information). Although this nmb-SO42− corresponds to only 10% of annual nss-SO42− in the present Antarctic atmosphere, future work to clarify its source is necessary for interpretation of the deep ice core record since it is suggested that the dominant sulfur source shifted to nmb-SO42− during the glacial period, as discussed in the next section.

Relation between [210Pb] (μBq m−3) and [SO42−]nmb (ng m−3) at Dome C in 2011. Red circles represent data obtained during October–December, one month before and after the [SO42−]nmb increase in November. Grey circles and dotted line are the case without the highest [SO42−]nmb. Data for other periods are presented as red diamonds.
Implication for sulfur sources through glacial-interglacial changes
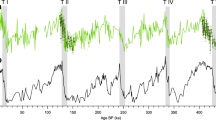
We further applied the above calculation to estimate the shift in sulfur sources during glacial ages using the reported δ34Snss values in snow and ice cores7,8,9,10,11 (Fig. 4). Alexander et al.11 reported δ34Snss values in deep ice cores showing a decrease from 12.2 ± 1.8‰ in the warm periods (Holocene and Eemian) to 10.2 ± 1.6‰ in the last glacial period. These δ34Snss values are clearly lower than our observation of 16.6 ± 0.3‰ on average for the present aerosol samples and 14.2 ± 1.8‰ for the other shallow ice cores. Such difference among the δ34Snss values for interglacial samples is possibly because of volcanic influence as discussed in Supplementary Information, and here we consider δ34Snss values reported by Alexander et al.11 as a representative for the interglacial values. This shift in δ34Snss values corresponds to a gradual decrease in fmb (the fraction of marine biogenic sulfate over the non-sea-salt sulfate) from 86 ± 3% at present to 59 ± 11% during the interglacial and to 48 ± 10% in the last glacial period (Fig. 4).

Observed δ34Snss values in aerosol SO42− (athis study) and in snow and ice core SO42− (bUemura et al.7, cJonsell et al.8, dBaroni et al.9, ePatris et al.10, fAlexander et al.11) shown with the estimated relative contribution of marine biogenic (fmb, green) and nonmarine biogenic (fnmb, gray) SO42−. Note that the δ34Snss values in ice core samples with obvious volcanic inputs are excluded from this estimate. The box plot indicates the interquartile range (box) and the average (line), and the whiskers indicate minimum and maximum values.
Thus, the δ34Snss record in deep ice cores11 combined with the constant nss-SO42− flux record3 imply that marine biologically produced SO42− is decreased during the glacial period. This conclusion is more consistent with the marine sediment core records that show a decrease in biological carbon export production during glacial times around the vicinity of Antarctica at latitudes higher than 50°S6. As a consequence, our result tends to reconciliate conclusions drawn from ice core records and the marine sediment core records, with both suggesting a decrease in marine productivity around Antarctica during cold climates. At the same time, the estimated result shows that nmb-SO42− increased during the glacial period relative to the interglacial. Given that the mineral dust fluxes to Antarctica are controlled by changes in transport efficiency associated with the hydrological cycle20, it is likely that the increased nmb-SO42− during the glacial period was also sourced from other continents, although its sulfur source remains uncertain. The recent work based on the SO42− and Ca2+ ion concentrations over eight glacial cycles21 estimated that the proportional contributions of biogenic sulfur during cold periods could decrease to 24% or 52% in total nss-SO42−, which depends on either assuming nss-Ca2+ originated only from primary terrestrial CaSO4 or from partial contribution of secondarily produced CaSO4 via the reaction between dust-sourced CaCO3 and marine biogenic sulfur. The latter case is in good agreement with our estimate of 48 ± 10% in the last glacial period (Fig. 4), suggesting the nss-SO4 during glacial periods may include both primary and secondary products.
Our estimate that the relative importance of nmb-SO42− had increased during the glacial period advances the understanding of the radiative cooling through the past climate change. Assuming that the micron-sized CaSO4 salt in Antarctic deep ice cores was secondarily produced during their transport from South America, Iizuka et al.22 concluded that the radiative cooling by marine biogenic sulfur had increased during the glacial periods, which is against the CLAW hypothesis proposed by Charlson et al.23. By contrast, our result suggests the sulfur source during glacial periods is not entirely marine biogenic but rather includes continental SO42−. If CaSO4 during glacial period contained continental sulfur, those particles would have brought the radiative cooling to broad areas including a part of the continents in the mid-latitude of the Southern Hemisphere, while there would not have been influenced when assuming marine biogenic sulfur only. Therefore, the radiative cooling by sulfate may have been stronger than the previous estimate that had assumed a marine biogenic sulfur source alone22. Quantitatively, it is worth noting that fnmb during inter-glacial and glacial periods were 41 ± 11% and 52 ± 10%, respectively, based on δ34Snss (Fig. 4), and this difference is smaller than the relative increase in CaSO4 from 7 ± 4% during inter-glacial period to 56 ± 14% during glacial period22. Therefore, although it is true that the increased CaSO4 in the glacial period includes both primary and secondarily products, the changes in sulfur source should be taken into account since it would consequently affect the radiative forcing.
Methods
Sampling and ion quantification
Aerosol samples used for this study were collected in 2011 at Dome C (75°10′S, 123°30′E; 3233 m a.s.l.) and DDU (66°40′S, 140°01′E; 40 m a.s.l.). Sampling site details are presented in the Supplementary Information. Concentration data of Na+, MS−, Cl−, Br−, and C2O42− at Dome C were reported by Legrand et al.14. Additionally, all ion concentration data at DDU were reported by Ishino et al.24, where the sampling and quantification processes are detailed. Coarse (>1 μm) and fine (<1 μm) mode particles for DDU samples and total suspended particles for Dome C samples were collected using a high-volume air sampler at 1.5–1.7 m3 min−1 with a time resolution of 1–2 weeks at both sites. The aerosol loaded filters were kept frozen and were transported to Grenoble, where the ions were extracted to 40 mL of ultrapure water. Quantification of anions (NO3−, SO42−) and cations (K+, Mg2+, Ca2+) was performed using the ion chromatography systems described by Savarino et al.25 and by Jourdain and Legrand26. The measured ion concentrations were corrected for blank values and were reported as the atmospheric concentration in standard temperature and pressure (T = 273.15 K, p = 101,325 Pa) based on meteorological data of Dome C and DDU provided by the IPEV/PNRA Project “Routine Meteorological Observation at Station Concordia” – (www.climantartide.it) and Meteo France. Uncertainties were estimated based on the typical uncertainty of the ion chromatography analyses (5%).
Concentrations of nss-SO42− were calculated by subtracting the sea salt fraction based on the Na+ concentration using the following equation, where k represents the [SO42−]/[Na+] mass ratio in sea salt particles.
A k value in seawater of 0.2527 is generally used for this calculation. However, the sea salt emitted from the sea ice surface at low temperatures below −8 °C is depleted in SO42− relative to Na+ because of the precipitation of mirabilite28. The mixing of those sea salts emitted from the open ocean and sea ice surface results in k values at Dome C and DDU of 0.16 ± 0.0929 and 0.13 ± 0.0426, respectively. We applied those shifted values for samples collected during May–October.
Sulfur isotope analyses
Sulfur isotopic compositions are expressed in delta notation defined with the following equation with respect to Vienna Canyon Diablo Troilite (VCDT) as a reference.
After ion quantification, samples were stored in a freezer at −20 °C before isotopic measurements. A total of 400 nmol and 2 μmol of SO42− were separated from other ions in each Dome C and DDU sample solution using ion chromatography, as described by Ishino et al.24. In this procedure, the yields of 100% of SO42− ensure no isotopic fractionation. We confirmed that the shifts in the δ34S values through this step were smaller than the analytical uncertainties in the subsequent procedures. Note that we analyzed the δ34S value for only the fine mode particles for the DDU samples because a large fraction of sea salt in the coarse mode particles (approximately 40% in summer to 100% in winter) leads to a large uncertainty. We used two methods for the sulfur isotope analyses of the Dome C and DDU samples because of sample size limitations.
For the Dome C samples, the δ34S values of sulfate were then measured using a multiple-collector inductively coupled plasma mass spectrometer (MC-ICP-MS; Neptune Plus, Thermo Fisher Scientific Inc.), as described by Albalat et al.30. Measurements were calibrated to values relative to VCDT with an external reproducibility of the in-house standard materials of ±0.12‰.
The DDU samples were processed through chemical conversion into Ag2S, as described in Geng et al.31, and then into SF6 in a method similar to that described by Ono et al.32, with modification as described by Hattori et al.33. We measured δ34S with a dual inlet system of an isotope ratio mass spectrometer (IRMS; Finnigan MAT 253, Thermo Fisher Scientific Inc.). The uncertainty in the measurement was estimated as ±0.2‰ based on replicate measurements of international standard materials (IAEA S1, S2, and NBS127).
The isotopic compositions of nss-SO42− for each sample were calculated with a simple mass balance equation with the sea salt SO42− fraction and δ34Sss = 21.0‰34.
Because of high sea salt loading on coarse mode particles at DDU, we used only fine mode particles for the isotope analyses.
Sulfur source apportionment
The δ34Snss values are determined by the relative contributions from various sulfur sources via the following equations:
where fi, δ34Si and [SO42−]i correspond to the relative contribution, the isotopic composition, and the concentration of sulfur source i, respectively. Here, we considered DMS emitted by marine biogenic activity (mb-), stratospheric SO42− inputs through vertical stratosphere-troposphere mixing or deposition of polar stratospheric clouds (st-), volcanic gaseous sulfur emissions (vl-), and anthropogenic sources, including those in the Antarctic continent and long-range transport from other continents (anth-), as possible sulfur sources15. Because the δ34S values of st-, vl-, and anth-SO42− are mutually overlapping, we designated them as nonmarine biogenic sources and assumed δ34Snmb values as their sum (0 to 5‰)35,36,37. Since these δ34Snmb values are distinguishable from the possible range of δ34Smb values (16.6 to 20.3‰)38,39,40, the fmb and fnmb can be estimated by Eqs (4) and (5), with the assumption of mixing of the two endmembers. Note that it has been recently observed that biologically produced dimethylsulfoniopropionate (DMSP) in Antarctic sea ice possesses δ34S values largely ranging from 10.6 to 23.6‰, whose lowest values were observed in only the extreme physiochemical conditions of isolated brine pockets40. However, given that such low δ34S values are spatially limited and that the mean δ34S value of DMSP for the corresponding sea ice core sample was 17‰40, this sulfur source is unlikely to go beyond the range of general δ34S values of mb-SO42−. The selection of δ34Si values of each source and the validity of the assumption are discussed in detail in the Supplementary Information.
References
Penner, J. E., Hegg, D. & Leaitch, R. Unraveling the role of aerosols in climate change. Environmental Science & Technology 35, 332a–340a, https://doi.org/10.1021/es0124414 (2001).
Cosme, E., Hourdin, F., Genthon, C. & Martinerie, P. Origin of dimethylsulfide, non-sea-salt sulfate, and methanesulfonic acid in eastern Antarctica. J Geophys Res-Atmos 110, D03302, https://doi.org/10.1029/2004jd004881 (2005).
Wolff, E. W. et al. Southern Ocean sea-ice extent, productivity and iron flux over the past eight glacial cycles. Nature 440, 491–496, https://doi.org/10.1038/nature04614 (2006).
Kaufmann, P. et al. Ammonium and non-sea salt sulfate in the EPICA ice cores as indicator of biological activity in the Southern Ocean. Quaternary Science Reviews 29, 313–323, https://doi.org/10.1016/j.quascirev.2009.11.009 (2010).
Legrand, M. et al. Ice-Core Record of Oceanic Emissions of Dimethylsulfide during the Last Climate Cycle. Nature 350, 144–146, https://doi.org/10.1038/350144a0 (1991).
Kohfeld, K. E., Le Quere, C., Harrison, S. P. & Anderson, R. F. Role of marine biology in glacial-interglacial CO2 cycles. Science 308, 74–78, https://doi.org/10.1126/science.1105375 (2005).
Uemura, R. et al. Sulfur isotopic composition of surface snow along a latitudinal transect in East Antarctica. Geophysical Research Letters 43, 5878–5885, https://doi.org/10.1002/2016gl069482 (2016).
Jonsell, U., Hansson, M. E., Morth, C. M. & Torssander, P. Sulfur isotopic signals in two shallow ice cores from Dronning Maud Land, Antarctica. Tellus B 57, 341–350, https://doi.org/10.1111/j.1600-0889.2005.00157.x (2005).
Baroni, M., Thiemens, M. H., Delmas, R. J. & Savarino, J. Mass-Independent Sulfur Isotopic Compositions in Stratospheric Volcanic Eruptions. Science 315, 84–87, https://doi.org/10.1126/science.1131754 (2007).
Patris, N., Mihalopoulos, N., Baboukas, E. D. & Jouzel, J. Isotopic composition of sulfur in size-resolved marine aerosols above the Atlantic Ocean. Journal of Geophysical Research: Atmospheres 105, 14449–14457, https://doi.org/10.1029/1999jd901101 (2000).
Alexander, B. et al. East Antarctic ice core sulfur isotope measurements over a complete glacial-interglacial cycle. Journal of Geophysical Research: Atmospheres 108, 4876, https://doi.org/10.1029/2003jd003513 (2003).
Leung, F. Y., Colussi, A. J. & Hoffmann, M. R. Sulfur isotopic fractionation in the gas-phase oxidation of sulfur dioxide initiated by hydroxyl radicals. J Phys Chem A 105, 8073–8076, https://doi.org/10.1021/jp011014+ (2001).
Preunkert, S. et al. Seasonality of sulfur species (dimethyl sulfide, sulfate, and methanesulfonate) in Antarctica: Inland versus coastal regions. Journal of Geophysical Research 113, https://doi.org/10.1029/2008jd009937 (2008).
Legrand, M. et al. Year-round record of bulk and size-segregated aerosol composition in central Antarctica (Concordia site) – Part 2: Biogenic sulfur (sulfate and methanesulfonate) aerosol. Atmospheric Chemistry and Physics 17, 14055–14073, https://doi.org/10.5194/acp-17-14055-2017 (2017).
Minikin, A. et al. Sulfur-containing species (sulfate and methanesulfonate) in coastal Antarctic aerosol and precipitation. Journal of Geophysical Research: Atmospheres 103, 10975–10990, https://doi.org/10.1029/98jd00249 (1998).
Preunkert, S. et al. Interannual variability of dimethylsulfide in air and seawater and its atmospheric oxidation by-products (methanesulfonate and sulfate) at Dumont d’Urville, coastal Antarctica (1999–2003). Journal of Geophysical Research 112, https://doi.org/10.1029/2006jd007585 (2007).
Global Volcanism Program, Report on Erebus (Antarctica). In: Venzke, E (ed.), Bulletin of the Global Volcanism Network, 42:6. Smithsonian Institution (2017).
Shirsat, S. V. & Graf, H. F. An emission inventory of sulfur from anthropogenic sources in Antarctica. Atmospheric Chemistry and Physics 9, 3397–3408, https://doi.org/10.5194/acp-9-3397-2009 (2009).
Elsässer, C. et al. Continuous 25-yr aerosol records at coastal Antarctica Part 2: variability of the radionuclides Be-7, Be-10 and Pb-210. Tellus B 63, 920–934, https://doi.org/10.1111/j.1600-0889.2011.00543.x (2011).
Markle, B. R., Steig, E. J., Roe, G. H., Winckler, G. & McConnell, J. R. Concomitant variability in high-latitude aerosols, water isotopes and the hydrologic cycle. Nature Geoscience 11, 853-+, https://doi.org/10.1038/s41561-018-0210-9 (2018).
Goto-Azuma, K. et al. Reduced marine phytoplankton sulphur emissions in the Southern Ocean during the past seven glacials. Nature Communications 10, 3247, https://doi.org/10.1038/s41467-019-11128-6 (2019).
Iizuka, Y. et al. Sulphate-climate coupling over the past 300,000 years in inland Antarctica. Nature 490, 81–84, https://doi.org/10.1038/nature11359 (2012).
Charlson, R. J., Lovelock, J. E., Andreae, M. O. & Warren, S. G. Oceanic Phytoplankton, Atmospheric Sulfur, Cloud Albedo and Climate. Nature 326, 655–661, https://doi.org/10.1038/326655a0 (1987).
Ishino, S. et al. Seasonal variations of triple oxygen isotopic compositions of atmospheric sulfate, nitrate, and ozone at Dumont d’Urville, coastal Antarctica. Atmospheric Chemistry and Physics 17, 3713–3727, https://doi.org/10.5194/acp-17-3713-2017 (2017).
Savarino, J., Kaiser, J., Morin, S., Sigman, D. M. & Thiemens, M. H. Nitrogen and oxygen isotopic constraints on the origin of atmospheric nitrate in coastal Antarctica. Atmospheric Chemistry and Physics 7, 1925–1945, https://doi.org/10.5194/acp-7-1925-2007 (2007).
Jourdain, B. & Legrand, M. Year-round records of bulk and size-segregated aerosol composition and HCl and HNO3 levels in the Dumont d’Urville (coastal Antarctica) atmosphere: Implications for sea-salt aerosol fractionation in the winter and summer. J Geophys Res-Atmos 107, 4645, https://doi.org/10.1029/2002jd002471 (2002).
Holland, H. D., Lazar, B. & Mccaffrey, M. Evolution of the Atmosphere and Oceans. Nature 320, 27–33, https://doi.org/10.1038/320027a0 (1986).
Wagenbach, D. et al. Sea-salt aerosol in coastal Antarctic regions. Journal of Geophysical Research: Atmospheres 103, 10961–10974, https://doi.org/10.1029/97jd01804 (1998).
Legrand, M. et al. Year-round records of bulk and size-segregated aerosol composition in central Antarctica (Concordia site) – Part 1: Fractionation of sea-salt particles. Atmospheric Chemistry and Physics 17, 14039–14054, https://doi.org/10.5194/acp-17-14039-2017 (2017).
Albalat, E. et al. Sulfur isotope analysis by MC-ICP-MS and application to small medical samples. Journal of Analytical Atomic Spectrometry 31, 1002–1011, https://doi.org/10.1039/c5ja00489f (2016).
Geng, L. et al. A simple and reliable method reducing sulfate to sulfide for multiple sulfur isotope analysis. Rapid Commun Mass Spectrom 32, 333–341, https://doi.org/10.1002/rcm.8048 (2018).
Ono, S., Wing, B., Johnston, D., Farquhar, J. & Rumble, D. Mass-dependent fractionation of quadruple stable sulfur isotope system as a new tracer of sulfur biogeochemical cycles. Geochimica et Cosmochimica Acta 70, 2238–2252, https://doi.org/10.1016/j.gca.2006.01.022 (2006).
Hattori, S. et al. Determination of the sulfur isotope ratio in carbonyl sulfide using gas chromatography/isotope ratio mass spectrometry on fragment ions 32S+, 33S+, and 34S+. Anal Chem 87, 477–484, https://doi.org/10.1021/ac502704d (2015).
Masterson, A. L., Wing, B. A., Paytan, A., Farquhar, J. & Johnston, D. T. The minor sulfur isotope composition of Cretaceous and Cenozoic seawater sulfate. Paleoceanography 31, 779–788, https://doi.org/10.1002/2016pa002945 (2016).
Castleman, A. W., Munkelwitz, H. R. & Manowitz, B. Isotopic Studies of Sulfur Component of Stratospheric Aerosol Layer. Tellus 26, 222–234, https://doi.org/10.1111/j.2153-3490.1974.tb01970.x (1974).
Nielsen, H. et al. Lithospheric Sources of Sulphur. Stable Isotopes in the Assessment of Natural and Anthropogenic Sulphur in the Environment, 65–132 (1991).
Rempillo, O. et al. Dimethyl sulfide air-sea fluxes and biogenic sulfur as a source of new aerosols in the Arctic fall. Journal of Geophysical Research 116, https://doi.org/10.1029/2011jd016336 (2011).
Amrani, A., Said-Ahmad, W., Shaked, Y. & Kiene, R. P. Sulfur isotope homogeneity of oceanic DMSP and DMS. Proc Natl Acad Sci USA 110, 18413–18418, https://doi.org/10.1073/pnas.1312956110 (2013).
Oduro, H., Van Alstyne, K. L. & Farquhar, J. Sulfur isotope variability of oceanic DMSP generation and its contributions to marine biogenic sulfur emissions. Proc Natl Acad Sci USA 109, 9012–9016, https://doi.org/10.1073/pnas.1117691109 (2012).
Carnat, G. et al. Variability in sulfur isotope composition suggests homogenous dimethylsulfoniopropionate cycling and microalgae metabolism in Antarctic sea ice. Communications Biology 1, 1–9, https://doi.org/10.1038/s42003-018-0228-y (2019).
Acknowledgements
We thank Dr. I. Bourgeois and Basile de Fleurian, who conducted winter sampling at Dome C and DDU in 2011, and to Dr. M. Lin for helpful discussions for interpretation of the data. We appreciate financial support and field supplies for winter and summer campaigns at Dome C and DDU by the Institut Polaire Français Paul Emile Victor from Program 414, program 1011 (SUNITEDC) and program 1177 (CAPOXI 35–75), and the French Environmental Observation Service CESOA (Etude du cycle atmosphérique du Soufre en relation avec le climat aux moyennes et hautes latitudes Sud) by the National Centre for Scientific Research (CNRS) Institute for Earth Sciences and Astronomy. This study was supported by JSPS KAKENHI (JP17J08978 (S.I.), JP19J00682 (S.I.), JP16H05884 (S.H.), JP18H05050 (S.H.), and JP17H06105 (N.Y. and S.H.)) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan, and by a grant from Labex OSUG@2020 (Investissements d’avenir – ANR10 LABX56) and by the French Agence Nationale de la Recherche (ANR) FOFAMIFS project (grant ANR—14-CE33-0009-01) and EAIIST project (grant ANR—16-CE01-0011-01). S.H. appreciates support for this project from JSPS and CNRS under the JSPS–CNRS Joint Research Program. J.S. thanks the CNRS/INSU (PRC program 207394) and the PH-SAKURA program of the French Embassy in Japan (project 31897PM) for financial support. S.I. thanks the Academy for Co-creative Education of Environment and Energy Science for financial support. Meteo France is acknowledged for providing meteorological data. We also acknowledge the Air Resources Laboratory (ARL) for the use of their HYSPLIT transport and dispersion model.
Author information
Authors and Affiliations
Contributions
S.I., S.H. and J.S. designed the research. J.S., M.L., S.P. and B.J. performed field samplings and provided meteorological and chemical observation data. S.I., S.H., J.S., E.A. and F.A. took chemical/isotopic measurements. S.I., S.H., J.S., M.L. and N.Y. interpreted the data. S.I. and S.H. wrote the paper with contributions from all coauthors.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ishino, S., Hattori, S., Savarino, J. et al. Homogeneous sulfur isotope signature in East Antarctica and implication for sulfur source shifts through the last glacial-interglacial cycle. Sci Rep 9, 12378 (2019). https://doi.org/10.1038/s41598-019-48801-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48801-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
