
Abstract
Adenosine triphosphate (ATP) is a crucial substrate and energy source commonly used in enzyme reactions. However, we demonstrated that the addition of this acidic compound to enzyme assay buffers can serve as a source of unnoticed pH changes. Even relatively low concentrations of ATP (up to 5 mM) shifted pH of reaction mixtures to acidic values. For example, Tris buffer lost buffering capacity at pH 7.46 by adding ATP at a concentration higher than 2 mM. In addition to the buffering capacity, the pH shifts differed with respect to the buffer concentration. High ATP concentrations are commonly used in hexokinase assays. We demonstrated how the presence of ATP affects pH of widely used enzyme assay buffers and inversely affected KM of human hexokinase 2 and S0.5 of human glucokinase. The pH optimum of human glucokinase was never reported before. We found that previously reported optimum of mammalian glucokinase was incorrect, affected by the ATP-induced pH shifts. The pH optimum of human glucokinase is at pH 8.5–8.7. Suggested is the full disclosure of reaction conditions, including the measurement of pH of the whole reaction mixtures instead of measuring pH prior to the addition of all the components.
Similar content being viewed by others


Introduction
Enzyme assays are an integral part of research, since enzymes are fundamental for maintaining the life functions of organisms. In the course of the development of enzyme assays, we must overcome many obstacles, since every enzyme represents its own individuality, which manifests by different requirements of storage, expression and purification processes, pH and temperature stability. Therefore, an adequate and well-defined environment for every enzyme is needed in order to accurately interpret outcomes of individual enzyme assays.
Many enzyme assays possess a high potential of biased outcomes caused by an inaccurately defined pH of the reaction. The well-defined pH of the reaction depends on both a reaction buffer and other components that are necessary for the reaction to proceed1. In this regard, the addition of an acidic substance, such as adenosine triphosphate (ATP), can serve as a source of unnoticed pH changes in enzymatic reactions. For example, high ATP concentrations are characteristic of hexokinase (HK) assays, where they have the potential to affect the Michaelis constant KM of HKs as well as the S0.5 of glucokinase (GCK). Both constants, KM and S0.5, define the values of the substrate concentration at which the rate of reaction is half of the limiting rate. Both enzymes have been subjected to pharmacological studies, in which interpretations of the substrate or the inhibitor/activator affinity and imitation of physiological conditions played a prominent role2,3,4. Surprisingly, many studies on HKs and other enzymes provide little information regarding the use of ATP in their reaction mixtures, or they often disclose pH of the buffer that has been measured only before the subsequent addition of other components, including ATP (Tables S1, S2). Moreover, the pH optimum of human GCK was never determined experimentally and was only inferred from seminal studies by Salas et al.5 that used rabbit GCK instead of its human ortholog (Table S1).
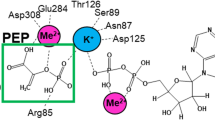
HKs appear to be sensitive to pH-induced changes. Available HK structures suggest that their catalytic domains possess deep crevice between the large and small sub-domains. Glucose binds to the bottom of this crevice using hydrogen bonds to both the subdomains and their connecting region. The interface between the two subdomains is rich in acidic residues and, therefore, susceptible to pH-mediated regulation6. Only relatively few studies on pH kinetics of HKs were previously published; particularly scarce are data on the impact of pH on enzyme activity due to protonation/deprotonation of active site residues. In this regard, the study of calorimetric profiles of yeast HK A revealed a single thermal transition in the acidic pH and two independent transitions in the alkaline pH6. One of the transitions at the alkaline pH corresponds to the small subdomain and the second transition at the alkaline pH corresponds to the large subdomain. The ionization state of the acidic residues at the active site likely regulates domain movements, induce the cleft closure and therefore cause the inaccessibility of active site to glucose6. In bovine brain HK (i.e., HK1), the −log V1 and −log V1/KM profiles displayed slopes of −1 when testing their pH kinetics with glucose and Mg ATP as substrate7. This is consistent with the protonation of a single group on the enzyme. Two ionizable groups were suggested to be involved in the reaction, one employed in the ATP binding and catalysis, and another playing a role in glucose binding7.
In the present study, we focus on enzyme assays, in which the outcomes are sensitive to the pH of the reaction. As a proof of principle, we investigated HK assays, in which the addition of ATP can serve as a source of unnoticed pH changes in enzymatic reactions. We showed how ATP concentration affects pH of different buffers and demonstrated the influence of pH on changes to the KM values of human HK2 as well as the S0.5 values of human GCK.
Methods
To test the buffering capacity of commonly used enzyme assay buffers according to changing ATP concentrations, we prepared the reaction mixtures as follows: 1 mL of the respective buffer; 0.4 mL of the GST-GCK elution buffer, with or without the tested enzyme; 0.1 M ATP in various volumes; and dH2O added to adjust the total volume to 2 mL. The composition of the elution buffer was as follows: 2.6 mM NADP, 0.1 mL 1 M glucose, 0.2 mL 50 mM Tris, 200 mM KCl, and 5 mM DTT; pH adjusted to 8.0. We kept all solutions at 23 °C, except for ATP and NADP, which were kept on ice. In some cases, we observed the shift in pH towards more acidic values after the addition of NADP. The amount of NADP was constant in all mixtures; therefore, the observed changes in pH were caused only by changing ATP concentration. The ATP solution was added to the buffers in a form of a 100 mM aqueous solution that was prepared directly from the ATP disodium salt hydrate powder, without any adjustment of its pH and without the addition of any salts. ATP was always added shortly before the experiments to avoid any potential issues with its stability. All chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
We prepared GST-GCK as described previously8,9 by a one-step purification using GSTrap HP (GE Healthcare, Chicago, IL). In the case of HK2, we introduced the insert encoding HK2 into pET-28a(+) and expressed HK2 in BL21(DE3)pLysS E. coli. We induced HK2 expression by the addition of 1 mM IPTG and subsequently cultivated the cells for 16 h at 22 °C. Afterwards, we purified HK2 using HisTrap HP (GE Healthcare, Chicago, IL). We measured the pH optimum of the purified GST-GCK using a coupled reaction with glucose-6-phosphate dehydrogenase as described previously10. We conducted measurements at 1 mM ATP, 50 mM glucose, 100 mM Tris, for pH range of 7.5–8.8 or 100 mM glycine for pH range of 8.6–10.3. We measured HK2 and GST-GCK activity using a coupled reaction with glucose-6-phosphate dehydrogenase as described previously10,11. In the case of HK2, we measured enzymatic activity in the range of 0–2 mM glucose, unlike GST-GCK, in which the activity we measured was in the range of 0–150 mM. We prepared all the buffers and measured the enzyme kinetics at 23 °C, thereby excluding effects of temperature on pH of the solutions used.
We performed all measurements in three or more independent experiments, each performed in triplicate. We analyzed the obtained curves by nonlinear regression using SigmaPlot 12.0.
Results and Discussion
Even relatively low concentrations of ATP (up to 5 mM) shifted pH of reaction mixtures to acidic values (Fig. 1). The ATP-induced pH shifts differed with respect to the buffering capacity, which is maximal in pKa of the respective substance, with Tris having its pKa at 8.07 and glycine at 9.8. Tris buffer lost its buffering capacity at pH 7.46 by adding ATP at a concentration higher than 2 mM (Fig. 1A). Similarly, glycine buffer lost its buffering capacity with the increasing difference from pKa of glycine (Fig. 1C,D). In addition to the buffering capacity, the pH shifts differed with respect to the buffer concentration, with lower buffer concentrations leading to more prominent ATP-induced pH shifts (Fig. 1B).

ATP addition affects pH of enzyme assay buffers and affects the measurement of the pH optimum as demonstrated in the example of GCK. (A) Effects of ATP addition to the buffer composed of 200 mM Tris, 300 mM KCl and 12 mM MgCl2, pH 7.46, 7.80, 8.09 and 8.68 as measured prior to the ATP addition, and tested before and after the addition of up to 5 mM ATP. (B) Effects of buffer concentration on buffer capacity demonstrated as effects of ATP addition to the buffer containing either 250 mM HEPES, 375 mM KCl and 15 mM MgCl2 or 50 mM HEPES, 50 mM KCl and 15 mM MgCl2, pH 7.26 and 7.35, respectively, as measured prior to the ATP addition and tested before and after the addition of up to 5 mM ATP. (C) Effects of ATP addition to the buffer composed from 200 mM glycine and 12 mM MgCl2, pH 8.34, 8.68, 9.51 and 9.96 as measured prior to the ATP addition and tested before and after the addition of up to 5 mM ATP. (D) Increase in buffering capacity of the glycine buffer demonstrated as the decrease in ΔpH of 200 mM glycine and 12 mM MgCl2 following the increase of pH of the buffer without ATP closer to its pKa. The demonstrated pH range reflects the range of the use of glycine buffer by Salas et al.5. (E) The pH optimum curve of mammalian GCK reprinted with permission from Salas et al.5. Note the use of glycine buffer in the range where it is out of its buffering capacity. Republished with permission of American Society for Biochemistry and Molecular Biology, from Salas, J., Salas, M., Vinuela, E. & Sols, A.: Glucokinase of rabbit liver, J. Biol. Chem. 240, edition 1, 1965, pp. 1014–1018; permission conveyed through Copyright Clearance Center, Inc. (F) The pH optimum curve of human GCK generated during the course of the present study in reaction buffers containing either 200 mM Tris (pH range up to 8.5) or 200 mM glycine (pH range from 8.1).
To demonstrate that the ATP-induced pH shifts may severely affect outcomes of enzyme kinetics measurements, we investigated HK assays, in which high ATP concentrations are commonly used. First, we re-evaluated the pH optimum of GCK that was previously published by Salas et al.5 (Fig. 1E) based on the rabbit GCK ortholog. This is still the only reference curve of the pH optimum of mammalian GCK despite the issues reported below; the pH optimum of human GCK itself was never tested. We particularly contradicted the use of glycine buffer in the published pH range, since a pH lower than 8.0 is outside of the reliable glycine buffering range. Thus, the addition of 5 mM ATP increased the pH of the reaction mixture outside of the desired pH values (Fig. 1C). Therefore, we measured the pH optimum of human GCK in buffers that were devoid of these issues (200 mM Tris and 200 mM glycine) and reflected the pH of the whole reaction mixtures (Fig. 1F), setting the pH optimum of human GCK to pH 8.5–8.7, high above physiological intracellular pH.
The pH optimum higher than physiological intracellular pH of adult differentiated cells is already known from some other glycolytic enzymes and is likely related to increased glycolysis in cancer cells. Similarly to the present data on GCK (Fig. 1), phosphofructokinase, a key rate-limiting glycolysis enzyme, has pH optimum at values higher than those considered physiological intracellular pH of adult differentiated cells. Moreover, the activity of phosphofructokinase drops by over two orders of magnitude when the pH decreases from 7.5 to 7.012,13. Phosphofructokinase studies revealed that its inhibition by protons is allosterically modified by ATP, and the inhibitory effects are lower when lower ATP concentrations are present. The inhibitory effects of ATP at low pH can be reversed by increased concentrations of AMP14,15,16,17,18. Another glycolytic enzyme with higher than expected pH optimum is lactate dehydrogenase, which regenerates NAD+ for glycolysis, and which has its pH optimum at pH 7.519. Consistent with the above findings, the acidosis is well known to decrease glycolytic activity both in vitro and in vivo20,21,22. Importantly, tumors have been repeatedly reported to have a higher intracellular (and lower extracellular) pH compared to normal differentiated adult cells23,24. The intracellular pH of cancer cells is usually between 7.3 and 7.6, whereas the normal differentiated adult cells have an intracellular pH ~7.224, which applies to cancer cells from their early developmental stages25, is sufficient to induce dysplasia26, and is further facilitated during their neoplastic progression27. The role of the dynamics of intracellular pH remains in regulating cell fate decisions and cancer progression remains understudied28 as the previous biochemical studies were only recently supported by an emerging evidence of transient changes in intracellular pH during key cellular processes, including cell cycle progression29, migration30,31, or differentiation32,33. As a large part of previously reported data on HK kinetics is likely affected by the artifacts introduced by the addition of ATP, trusting in the buffering capacity of the respective buffers, we next focused on whether the effects of the pH changes could be generalized to all HKs. We compared the data obtained using human GCK with those obtained using HK2. The S0.5 of GCK gradually decreased by over one half with a pH increase from 6.6 to 8.4 (Fig. 2a). In contrast, the KM value of HK2 gradually increased by one half with a pH increase from 6.9 to 8.5 (Fig. 2b). During the same treatment, the cooperativity of GCK remained stable or slightly increased at the highest pH interval that was tested (Fig. 2a). In contrast, the relative specific activity of HK2 was stable only in a narrow interval of pH 7.3–7.9, while it decreased substantially at pH 6.9 and increased at pH 8.2 and higher (Fig. 2b). Collectively, these data suggest that HKs do not respond to pH changes uniformly. Their pH-dependent changes in activity may be of particular importance in emerging role of HKs in dysregulated cancer metabolism34,35, including their involvement in pH-induced changes of cancer metabolism36,37,38.

The effects of the pH on enzyme kinetics of human GCK and HK2. (a) S0.5 and the Hill index (h) of human GCK as measured in the pH range from 6.6 to 8.4. (b) KM and relative specific activity of human HK2 as measured in the pH range from 6.9 to 8.5.
We proved that even low concentrations of reaction components, such as ATP, may shift the pH of the reaction to undesired values and adversely influence outcomes. We suggest paying more attention to the choice of appropriate assay buffer, its concentration, buffering capacity and the influence of other substances needed for enzyme assays, since enzymes are very susceptible to pH changes. We demonstrated the problem of the sufficient choice of buffer on the pH optimum of GCK, previously measured by Salas et al.5. Due to the consideration of the pH of the whole reaction, we identified a more alkaline pH optimum of human GCK (pH 8.5–8.7) compared to the previously reported optimum of rabbit GCK (pH 7.5–8.0), which was measured under likely irreproducible conditions5. We must admit that the effect of ATP could be eliminated based on the use of a prebuffered ATP solution, which can be purchased or prepared homemade. Nevertheless, the use of such a solution instead of pure ATP has not been mentioned in any of the publications reporting HK assays.
The topic of pH reliability, biased assay outcomes and confusing descriptions of enzyme conditions belong to underestimated but important issues related to research integrity and reproducibility. As an example of a good practice, we would like to cite F. M. Matschinsky and colleagues10, who described the GCK assay as follows: “Glucokinase activity was measured spectrophotometrically using an NADP+ coupled assay with glucose-6-phosphate dehydrogenase as described39. The pH of all assays was 7.4, except for assays which assessed the inhibition of glucokinase by glucokinase regulatory protein (GKRP) where a pH of 7.1 was used.” Although Matschinsky and colleagues referred to a previous paper with regards to the method used, they completed the description of the enzyme assay with information about the pH. Notwithstanding, the common practice is rather to introduce only individual components of the reaction mixture (Table S1). Therefore, we suggest the practice of a full disclosure of reaction conditions of the experiments, including the measurement of the pH of the whole reaction mixtures.
Data Availability
All data are available in the main text or in the supplementary materials. Figure 1E is reprinted with permission from Salas et al.5.
References
Ferreira, C. M., Pinto, I. S., Soares, E. V. & Soares, H. M. Un)suitability of the use of pH buffers in biological, biochemical and environmental studies and their interaction with metal ions – a review. RSC Adv. 5, 30989–31003 (2015).
Lin, H. et al. Discovery of a novel 2,6-disubstituted glucosamine series of potent and selective hexokinase 2 inhibitors. ACS Med. Chem. Lett. 7, 217–222 (2015).
Zhang, H. N. et al. Systematic identification of arsenic-binding proteins reveals that hexokinase-2 is inhibited by arsenic. Proc. Natl. Acad. Sci. USA 112, 15084–15089 (2015).
Fujieda, H. et al. Discovery of a potent glucokinase activator with a favorable liver and pancreas distribution pattern for the treatment of type 2 diabetes mellitus. Eur. J. Med. Chem. 156, 269–294 (2018).
Salas, J., Salas, M., Vinuela, E. & Sols, A. Glucokinase of rabbit liver. J. Biol. Chem. 240, 1014–1018 (1965).
Kumar, D. P., Tiwari, A. & Bhat, R. Effect of pH on the stability and structure of yeast hexokinase A. Acidic amino acid residues in the cleft region are critical for the opening and the closing of the structure. J. Biol. Chem. 279, 32093–32099 (2004).
Solheim, L. P. & Fromm, H. J. pH kinetic studies of bovine brain hexokinase. Biochemistry 19, 6074–6080 (1980).
Šimčíková, D., Kocková, L., Vackářová, K., Těšínský, M. & Heneberg, P. Evidence-based tailoring of bioinformatics approaches to optimize methods that predict the effects of nonsynonymous amino acid substitutions in glucokinase. Sci. Rep. 7, 9499 (2017).
García-Herrero, C. M. et al. Functional analysis of human glucokinase gene mutations causing MODY2: exploring the regulatory mechanisms of glucokinase activity. Diabetologia 50, 325–333 (2007).
Davis, E. A. et al. Mutants of glucokinase cause hypoglycaemia- and hyperglycaemia syndromes and their analysis illuminates fundamental quantitative concepts of glucose homeostasis. Diabetologia 42, 1175–1186 (1999).
Nawaz, M. H. et al. The catalytic inactivation of the N-half of human hexokinase 2 and structural and biochemical characterization of its mitochondrial conformation. Biosci. Rep. 38, BSR20171666 (2018).
Fidelman, M. L., Seeholzer, S. H., Walsh, K. B. & Moore, R. D. Intracellular pH mediates action of insulin on glycolysis in frog skeletal muscle. Am. J. Physiol. 124, 87–93 (1982).
Ui, M. A role of phosphofructokinase in pH-dependent regulation of glycolysis. Biochim. Biophys. Acta 124, 310–322 (1966).
Trivedi, B. & Danforth, W. H. Effect of pH on the kinetics of frog muscle phosphofructokinase. J. Biol. Chem. 241, 4110–4112 (1966).
Dobson, G. P., Yamamoto, E. & Hochachka, P. W. Phosphofructokinase control in muscle: nature and reversal of pH-dependent ATP inhibition. Am. J. Physiol. 250, R71–R76 (1986).
Erecińska, M., Deas, J. & Silver, I. A. The effect of pH on glycolysis and phosphofructokinase activity in cultured cells and synaptosomes. J. Neurochem. 65, 2765–2772 (1995).
Frieden, C., Gilbert, H. R. & Bock, P. E. Phosphofructokinase III. Correlation of the regulatory kinetic and molecular properties of the rabbit muscle enzyme. J. Biol. Chem. 251, 5644–5647 (1976).
Andrés, V., Carreras, J. & Cussó, R. Regulation of muscle phosphofructokinase by physiological concentrations of bisphosphorylated hexoses: effect of alkalinization. Biochem. Biophys. Res. Commun. 172, 328–334 (1990).
Gray, J. A. Kinetics of enamel dissolution during formation of incipient caries-like lesions. Arch. Oral Biol. 11, 397–422 (1966).
Seglen, P. O. The effect of perfusate pH on respiration and glycolysis in the isolated rat liver perfused with an erythrocyte- and protein-free medium. Biochim. Biophys. Acta 264, 398–410 (1972).
Wu, T. F. & Davis, E. J. Regulation of glycolytic flux in an energetically controlled cell-free system: the effects of adenine nucleotide ratios, inorganic phosphate, pH, and citrate. Arch. Biochem. Biophys. 209, 85–89 (1981).
Folbergrová, J., MacMillan, V. & Siesjö, B. K. The effect of hypercapnic acidosis upon some glycolytic and Krebs cycle-associated intermediates in the rat brain. J. Neurochem. 19, 2507–2517 (1972).
Webb, B. A., Chimenti, M., Jacobson, M. P. & Barber, D. L. Dysregulated pH: a perfect storm for cancer progression. Nat. Rev. Cancer 11, 671–677 (2011).
White, K. A., Grillo-Hill, B. K. & Barber, D. L. Cancer cell behaviors mediated by dysregulated pH dynamics at a glance. J. Cell Sci. 130, 663–669 (2017).
Reshkin, S. J. et al. Na+/H+ exchanger-dependent intracellular alkalinization is an early event in malignant transformation and plays an essential role in the development of subsequent transformation-associated phenotypes. FASEB J. 14, 2185–2197 (2000).
Grillo-Hill, B. K., Choi, C., Jimenez-Vidal, M. & Barber, D. L. Increased H+ efflux is sufficient to induce dysplasia and necessary for viability with oncogene expression. eLife 4, e03270 (2015).
Cardone, R. A. et al. A novel NHE1-centered signaling cassette drives epidermal growth factor receptor-dependent pancreatic tumor metastasis and is a target for combination therapy. Neoplasia 17, 155–166 (2015).
Tatapudy, S., Aloisio, F., Barber, D. & Nystul, T. Cell fate decisions: emerging roles for metabolic signals and cell morphology. EMBO Rep. 18, 2105–2118 (2017).
Putney, L. K. & Barber, D. L. Na-H exchange-dependent increase in intracellular pH times G2/M entry and transition. J. Biol. Chem. 278, 44645–44649 (2003).
Denker, S. P. & Barber, D. L. Cell migration requires both ion translocation and cytoskeletal anchoring by the Na-H exchanger NHE1. J. Cell Biol. 159, 1087–1096 (2002).
Stock, C. & Schwab, A. Protons make tumor cells move like clockwork. Pflugers Arch. 458, 981–992 (2009).
Ulmschneider, B. et al. Increased intracellular pH is necessary for adult epithelial and embryonic stem cell differentiation. J. Cell Biol. 215, 345–355 (2016).
Singh, Y. et al. Alkaline cytosolic pH and high sodium hydrogen exchanger 1 (NHE1) activity in Th9 cells. J. Biol. Chem. 291, 23662–23671 (2016).
Hay, N. Reprogramming glucose metabolism in cancer: can it be exploited for cancer therapy? Nat. Rev. Canc. 16, 635–649 (2016).
Wang, L. et al. Hexokinase 2-mediated Warburg effect is required for PTEN- and p53-deficiency-driven prostate cancer growth. Cell Rep. 8, 1461–1474 (2014).
Quach, C. H. et al. Mild alkalization acutely triggers the Warburg effect by enhancing hexokinase activity via voltage-dependent anion channel binding. PLoS ONE 11, e0159529 (2016).
Harduindey, S. et al. Cellular acidification as a new approach to cancer treatment and to the understanding and therapeutics of neurodegenerative diseases. Semin. Canc. Biol. 43, 157–179 (2017).
Hardonniere, K., Huc, L., Sergent, O., Holme, J. A. & Lagadic-Gossmann, D. Environmental carcinogenesis and pH homeostasis: Not only a matter of dysregulated metabolism. Semin. Canc. Biol. 43, 49–65 (2017).
Liang, Y. et al. Variable effects of maturity-onset-diabetes-of-youth (MODY)-associated glucokinase mutations on substrate interactions and stability of the enzyme. Biochem. J. 309, 167–173 (1995).
Acknowledgements
We thank Maria Angeles Navas (Complutense University of Madrid) for the GST-GCK construct. The study was supported by the Czech Science Foundation project 15-03834Y and Charles University in Prague projects Primus/MED/32, GA UK 1428218 and 260387/SVV/2017.
Author information
Authors and Affiliations
Contributions
D.S. performed the experiments. D.S. and P.H. conceived and designed the experiments, analyzed the data, wrote the paper, are responsible for the integrity of this work, revised the article’s intellectual content and approved the final version.
Corresponding author
Ethics declarations
Competing Interests
D.S. and P.H. have been funded by the Czech Science Foundation project 15-03834Y and Charles University in Prague projects Primus/MED/32, GA UK 1428218 and 260387/SVV/2017. The authors declare no other competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Šimčíková, D., Heneberg, P. Identification of alkaline pH optimum of human glucokinase because of ATP-mediated bias correction in outcomes of enzyme assays. Sci Rep 9, 11422 (2019). https://doi.org/10.1038/s41598-019-47883-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47883-1
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
