
Abstract
Staphylococcus hominis is part of the normal human microbiome. Two subspecies, S. hominis hominis (Shh) and S. hominis novobiosepticus (Shn), have clinical significance. Forty-nine S. hominis isolates were analyzed by the MicroScan automated system, SDS-PAGE and MALDI-TOF methods, followed by partial sequencing of the 16S rDNA gene. The trehalose fermentation test, disk diffusion and broth microdilution tests were used to identify (novobiocin test) and access the susceptibility to oxacillin and vancomycin of isolates. The SCCmec elements and genomic diversity were evaluated by PCR and PFGE methods, respectively. Profiles of 28 (57%; 8 Shh and 20 Shn) isolates corroborated with the results found in all the applied methods of identification. The remaining 21 (43%) isolates were phenotypically identified as Shh by MicroScan; however, they were identified as Shn by SDS-PAGE and mass spectral, and confirmed by 16S rDNA sequencing. Among 41 isolates identified as Shn by the molecular and mass spectrometry methods, 19 (41%) were novobiocin-sensitive, and the trehalose test indicated 11 positive isolates, which are considered atypical phenotypic results for this subspecies. In addition, 92.7% of the isolates identified as Shn by these methods carried mecA gene, while only 12.5% of the Shh isolates were positive. Together, the results highlighted the SDS-PAGE and MALDI-TOF MS methods as promising tools for discriminating S. hominis subspecies.
Similar content being viewed by others

Introduction
Coagulase-negative Staphylococcus (CoNS) is an important group of gram-positive coccoid bacteria. They are mainly associated with human and animal biota and are also detected in blood cultures of hospitalized patients1,2,3. The main concern with those microorganisms is the possibility of serving as a reservoir for resistance and virulence genes4. Among CoNS species, Staphylococcus hominis is the third most frequent one, is regarded as a causal agent of bacteremia and endocarditis2.
As described amongst staphylococcal isolates, resistance to oxacillin (or methicillin) in S. hominis occurs because of the acquisition of the mecA gene, which encodes an altered penicillin-binding protein with low affinity to beta-lactam drugs5. This gene is located in a mobile genetic element, called Staphylococcal Cassette Chromosome mec (SCCmec), which is present in Staphylococcus genus and is highly conserved among its species6. S. hominis isolates have also been reported to show decreased susceptibility to vancomycin7, as well as resistance to linezolid8.
Amongst S. hominis isolates, two subspecies have been described: S. hominis hominis (Shh) and S. hominis novobiosepticus (Shn). The latter is usually characterized as novobiocin-resistant and unable to form acid aerobically, even by using trehalose or N-acetylglucosamine9. In addition, Shn clinical isolates are usually described as multiresistant and are frequently isolated in bacteremia cases, compared to other subspecies2,7,10,11,12. However, studies have demonstrated that Shn isolates are usually misidentified when only biochemical or automated methods are used13,14, making its characterization unreliable. In addition, few studies have shown the genomic diversity of the S. hominis subspecies15,16, and a reliable standard method to identify the subspecies has not been made available yet. According to Zhang and coworkers16, a discrimination of the two subspecies may be difficult since Shn seems to form a paraphyletic taxon. However, tools that help in the better characterization of S. hominis isolates would contribute to generating more accurate clinical epidemiology data. Based thereon, this work aimed to evaluate different methods to try to improve the discrimination of Shh and Shn isolated from blood cultures, including their susceptibility to oxacillin and vancomycin and clonal profiles.
Results
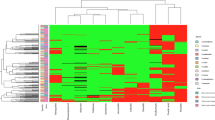
Forty-nine S. hominis isolates were initially identified at subspecies level by phenotypic tests by using the MicroScan WalkAway® automation and novobiocin susceptibility tests. Then, the SDS-PAGE and mass spectrometry (MALDI-TOF MS) methods were also applied for all isolates. All methods were concordant to identify an amount of 28 (57%) isolates, 8 S. hominis hominis (Shh) and 20 S. hominis novobiosepticus (Shn) (Groups I and II, Supplementary Material 1). The other 21 (43%) isolates (Groups III and IV) could not be properly discriminated, and by comparing all three methodologies (SDS-PAGE, mass spectrometry methods and by 16S RNA analysis), all were identified as Shn. In addition, 19 of these 21 isolates (Group III) were sensitive to the novobiocin disk and presented low minimum inhibitory concentration (MIC) values, ranging from <0.25 to 1.0 µg/ml (Fig. 1). The two remaining isolates (656 s and 670 s from Group IV) were resistant to novobiocin (MICs of 8.0 and 16 µg/mL).

Dendrogram of the pulsed-field gel electrophoresis (PFGE) profiles of SmaI and ApaI-digested genomic DNA of 49 Staphylococcus hominis spp. isolates and associated characteristics. Similarities percentage is identified on dendrogram derived from the unweighted pair group method using arithmetic averages and based on Dice coefficients; +: positive; −: negative; SCCmec: Staphylococcal chromosomal cassette mec; MIC: minimum inhibitory concentration in µg/mL; oxa: oxacillin; van: vancomycin; novob: novobiocin; na: not-applicable; NT: non-typeable; asubspecies identification based on total proteins profile by SDS-PAGE and MALDI-TOF, and by partial sequencing of the 16S rDNA gene; bisolates submitted to partial rDNA 16S sequencing; cisolates identified as Shh by the Microscan automated system; dabsence of ccr complex.
The SDS-PAGE analysis14 showed common regions between Shh and Shn isolates (similarity index of 90.4%), although proteins with a molecular mass of about 49 kDa and 85 kDa were present only in Shh, while Shn presented exclusive proteins of about 15 kDa, 26 kDa, 37 kDa and 90 kDa (Fig. 2A). By using the same cell extract, the mass spectrometry analysis also showed a distinct protein profile spectrum, in a range from 2000 m/z to 13000 m/z, between Shn and Shh strains (Fig. 2B). In fact, based on the data analysis, with the peak list (Presence – 100%; absence – 0%) revealed different proteins peaks between Shh and Shn (Fig. 2C,D). In both S. hominis subspecies the different masses proteins (m/z) were concentrated in the range from 2559 Da to 5226 Da. Figure 2E presents a section of 350 bp amplicon fragment (bases 102 to 151) obtained by DNA sequencing showing the Single Nucleotide Polymorphism (SNP 112: G→A) of rDNA 16S amplicon, which discriminate the subspecies Shh and Shn. Thus, based on these data, all the 21 misidentified isolates were identified as Shn.

Overview of the mass spectra by MALDI-TOF and proteins profile in SDS-PAGE of S. hominis hominis in comparison to the mass spectrum of and S. hominis novobiosepticus. (A) SDS-PAGE showing the proteins profiles of S. hominis hominis (Shh) in comparison to the S. hominis novobiosepticus (Shn). Arrows (→) indicate the prominent proteins for each subspecies; (B) Distribution of the detected masses by MALDI-TOF mass according to m/z spectra peaks obtained from Shh and Shn. Values in bold means spectra peaks found in Shh or Shn; (E) Section of 350 bp amplicon fragment (base 102 to 151) obtained by DNA sequencing showing the Single Nucleotide Polymorphism (SNP 112:G→A) of rDNA 16S amplicon, which discriminate Shh and Shn. (C,D) *Each peak represents a different mass (m/z) of an intact protein detected in the analyses for Shh or Shn.
Susceptibility to methicillin was evaluated by the detection of the mecA gene, and 39 (79.6%) S. hominis isolates were positive. Out of the 41 isolates identified as Shn by the MALDI-TOF and SDS-PAGE methods (Fig. 1; Supplementary Material 1), 92.7% carried mecA gene, while only 12.5% of the Shh isolates were positive. The oxacillin MIC ranged from <0.125 to 4 µg/mL for Shh, and from <0.125 to >256 µg/mL for Shn when the classification by MALDI-TOF and SDS-PAGE was considered. Similarly, vancomycin MIC was also evaluated and the values ranged from 0.5 to 2 µg/mL for Shn isolates (identified by MALDI-TOF and SDS-PAGE methods), but 85.4% of them presented MIC from 1 to 2 μg/mL. On the other hand, Shh isolates (also identified by MALDI-TOF and SDS-PAGE methods) presented vancomycin MIC values ≤ 1 µg/mL.
Considering the PFGE results, the dendrogram obtained by Bionumerics analysis of the 49 Shn isolates, showed no clonal relationship between S. hominis subspecies isolates (Less than 50% of similarity). Among Shh isolates (PFGE types “a” to “e”) one isolate was positive for the mecA gene, but the SCCmec complex was non-typeable (NT). The 41 Shn isolates were clustered into 17 PFGE types, alphabetically labeled “A” to “Q”, and just two PFGE types (“A” and “B”) comprised 44% of the isolates. Among the Shn isolates, 38 (92.7%) were mecA positive, and only two showed typeable SCCmec elements (type III). The other 36 mecA positive Shn isolates were included in eight NT SCCmec profiles, as it follows: NT1–12 isolates (ccrC + mec complex A); NT2–11 (ccr absent and mec complex A); NT3–7 (ccr1 + mec complex A); NT4–2 (ccr1 + ccr3 + mec complex A); NT5–1 (ccr1 + ccr2 + ccr5 + mec complex A); NT6–1 (ccr1 + ccr2 + mec complex A); NT7–1 (ccr1 + ccr3 + ccr5 + mec complex A); and NT8–1 (ccr2 + ccr4 + mec complex A).
Discussion
Staphylococcus hominis is the third species of CoNS identified in bloodstream infections4,17. This identification is usually based on automated (eg. MicroScan) and/or biochemical tests, which discriminates the species into two subspecies: S. hominis hominis (Shh) and S. hominis novobiosepticus (Shn). Resistance to novobiocin and failure to produce acid aerobically from D-trehalose and N-acetyl-D-glucosamine were pointed out as the main distinguishable characteristics of Shn, when S. hominis subspecies were described in 19989. However, several authors have described how complicated it is to differentiate S. hominis isolates into subspecies18,19. Recently, a study16 aimed to genetically characterize S. hominis isolates by using MLST, showing that phylogenetic analyses indicate that Shn sequence-types (STs) do not form a single, well-supported cluster, and maybe a paraphyletic taxon. In the present study, we evaluated 49 S. hominis isolates from blood obtained at a tertiary hospital in Rio de Janeiro and 28 (57%) of them were concordant with all the identification methods performed. We used the SDS-PAGE method as a tool to discriminate the S. hominis subspecies, as recommended previously14. We also included a MALDI-TOF MS analysis, in order to obtain a better characterization of these isolates. Our data show that both SDS-PAGE and MALDI-TOF MS analyses helped confirmed MicroScan/Novobiocin resistance tests results for the majority of the isolates, showing they are useful tools to differentiate S. hominis subspecies. The MALDI-TOF MS method highlighted 13 specific peaks for Shn or Shh, and seven of them were discriminatory, which can potentially be used to distinguish these two subspecies. The SDS-PAGE showed six different protein bands, and these subspecies could be visually distinguishable by using reference strains for comparing.
In relation to the other 21 isolates, the results of the phenotypic tests were not concordant with those of the SDS-PAGE and MALDI-TOF MS tests. The MicroScan automated test identified these isolates as Shh. However, SDS-PAGE and MALDI-TOF identification showed that these isolates belonged to the Shn subspecies. In addition, SDS-PAGE and MALDI-TOF data are in disagreement with the results of the biochemical methods applied (trehalose fermentation and novobiocin resistance tests). Since 19 of these 21 isolates identified as Shn by SDS-PAGE and MALDI-TOF were proven to be novobiocin-sensitive, this result suggests/confirms that phenotypical analysis is not reliable to distinguish S. hominis subspecies, and nor is the trehalose test, which indicated 11 of all Shn isolates (identified by the SDS-PAGE and MALDI-TOF MS) as positives for this test. It is recognized that automated methods (e.g. MicroScan) show several inconsistences with regard to bacterial phenotypical characterization, as described previously20. The failure of the novobiocin-resistance test has also been described before18,19. Zhang and coworkers16 described a single-nucleotide polymorphism (SNP) in the gyrB gene that encodes a bacterial DNA gyrase, which is the target of novobiocin resistance. They evaluated 108 S. hominis isolates, and out of 38 novobiocin-resistant isolates, all of them showed the SNP 431: G→T, resulting in a predicted amino acid polymorphism, 144:R→L. All 70 novobiocin-sensitive isolates had the G431 allele, and all 38 novobiocin-resistant isolates had the T431 allele. These data demonstrated the relation between SNP polymorphism and novobiocin resistance, but not regarding the identification of S. hominis subspecies, indicating that this phenotypic test is not useful to discriminate Shn and Shh, as shown in the present study. The authors used the trehalose degradation test as a discriminatory test to separate both S. hominis subspecies. What drew our attention was that 11 (27%) out of 41 Shn isolates from our collection were positive for this test. Using the parameters described by Zhang and coworkers16 to evaluate our results, 11 isolates of Shn would be identified as Shh and one isolate of Shh would be identified as Shn.
In view of the results disclosed herein, chances are that the present study is the first one to use MALDI-TOF MS data to differentiate these subspecies, bringing a higher specificity to Shh and Shn discrimination. MALDI-TOF MS is believed to be the future of bacterial identification, in view of its high specificity and short analytical time. In addition, it is an excellent tool for identifying pathogens and it inaugurates a new era in modern microbiology21. In order to confirm our protein profile data, 15 isolates from different phenotypical groups had their rDNA 16S sequenced, and the results were found to be concordant with the MALDI TOF MS analyses.
In Brazil, some studies have reported oxacillin resistance rates of 86% among CoNS isolated from bacteremia22,23. Evaluating the resistance rates between S. hominis subspecies, Palazzo and colleagues10 found 83.3% of oxacillin resistance among Shn isolates, and Caierão and coworkers24 detected oxacillin resistance among all Shn isolates. In the present work, 97.4% of the mecA positive isolates were identified as Shn and the majority of chromosome cassettes were non-typeable, as already described25. The detection of oxacillin/methicillin resistance is extremely relevant since its early and accurate observation can assist in establishing the correct antibiotic therapy to combat infection. In our study, a high prevalence of oxacillin resistance was observed among the Shn isolates (92.7%), while only 12.5% of the Shh isolates were oxacillin-resistant, which confirms the existence of higher resistance rates among the isolates of the Shn subspecies. The Shn isolates also expressed higher MICs to vancomycin (85.4% presented MIC from 1 to 2 µg/mL), as already described by Sorlozano and coworkers for this subspecies26. On the other hand, all Shh isolates had vancomycin MIC ≤ 1 µg/mL. Therefore, we may conclude that an analysis of the total protein profile, by SDS-PAGE or MALDI-TOF MS, of S. hominis subspecies together with the evaluation of their oxacillin and vancomycin susceptibilities, could provide us with better discrimination between the Shh and Shn subspecies.
Studies in the literature concerning CoNS genomic diversity describe the high genomic diversity of isolates, including the S. hominis subspecies2. We analyzed the S. hominis isolates by PFGE to determine the different PFGE types, and each subspecies was grouped independently and the dendrogram showed no clonal relationship between them. Among Shh isolates, related profiles were found. For 41 Shn isolates, despite the clonal diversity, 44% of the isolates belonged to only two clonal groups (“A” and “B”). Moreover, it should be noted that the isolates of genotype “A” (subtypes A1, A1, A2, A4 and A8) were obtained in the same period from the same hospital (data not shown), confirming the ability of this S. hominis subspecies to spread and stay in this setting.
In conclusion, our results showed that the phenotypical methods normally used for discrimination of S. hominis subspecies are not reliable. Hence, we suggest that protein profile analysis and/or MALDI-TOF MS analysis might be complementary and useful tools to discriminate these subspecies more accurately, contributing to understanding its clinical epidemiology.
Methods
Bacterial isolates and reference strains
Forty-nine S. hominis isolates, obtained from blood cultures at a tertiary care hospital located in Rio de Janeiro, Brazil, and belonging to our culture laboratory collection, were used for this study. The hospital is an institution with 532 beds, 35 of which are in an Intensive Care Unit (ICU). All isolates were collected between 1998 and 2002 (17 isolates); and from 2005 to 2009 (32 isolates). One ATCC 27844 S. hominis hominis and a clinical isolate 34Hp S. hominis novobiosepticus14 were included as control strains. ATCC 29213 S. aureus and ATCC 33591 strains were also included as controls for the methicillin and vancomycin susceptibility tests.
Isolates identification
All S. hominis isolates were initially identified phenotypically by the Microscan WalkAway automated system (Dade Behring Microscan Inc. Division, West Sacramento, USA). Additional tests were performed as follows:
Novobiocin susceptibility and trehalose tests
Novobiocin (Sigma-Aldrich Company, St. Louis, USA) susceptibility determination by using the disk diffusion and broth microdilution methods was performed in accordance with Bannerman and Peacock (2007)27 and the Clinical and Laboratory Standards Institute (CLSI)28. Concentration ranged from 0.250 to 16 µg/mL. Acid production from D-trehalose was determined according to Iorio and coworkers13
Protein profile analysis
In order to discriminate Shh from Shn isolates, whole-cells protein extracts were prepared according to Santos and coworkers14. Briefly, to obtain the protein extracts the cells were cultured overnight until they reached a concentration of 107 CFU/mL Cells were collected by centrifugation at 12,000 × g for 5 min. After being washed twice in 0.85% NaCl, the cells were incubated in the presence of 0.2 mg/mL lysostaphin for 2 h at 37 °C. After the incubation period, equal volumes of 0.5 M Tris-HCl (pH 7.2) buffer containing 4% SDS, 10% Beta-mercaptoethanol, 20% glycerol, and 0.1% bromophenol blue were added, and the samples were boiled for 5 min. Cellular extracts were submitted to Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) analysis. The obtained band patterns were analyzed through the Bionumerics program, version 6.0 (Applied Maths, Belgium), and similar percentages were derived from the unweighted pair group method using arithmetic averages and based on Dice coefficients.
Maldi-tof Ms
Mass spectrometry technology through the Matrix Assisted Laser Desorption Technology/Ionization Time of Flight Mass Spectrometry (MALDI-TOF MS) was used in accordance with Alatoom and coworkers29 to identify S. hominis subspecies. All strains of the groups III and IV, seven of the group II and two of the group I were used to construct the database (Supplementary Material 1). Two reference strains were used: 238 s (Shn) and 72 Hp (Shh) (Santos et al., 2009). Five bacterial colonies of isolates previously identified as Shh and Shn by SDS PAGE method were dissolved in 300 µL of HPLC water (Tedia) in microtubes and mixed. Then, 900 µL of ethanol P.A. (Sigma-Aldrich) was added and it was vigorously homogenized. The suspension was then centrifuged at 16,000 × g for 2 min and the supernatant discarded. The pellet was allowed to dry at room temperature for approximately 20 min. After dried, 20 µL of 70% formic acid (Sigma-Aldrich) and 20 µL of acetonitrile P.A. (Sigma-Aldrich) was added to the pellet and gently homogenized, centrifuged and the supernatant transferred to a differently labelled microtube for further MS analysis. All tubes were kept at −80 °C for two days until the moment of use and 10 days maximum
For the mass spectrometry analysis, 1 µL of the extraction solution was added onto the spot a MALDI-TOF MS plate (MSP 96 target polished steel BC, Bruker Daltonics). Samples were allowed to dry and each spot was covered with 1 µL of 10 g/L CHCA matrix (Cyano-4-hydroxycinnamic acid (Bruker Daltonics) in 50% acetonitrile (Tedia) and 2.5% trifluoroacetic acid (Tedia). For every isolate, 27 spots were applied to produce 27 technical replicates of spectra. Spectra were obtained on Microflex LT mass spectrometer (Bruker Daltonics) and the FlexAnalysis software was used for default parameters (laser frequency of 60 Hz, ion source voltages of 2.0 and 1.8 kV, and lens voltage of 6 kV), generating spectra in a range of 2,000–20,000 m/z. All spectra from Biotyper were preprocessed and normalized using standard parameters (baseline subtraction by running ruling disc and peak detection using a signal to noise ratio of 10). After the preprocessing, a peak match was performed using a tolerance of ±0.002 m/z. Subsequently, by using the MALDI Biotyper 3 software (flex control and flex analysis software) the 27 spectra were compiled to form a single spectrum and the most prevalent and with the highest masses (m/z) were considered the ideal fingerprint spectrum to differentiate the subspecies.
rDNA single nucleotide polymorphism analysis
To discriminate Shh from Shn, 15 S. hominis isolates from different phenotypical groups were selected and, a partial sequencing of the 16S rDNA gene, which includes a single nucleotide polymorphism (SNP), was performed14. For the analysis, primers were designed by using the Oligo Analyzer (Integrated DNA Technologies, Coralville, IA, USA) and BioEdit (Ibis Biosciences, Carlsbad, CA, USA), based on the GenBank sequences database (Accession Numbers: KU364060; MG255965; MF327693 for Shh, and MG201782; KY218859 for Shn). The following primers were designed to amplify a 350 bp fragment of S. hominis 16S rDNA gene.: SH16S-F (5′ GCTTGCTCCTTTGACGTTAG 3′) and SH16S-R (5′ CGAAGACCTTCATCACTCAC 3′). Oligonucleotides were purchased from Integrated DNA Technologies, Inc. (Coralville, IA, USA).
DNA was obtained according Walsh, Metzer & Higuchi30 as previously described. Amplification was performed on a Programmable Thermal Controller (Eppendorf Mastercycler Gradient, Hamburg, Germany) with the following conditions were: denaturation for 5 min at 94 °C, followed by 35 cycles of 94 °C for 1 min, 58 °C for 1 min and 72 °C for 1 min, with a final extension at 72 °C for 7 min. Amplified products were analyzed on 1% agarose gels, stained with UniSafe (Uniscience, São Paulo, Brazil) and visualized on a UV transilluminator. PCR band products of each gene were purified by using GTX PCR, according to the manufacturer’s instructions (GE 50 Healthcare, Buckinghamshire, England). Sequencing was made in an automated DNA Mega BACE1000 (Biotech, EUA), using the DYEnamic ET Dye Terminator system (GE Healthcare, England). Both DNA strands (forward and reverse) were sequenced and the reverse one was analyzed. To identify the previously mentioned mutation point (112: G→A), sequences obtained were analyzed by using Bioedit (Carlsbad, USA).
Oxacillin and vancomycin MIC tests
Minimum inhibitory concentrations (MICs) to oxacillin and vancomycin (Sigma-Aldrich Company) were determined by broth microdilution tests according to CLSI 201228. The concentrations ranged from 0.125 to 256 µg/mL for oxacillin and from 0.125 to 16 µg/mL for vancomycin. The ATCC strain 29213 was used as control for the MIC tests.
Detection of mecA gene, SCCmec and PFGE typing
The methodology used for the mecA gene detection and SCCmec typing were performed according to Del Vecchio and coworkers31 and Kondo and coworkers32, respectively. After DNA extraction33 the SCCmec typing was performed through two multiplex PCR reactions to detect the ccr complex and the mec gene class. The combination of the types of ccr and mec class allowed the identification of the types of SCCmec (I to VI).
All isolates were typed by pulsed-field gel electrophoresis (PFGE) after digesting whole cell DNA with SmaI and ApaI to obtain specific fragments26 in a CHEF-DRIII system (Bio-Rad, Richmond, CA, USA), as previously described34 with some modifications. Briefly, the S. hominis immobilized DNA in agarose blocks were transferred to a solution containing 10 U of ApaI (Biolabs), supplemented with 1.5 µL of BSA (Bovine Serum Albumin) at 10 mg/mL, for 18–24 h at 25 °C, followed by incubation with SmaI (4 h at 25 °C). To determine the similarity index between the isolates the PFGE fingerprints were analyzed by the BioNumerics program version 6.0 (Applied Maths, Belgium) using the Dice correlation coefficient and the unweighted pair-group method with arithmetic mean (UPGMA) clustering analysis. Isolates with four or fewer bands of difference or a minimum of 80% similarity were designated as the same genotype35.
References
Mendoza-Olazarán, S. et al. Antibiotic susceptibility of biofilm cells and molecular characterization of Staphylococcus hominis isolates from blood. PLoS One 10, e0144684, https://doi.org/10.1371/journal.pone.0144684 (2015).
Mendonza-Olazarán et al. Microbiological and molecular characterization of Staphylococcus hominis isolates from blood. PLoS One 8, e61161, https://doi.org/10.1371/journal.pone.0061161 (2013).
Pradipta, I. S. et al. Antibiotic Resistance in Sepsis Patients: Evaluation and Recommendation of Antibiotic Use. N. Am. J. Med. Sci. 5, 344–352, https://doi.org/10.4103/1947-2714.114165 (2013).
Becker, K., Heilmann, C. & Peters, G. Coagulase-negative staphylococci. Clin. Microbiol. Rev. 27, 870–926, https://doi.org/10.1128/CMR.00109-13 (2014).
Ito, T. et al. Structural comparison of three types of staphylococcal cassette chromosome mec integrated in the chromosome in methicillin-resistant Staphylocccus aureus. Antimicrob. Agents Chemother. 45, 1323–1336, https://doi.org/10.1128/AAC.45.5.1323-1336.2001 (2001).
Ito, T., Okuma, K., Ma, X. X., Yuzama, H. & Hiramatsu, K. Insights on antibiotic resistance of Staphylococcus aureus from it whole genome: genomic island SCC. Drug. Resist. Update 6, 41–52, https://doi.org/10.1016/S1368-7646(03)00003-7 (2004).
Palazzo, I. C. V., D’azevedo, P. A., Pignatari, C. S. A. C. C. & Darini, A. L. C. Staphylococcus hominis subsp. novobiosepticus strains causing nosocomial bloodstream infection in Brazil. J. Antimicrob. Chemother. 62, 1222–1226, https://doi.org/10.1093/jac/dkn375 (2008).
Chamon, R. C. et al. Linezolid-resistant Staphylococcus haemolyticus and Staphylococcus hominis: single and double mutations at the domain V of 23S rRNA among isolates from a Rio de Janeiro hospital. Diagn. Microbiol. Infect. Dis. 80, 307–310, https://doi.org/10.1016/j.diagmicrobio.2014.09.011 (2014).
Kloos, W. E. et al. Staphylococcus hominis subsp. novobiosepticus, a novel trehalose and N-acetyl-Dglucosamine-negative, novobiocin and multiple antibiotic resistant subspecies isolated from human blood cultures. Int. J. Syst. Bacteriol. 48, 799–812, https://doi.org/10.1099/00207713-48-3-799 (1998).
Chaves, F., Garci´A-Alvarez, M., Sanz, F., Alba, C. & Otero, J. Nosocomial spread of a Staphylococcus hominis subsp. novobiosepticus strain causing sepsis in a neonatal care unit. J. Clin. Microbiol. 43, 4877–4879, https://doi.org/10.1128/JCM.43.9.4877-4879.2005 (2005).
Ahmed, N. H., Baruah, F. K. & Grover, R. K. Staphylococcus hominis subsp. novobiosepticus, an emerging multidrug-resistant bacterium, as a causative agent of septicaemia in cancer patients. Ind. J. Med. Res. 146, 420–425, https://doi.org/10.4103/ijmr.IJMR_1362_15 (2017).
Szczuka, E., Trawczyński, K., Bogucka, N. & Kaznowski, A. Multifactorial mechanisms of the pathogenesis of Staphylococcus hominis isolated from bloodstream infections. Antoine van Leeuwenhoek. 111, 1259–1265, https://doi.org/10.1007/s10482-017-1007-3 (2018).
Iorio, N. L. P. et al. Simplified and reliable scheme for species-level identification of Staphylococcus clinical isolates. J. Clin. Microbiol. 45, 2564–2569, https://doi.org/10.1128/JCM.00679-07 (2007).
Santos, O. C. et al. Reliable identification of clinically prevalent species and subspecies of staphylococci by sodium dodecyl sulfate-polyacrylamide gel electrophoresis analysis. Diagn. Microbiol. Infect. Dis. 64, 1–5, https://doi.org/10.1016/j.diagmicrobio.2008.12.017 (2009).
Szczuka, E., Trawczyński, K. & Kaznowski, A. Clonal analysis of Staphylococcus hominis strains isolated from hospitalized patients. Pol. J. Microbiol. 63, 349–354 (2014).
Zhang, L. et al. Multilocus sequence typing and further genetic characterization of the enigmatic pathogen, Staphylococcus hominis. PLoS One. 8, e66496, https://doi.org/10.1371/journal.pone.0066496 (2013).
Bouchami, O., Achour, W., Mekni, M. A., Rolo, J. & Ben Hassen, A. Antibiotic resistance and molecular characterization of clinical isolates of methicillin-resistant coagulase-negative staphylococcal isolated from bacteremic patients in oncohematology. Folia Microbiol. 56, 122–130, https://doi.org/10.1007/s12223-011-0017-1 (2011).
Weinstein, M. P. et al. Clinical importance of identifying coagulase-negative staphylococci isolated from blood cultures: evaluation of microscan rapid and dried overnight gram-positive panels versus a conventional reference method. J. Clin. Microbiol. 36, 2089–2092 (1998).
Gilad, J. & Schwartz, D. Identification of Staphylococcus species with the VITEK 2 system: the case of Staphylococcus hominis. J. Clin. Microbiol. 45, 685–686, https://doi.org/10.1128/JCM.02228-06 (2007).
Kim, M. et al. Comparison of the MicroScan, VITEK 2, and Crystal GP with 16S rRNA sequencing and MicroSeq. 500 v2.0 analysis for coagulase-negative Staphylococci. BMC Microbiol. 23, 233, https://doi.org/10.1186/1471-2180-8-233 (2008).
Sandalakis, V., Goniotakis, I., Vranakis, I., Chochlakis, D. & Psaroulaki., A. Use of MALDI-TOF mass spectrometry in the battle against bacterial infectious diseases: recent achievements and future perspectives. Expert Rev. Proteomics. 14, 253–267, https://doi.org/10.1080/14789450.2017.1282825 (2017).
Miragaia, M. et al. Molecular characterization of methicillin-resistant Staphylococcus epidermidis clones: Evidence of geographic Dissemination. J. Clin. Microbiol. 40, 430–438, https://doi.org/10.1128/JCM.40.2.430-438.2002 (2002).
Marra, A. R. et al. Nosocomial Bloodstream Infections in Brazilian Hospitals: Analysis of 2,563 Cases from a Prospective Nationwide Surveillance Study. J. Clin. Microbiol. 8, 1866–1871, https://doi.org/10.1128/JCM.00376-11 (2011).
Caierão, J. et al. Evaluation of phenotypic methods for methicillin resistance characterization in coagulase-negative staphylococci (CNS). J. Med. Microbiol. 53, 1195–1199, https://doi.org/10.1099/jmm.0.45697-0 (2004).
Ibrahem, S. et al. Carriage of methicillin-resistant staphylococci and their SCCmec types in a long-term-care facility. J. Clin. Microbiol. 47, 32–37, https://doi.org/10.1128/JCM.01085-08 (2009).
Sorlozano, A. et al. Detection of new mutations conferring resistance to linezolid in glycopeptide-intermediate susceptibility Staphylococcus hominis subspecies hominis circulating in an intensive care unit. Eur. J. Clin. Microbiol. Infect. Dis. 29, 73–80, https://doi.org/10.1007/s10096-009-0823-4 (2010).
Bannerman, T. L & Peacock, S. Staphylococcus, Micrococcus and other catalase-positive cocci. In Murray, P. R., Barron, E. J., Jorgensen, J., Landry, M. L. & Pfaller, M. A. (eds) Manual of Clinical Microbiology, (10th ed, vol 1. ASM Press) 390–411 (Washington, D. C., 2007).
Clinical and Laboratory Standards Institute (CLSI). CLSI Document M07-A9. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically: Approved Standard. (19th Edition, CLSI) 54–56 (Wayne, 2012).
Alatoom, A. A., Cunningham, S. A., Ihde, S. M., Mandrekar, J. & Patel, R. Comparison of direct colony method versus extraction method for identification of gram-positive cocci by use of Bruker Biotyper matrix-assisted laser desorption ionization-time of flight mass spectrometry. J. Clin. Microbiol. 49, 2868–2873, https://doi.org/10.1128/JCM.00506-11 (2011).
Walsh, P. S., Metzger, D. A. & Higuchi, R. Chelex-100 as a medium for simple extraction of DNA for PCR-based typing from forensic material. Biotechniques. 10, 506–513, https://doi.org/10.2144/000114018 (1991).
Del Vecchio, V. G. et al. Molecular genotyping of methicillin-resistant Staphylococcus aureus via fluorophore-enhanced repetitive-sequence PCR. J. Clin. Microbiol. 33, 2141–2144 (1995).
Kondo, Y. et al. Combination of multiplex PCRs for staphylococcal cassette chromosome mec type assignment: rapid identification system for mec, ccr, and major differences in junkyard regions. Antimicrob. Agents. Chemother. 51, 264–274, https://doi.org/10.1128/AAC.00165-06 (2007).
Aires de Sousa, M. et al. Characterization of methicillin-resistant Staphylococcus aureus isolates from Portuguese hospitals by multiple genotyping methods. Microb. Drug Resist. 2, 331–341, https://doi.org/10.1089/mdr.1996.2.331 (1996).
Vivoni, A. M. et al. Clonal composition of Staphylococcus aureus isolates at a Brazilian University Hospital: identification of international circulation lineages. J. Clin. Microbiol. 44, 1686–1691, https://doi.org/10.1128/JCM.44.5.1686-1691.2006 (2006).
van Belkum, A. et al. Guidelines for the validation and application of typing methods for use in bacterial epidemiology. Clin. Microbiol. Infect. Dis. 13, 1–46, https://doi.org/10.1111/j.1469-0691.2007.01786.x (2007).
Acknowledgements
We thank Dr. Lesliê Vieira Mulico for kindly reviewing this manuscript. This study was supported by grants from: Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento Pessoal de Nível Superior - Brasil (CAPES) – Finance Code 001.
Author information
Authors and Affiliations
Contributions
E.M.P. and K.R.N.S. conceived both study and experiments (excluding MALDI-TOF); E.M.P., D.C.F. and M.S.L. conceived and conduced the rDNA sequencing experiment; M.S.L. conceived the SDS-PAGE experiment; C.S.M. and O.C.S. conduced the SDS-PAGE, phenotypical identification, susceptibility and PFGE experiments; T.L.R.O. conduced the rDNA sequencing experiment; E.O.F. conceived and conduced the MALDI-TOD identification.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pereira, E.M., de Mattos, C.S., dos Santos, O.C. et al. Staphylococcus hominis subspecies can be identified by SDS-PAGE or MALDI-TOF MS profiles. Sci Rep 9, 11736 (2019). https://doi.org/10.1038/s41598-019-48248-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-48248-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
