
Abstract
Thalidomide is widely used for several diseases; however, it causes malformations in embryos exposed during pregnancy. The complete understanding of the mechanisms by which thalidomide affects the embryo development has not yet been obtained. The phenotypic similarity makes TE a phenocopy of syndromes caused by mutations in ESCO2, SALL4 and TBX5 genes. Recently, SALL4 and TBX5 were demonstrated to be thalidomide targets. To understand if these genes act in the TE development, we sequenced them in 27 individuals with TE; we verified how thalidomide affect them in human pluripotent stem cells (hPSCs) through a differential gene expression (DGE) analysis from GSE63935; and we evaluated how these genes are functionally related through an interaction network analysis. We identified 8 variants in ESCO2, 15 in SALL4 and 15 in TBX5. We compared allelic frequencies with data from ExAC, 1000 Genomes and ABraOM databases; eight variants were significantly different (p < 0.05). Eleven variants in SALL4 and TBX5 were previously associated with cardiac diseases or malformations; however, in TE sample there was no association. Variant effect prediction tools showed 97% of the variants with potential to influence in these genes regulation. DGE analysis showed a significant reduction of ESCO2 in hPSCs after thalidomide exposure.
Similar content being viewed by others

Introduction
Thalidomide and its analogs – pomalidomide and lenalidomide - are drugs widely used worldwide for several conditions, such as erythema nodosum leprosum (ENL) - a skin condition related to Hansen’s disease (also known as leprosy) - and multiple myeloma1. These drugs have anti-inflammatory, immunomodulatory and antiangiogenic properties2,3; however, when used during pregnancy they cause Thalidomide Embryopathy (TE) in exposed embryos. From all these three drugs, only thalidomide is produced in Brazil4,5,6; the only country that still registers cases despite laws and regulations regarding its distribution7.
From epidemiological data collected in the 1960s, it is estimated that 20–50% of the embryos exposed to thalidomide present TE8. Thus, a large number do not develop an abnormal phenotype. The frequency of children born with a TE compatible phenotype has increased. From 1982 to 1999, in Brazilian hospitals, the frequency was around 1.92/10,000 births and from 2000 to 2008 around 3.10/10,000 births9. This phenomenon is probably explained by the higher availability of the drug for the treatment of ENL9. The molecular mechanisms of thalidomide teratogenicity are not fully elucidated, slowing down the development of a safe analog (i.e. without the teratogenic propriety). The embryonic genetic background is believed to act in the differential susceptibility to teratogen-induced damage7,10,11,12.
TE is considered a phenocopy of three genetic syndromes - Roberts syndrome, Duane-radial ray syndrome (also known as Okihiro syndrome) and Holt-Oram syndrome - since its features (caused by a non-genetic agent) resemble those phenotypes caused by mutations13 (Table 1). Roberts syndrome (RBS; MIM #268300), also known as Pseudothalidomide syndrome, is a rare autosomal recessive disorder caused by mutations in ESCO2 gene. ESCO2 protein is an acetyltransferase responsible for cohesion of sister chromatids during cell division process14,15. Duane-radial ray syndrome (DRRS; MIM #607323) and Holt-Oram syndrome (HOS; MIM #142900) are both autosomal dominant conditions caused by mutations in SALL4 and TBX5 genes, respectively. These two genes encode transcription factors that interact and act on limb and heart development16,17,18,19.
Some syndromes are caused by mutations in genes that encode proteins that are affected by some teratogens. Thus, the analysis of the molecular bases of some syndromes has been efficient in the understanding of teratogenic mechanisms20. TBX5 and SALL4 gene expression is reduced in wing buds of chicken embryos and primary human embryonic fibroblasts exposed to thalidomide21. TBX5 was recently demonstrated to be a direct target of thalidomide; in the presence of the drug, the protein dramatically reduces its DNA binding potential and the activation of its target genes. The drug also prevents TBX5 binding to HAND2, an important protein involved in the heart development22. Even more recently, two studies suggested a new hypothesis to thalidomide teratogenesis, highlighting the SALL4 protein degradation as the mechanism responsible for the malformations observed in TE. These studies demonstrated that SALL4 is degraded post-transcritionally in rabbits and different types of human cell lines after thalidomide or its analogs exposure, but not in models not sensitive to thalidomide, such as mice23,24.
Taking into account the phenotypic similarity between people with TE and individuals with the aforementioned genetic syndromes, and also the experimental studies showing that these genes are affected by thalidomide, the investigation of these genes might explain at least partially why some people are affected by TE and others are not. Thus, in order to evaluate the role of ESCO2, TBX5 and SALL4 genes in thalidomide teratogenesis we have sequenced these genes in individuals with TE and analyzed the variants found in relation to their potential to increase the risk to TE development. After that, we verified - from transcriptomes available in the Gene Expression Omnibus (GEO) database - how thalidomide affects their expression on human pluripotent stem-cells (hPSC) exposed to the drug. Finally, we verified - through systems biology databases - if and how these genes interact with each other.
Results
Gene panel sequencing of individuals with Thalidomide Embryopathy and in silico functional predictions
Twenty-seven Brazilian subjects with TE were included in our study. The characterization of these individuals, regarding their congenital anomalies and late outset diseases, is described in Table 2.
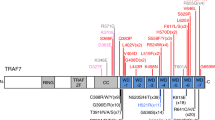
Using a targeted sequencing approach, we sequenced exons, flanking intronic regions, and untranslated regions of ESCO2, SALL4 and TBX5 genes. The average depth of coverage for the variants found was 491.5×. A coverage higher than 100x was obtained in 92.44% of the bases. Gene sequencing data has been deposited at the Sequence Read Archive (SRA) under accession number SRP160424. A novel variant has been submitted to Leiden Open Variation Database (LOVD). Figure 1 and Table 3 summarize all variants found and their position in each gene.

Position of variants found in exons, flanking introns and untranslated regions of ESCO2, SALL4 and TBX5 genes in people with Thalidomide Embryopathy. We identified 8 variants in ESCO2 gene, 15 in SALL4 gene and 15 in TBX5 gene (one of them – c.420 C > T (p. Asp140=) – a novel variant).
The frequency of all the variants was in accordance with the Hardy-Weinberg equilibrium, except for c.1013 + 35 G > A and c.*71_*74delTATT of ESCO2 gene. We observed differences in the allelic frequencies of eight variants between the TE group and the genomic databases (ExAC, 1000 Genomes and ABraOM) (Table 3), three in ESCO2, two in SALL4 and three in TBX5. Three of them (from SALL4 and TBX5 genes) were in coding sequences.
There was no association between the twelve rare variants identified (frequency < 0.01 in all databases) and the presence of some specific congenital anomaly or disease observed in the TE group.
Variants in the same gene demonstrated high linkage disequilibrium (D’ > 0.9 and LOD > 2) (Supplementary Table S2). The haplotypes inferred for each gene (Supplementary Table S3) were also not associated with specific congenital anomalies or diseases in the TE group.
According to the functional prediction tools, some variants potentially affect different aspects of the gene or the protein (Fig. 2) (Supplementary Table S4). They were also more frequent than expected in the TE group. Thus, they stood out, being more likely to influence in the TE susceptibility and in the risk of development of specific congenital anomalies identified in the affected individuals (Fig. 2) (Supplementary Tables S4 and S5).

Heatmap representing the potential impact of variants found in ESCO2, SALL4 and TBX5 genes in regulatory features of these genes, their proteins and in TE. In the lines are represented the variants, the scores assigned to them after functional predictions and the final score. The scores are presented by color tone variation; the higher the points the darker the color; the lower the points the lighter the color. Regarding the columns, the first ten represent variant effect prediction tools, the eleventh represents the formation of haplotypic blocks and the twelfth represents the statistically significant difference of some variant with the genomic databases. The variant effect prediction tools pointed out variants which altered the features evaluated by the tool, being the impact of lesser or greater degree. To haplotypic blocks, it received a point variants that formed block. Variants with a statistically significant difference of allelic frequencies between the TE sample and the databases were pointed out, being greater the score of variants which differed from more than one database.
ESCO2 gene revealed few variants, highlighting the regulatory ones
Eight variants were found in ESCO2 gene in all individuals (Fig. 1 and Table 3). Three variants located in regulatory regions stood out according to a score we developed (based in the variants effect according to prediction tools and their frequencies in the TE group): c.−151 G > A, c.1013 + 35 G > A and c.*71_*74delTATT (Fig. 2).
The 5′UTR c.−151 G > A was predicted as pathogenic, affecting the splicing, one CpG island and one miRNA binding site (hsa-miR-6858-3p) (Supplementary Table S4). The intronic variant c.1013 + 35 G > A, reported as benign in the ClinVar database25, was significantly more prevalent in the TE group than in the evaluated databases (Table 3) and was predicted to affect two transcription factors binding sites (TBP, which has a binding site in ESCO2 and POU6F1, which acts in heart development)25 (Supplementary Table S4). The 3′UTR c.*71_*74delTATT was also significantly more frequent in TE group (Table 3) and it affects splicing and two miRNAs binding sites (hsa-miR-606 and hsa-miR-3149) (Supplementary Table S4).
SALL4 gene presented many variants in coding regions with potential to affect splicing and transcription factors binding sites
We identified 15 variants in SALL4 gene, 10 of them in coding regions (Fig. 1 and Table 3). Five variants stood out by our score: c.2977 G > C (p.Gly993Arg), c.1520 T > G, c.*497 T > C, c.1860 A > G (p.Thr620=) and c.131-226 T > C (Fig. 2).
The missense c.2977 G > C (p.Gly993Arg) and the downstream c.*497 T > C were significantly more frequent in the TE group (Table 3); they were predicted as pathogenic and affecting splicing; the c.*497 T > C also affecting three miRNAs (hsa-miR-5095, hsa-miR-1254 and hsa-miR-5689) and one transcription factor binding site (PITX2, which acts in heart and limb development)25 (Supplementary Table S4). The missense c.1520 T > G (p.Leu507Arg) and the synonymous c.1860 A > G (p.Thr620=) were in a haplotype block and were predicted as affecting splicing; the first disrupts seven transcription factors binding sites (PAX5, PPARG and RXRB, which have binding sites in SALL4 and PPARA, PPARD, RARB and RXRA, that act in heart development)25; the last disrupts two CpG islands (Supplementary Table S4). The intronic c.131–226 T > C possibly affects the splicing, one miRNA (hsa-miR-615-5p) and two transcription factors binding sites (TFAP4 and ZBTB7B, which regulate in SALL4)25 (Supplementary Table S4).
In TBX5 a novel variant within the T-box domain was identified and many variants were predicted as pathogenic
We identified 15 variants in TBX5 gene, one of them – NM_000192:c.420 C > T (p. Asp140=) – has not been reported in genomic databases (Fig. 1 and Table 3). This novel variant was identified in heterozygosis in one individual, with a coverage of 497x in the sequencing (50% for each nucleotide). Four variants of TBX5 stood out by our score: c.787 G > A (p.Val263Met), c.−38-1865 G > A, c.511-56 T > C and the novel c.420 C > T (p. Asp140=) (Fig. 2).
The missense c.787 G > A (p.Val263Met), which was not located in the T-box domain, was significantly more frequent in the TE group (Table 3) and predicted as pathogenic and affecting splicing (Supplementary Table S4). In the TE group, two individuals had this variant; one of them presents angina (a chest pain due to reduced blood flow to the heart). The intronic variants c.-38-1865 G > A and c.511-56 T > C were predicted as pathogenic and affecting splicing. The first affects also one CpG island and one transcription factor binding site (NFIB, that has a binding site in TBX5)25. The second affects two transcription factors binding sites (GATA1 and GATA2, which act in heart development)25 (Supplementary Table S4).
A novel synonymous variant c.420 C > T (p. Asp140=) was located within the T-Box domain region of the gene. It was predicted as pathogenic and affecting splicing (Supplementary Table S4). Once it was never described, we classified it based on the classification criteria of the American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) Standards and Guidelines26. The included criteria were: (1) Absent from controls (or at least low frequency if recessive) in Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium and (2) Multiple lines of computational evidence support a deleterious effect on the gene or gene product (conservation, evolutionary, splicing impact, etc.). According ACMG/AMP system this alteration was classified as a variant of uncertain significance (VUS).
Differential Gene Expression (DGE) analysis from secondary data of the GEO database
The expression of ESCO2, SALL4 and TBX5 genes in human pluripotent stem-cells after thalidomide exposure was evaluated from secondary data obtained in the GSE63935 study, available in the GEO database27. The differential gene expression (DGE) analyses demonstrated a sharp reduction in ESCO2 expression after 2 and 6 days of thalidomide exposure (p = 1.39e-09 and 0.045699) (Table 4).
Interaction networks and gene ontology analysis
The interaction network analyses performed in STRING database showed that ESCO2, SALL4 and TBX5 proteins do not interact directly, although they are included in the same network through interactions with other proteins (Fig. 3). A Gene Ontology (GO) analysis identified 160 biological processes significantly enriched these genes’ network, mainly linked to cell cycle and DNA replication (Supplementary Table S6). SALL4 and TBX5 share, as expected, ontologies of embryonic, limb and heart development.

Protein–protein interaction network including ESCO2, SALL4 and TBX5 proteins. These three proteins do not interact directly, but through others secondary binding targets.
Discussion
Although the thalidomide teratogenic effect has been known for almost 60 years, new cases of TE are still reported in Brazil7. Studies have attempted to understand the mechanisms by which thalidomide affects the embryo development; however, the complete understanding has not yet been obtained. The discovery of genes or proteins that are affected by thalidomide or somehow influence the susceptibility of the thalidomide teratogenic action is extremely important for the better understanding of the teratogenic mechanisms of thalidomide.
In this study we investigated a new hypothesis regarding thalidomide teratogenesis. The identification of teratogenic mechanisms is extremely difficult since teratogenesis is a multifactorial process10. Some teratogenic mechanisms could be clarified through the analysis of the molecular bases of genetic disorders phenocopies of embryopathies due to drugs exposure20. Here we evaluated three genes that are the genetic basis of syndromes in which TE is a phenocopy.
The relationship of thalidomide with SALL4 and TBX5 proteins was recently demonstrated by three experimental studies22,23,24. Thalidomide is capable of binding to TBX5 protein22 and it is able to induce SALL4 degradation23,24. Both studies suggested that such capacities of thalidomide in SALL4 and TBX5 proteins could be the possible mechanisms by which thalidomide causes TE. In this way, our approach became even more relevant, providing new insights about the role of these genes in the genetic susceptibility to TE in humans.
In ESCO2 gene, the regulatory variants that we found showed to have the greatest pathogenic potential to increase the risk to TE. It is known that regulatory variants could increase the susceptibility to diseases since they can act in transcription, splicing and translation processes28; however, we did not find an association between their frequencies in the TE group and an increased risk for this condition.
Regarding the SALL4 gene, some variants were highlighted to their potential risk for TE or specific anomalies. The rare missense c.2977 G > C (p.Gly993Arg), predicted as pathogenic and affecting splicing, was present only in one individual in the TE group. Because of the absence of pathogenicity information for this variant in the literature and the scarcity of clinical information from this carrier, we did not consider it as causative of an anomaly or syndrome on its carrier. Two variants in coding regions – c.1520 T > G (p.Leu507Arg) and c.1860 A > G (pThr620=) – were in a haplotype block. Haplotypes have been already associated with a risk for teratogenesis29, including thalidomide teratogenesis11; however, despite some of these variants carriers present auditory defect (4/14 carriers), visual impairment (6/14) and cardiovascular diseases (3/14), these anomalies could be not associated with the frequency of this haplotype block. Finally, the 3′UTR c.*497 T > C appeared affecting the binding the miRNA hsa-miR-1254. This miRNA is expressed in human embryonic stem cells and it regulates genes of stem cells differentiation, development and transcriptional regulation30, compatible with SALL4 function. The carriers of this variant did not have any exclusive congenital anomaly that could be associated with its presence.
SALL4 protein was recently demonstrated to be degraded post-transcriptionally after thalidomide or its analogs exposure in different types of human cells23,24. SALL4 degradation is mediate through thalidomide-dependent Cereblon protein (CRBN)23,24, an integrant of the CRL4CRBN ubiquitin-ligase complex, previously described as the primary target of thalidomide31. The residue G416 in the second zinc finger domain of SALL4 was essential for its recruitment and degradation; also, the residue G600 of the fourth zinc finger domain had its importance highlighted23,24. Here, we did not identify any variation in these reported positions in the TE group. Similarly, there are no variants reported in humans for the residue G416 of SALL4 in the genomic databases (ExAC, 1000 Genomes and AbraOM). For the residue G600, just one variant – NM_020436:c.1798 G > A (p.Gly600Ser) - was identified (frequency < 0.003) in these databases. Thus, comparing our findings in the TE group and data from genomic databases with the studies aforementioned, it is not possible to explain based on the SALL4 variants why only 20–50% of the individuals exposed to thalidomide developed TE8.
Cardiac abnormalities are frequent in individuals with TE, with an estimated frequency of 8%32. SALL4 and TBX5 genes are essential to heart development19,33 and changes in them have been already associated to increased risk for cardiac diseases34,35,36,37,38,39. Here, eleven variants found in SALL4 and TBX5 were previously evaluated or associated with cardiac malformations or cardiovascular diseases (Supplementary Table S7)34,36,37,38. It is possible that the small sample size of this study did not allow us to find an association of these variants with increased risk for malformations or cardiovascular diseases in individuals with TE; however, it is quite likely that at least some of the variants found in these genes may play an important role in such conditions in these individuals, being important a further genetic and transcriptional evaluation of them in larger samples and also in experimental assays.
TBX5 action in heart development was demonstrated to be affected by thalidomide due to the drug impaired TBX5 and HAND2 proteins interaction22. Here, we did not identify in our TE sample any variant reported as important for TBX5 binding to DNA or to HAND2, which could be associated with cardiac malformations or related diseases. We found a missense variant c.787 G > A (p.Val263Met) in these gene in two individuals of the TE group. This variant was previously associated with bicuspid aortic valve37. One of the carriers has angina, but we cannot associate this phenotype with its presence since we observed it just in on individual.
Although being in the T-Box domain and having been predicted as pathogenic, the novel synonymous c.420 C > T (p. Asp140=) of TBX5 was classified as VUS using the ACMG criteria. Because of that, we have not considered it as causing Holt-Oram Syndrome in the carrier. Moreover, the carrier does not have any exclusive anomaly that could to be caused by this variant.
Changes in SALL4 and TBX5 genes expression and SALL4 and TBX5 proteins levels after thalidomide exposure have been demonstrated21,23,24; however, ESCO2 differential expression was not reported23. Here, we observed thalidomide drastically reducing ESCO2 expression in human pluripotent stem-cells. Because ESCO2 protein plays a key role during development, its reduction in cells directly affect the development, causing malformations such as those seen in Roberts Syndrome15. Taking this into account, it is possible to suggest that the effect of thalidomide on this gene expression helps in the TE development, and this would also explain the mechanism by which TE is a phenocopy of Roberts Syndrome.
The sample size of this work is a limitation of our investigation. This sample was recruited through a collaboration with the Brazilian Association of Patients with Thalidomide Syndrome (ABPST) and represents more than 10% of the live people with Thalidomide Embryopathy (TE) in Brazil. However, there is great difficulty to expand this sample, because the majority of the individuals with TE are elderly and live in different regions in Brazil, not favoring recruitments and not attending ABPST meetings. Taking into account that new cases are not expected due to birth control in women taking thalidomide, this in the only Brazilian resource of recruitment. The evaluation of a control group, including people who were exposed to thalidomide during pregnancy but did not develop TE, would be the best epidemiological design for comparison with the cases of our sample. However, the information about which mothers used thalidomide during pregnancy and had children without TE in 1960’s is very difficult to recover. Many of these mothers are already deceased, others are in advanced age and do not remember such information. Epidemic TE cases occurred more than 50 years ago, and even if we tried to include a control sample from that time, it would probably be a biased information due to the time elapsed since then. In order to provide a genetic background for comparison of the frequency of variants, we have used public genome databases from different ethnic groups, including healthy Brazilians. Nevertheless, this is the only sample of survivors of thalidomide embryopathy which were born in 1960´s that has been widely assessed regarding genomic and clinical features. This approach can provide different and valuable insights about mechanisms and pathways of thalidomide involved in embryology and therapeutic actions in humans. Another limiting factor is the expression of the genes investigated in adults. Previous studies show that SALL4 and TBX5 have a high expression during development and then decrease their expression and restrict it to only a few tissues40,41. For this reason, we decided to evaluate the effect of thalidomide on the expression of SALL4, TBX5 and ESCO2 in cells representing the stage of embryonic development. Finally, few studies in literature evaluated the sequence of these three genes in humans, increasing the difficulty to compare and make hypotheses about genetic variability and its impact on the proteins.
Here we investigated a different hypothesis of those traditionally studied in thalidomide teratogenesis, evaluating the role of the genes that cause syndromes whose TE is a phenocopy. From the sequencing data of this study, we cannot conclude that the genetic variants found in ESCO2, SALL4 and TBX5 in our sample of people with TE could act in the genetic susceptibility to the TE development; however, this approach showed that variants in SALL4 and TBX5 with a known impact on cardiac malformations or diseases are not uncommon in individuals with TE. We could also demonstrate that genetic variants recently described as associated with thalidomide-TBX5 binding and SALL4 degradation thalidomide-mediate22,23,24 - proposed as responsible for the thalidomide teratogenesis - do not appear in our TE group. Moreover, we described a novel synonymous variant in the T-Box domain of TBX5 gene, the most important domain of this transcription factor, and we classified it as VUS, being necessary its experimental validation. From the analysis of secondary gene expression data, which included a group of human pluripotent stem-cells exposed to thalidomide (cases) and an unexposed group (control), we have described for the first time that exposure to thalidomide is capable to affect ESCO2 gene expression, a gene essential for cell division. Such result points to the ESCO2 gene as a possible target for thalidomide, primary or secondary, that should be taken into account in future studies on thalidomide teratogenesis mechanisms as well as in investigations on thalidomide safe analogues.
Phenotypic evaluations are an important alternative to provide insights in molecular genetics researches. Regarding the current research, it is clear that further studies are necessary, although we demonstrated the importance of understanding how thalidomide mimics genetic syndromes to also comprehend its teratogenic property.
Materials and Methods
Ethical considerations
This study was approved by the Research Ethics Committee of the Hospital de Clínicas de Porto Alegre (number 10-0244). The whole research was performed in accordance with relevant guidelines and regulations. All subjects assigned a free and informed consent form.
Sample
The 27 subjects of our sample were recruited through the Brazilian Association of People Affected by Thalidomide Syndrome (ABPST). This sample represents about 10% of the live cases of TE in Brazil.
Molecular analysis
Saliva was collected through Oragene-DNA OG-500 (DNA Genotek, Canada) and DNA was obtained according to the manufacturer’s instructions.
A gene panel including ESCO2, SALL4 and TBX5 genes was designed through Ion AmpliseqTM Designer tool (Thermo Fisher Scientific, USA) covering the coding regions and 50 bp of adjacent introns of each gene. The targeted gene sequencing was performed in Ion PGM technology (Thermo Fisher Scientific, USA) at Hospital de Clínicas de Porto Alegre. The sequences obtained were analyzed through Ion Reporter v.5.2 (Thermo Fisher Scientific, USA) using the genome reference GRCh37. The sequences of reference transcripts used were: NM_001017420 to ESCO2, NM_020436 to SALL4 and NM_000192 to TBX5.
Statistical analysis
We compared the allelic frequencies found in the TE sample with data from three different databases: 1000 Genomes Project (European population), Exome Aggregation Consortium (ExAC) (Europeans non-finnish) and Arquivo Brasileiro Online de Mutações (ABraOM) (Brazilian population database) using Chi-square or Fisher’s Exact Test in SPSS® v.18 (SPSS, IBM, USA) software. The FDR correction was used to eliminate the false discover rate. A two-tailed p-value < 0.05 was considered significant.
Linkage disequilibrium between variants was estimated using the Haploview program v.4.2 (IBM, USA), and the haplotypes were obtained with Bayesian algorithm in the PHASE v.2.1.1 tool (University of Chicago, USA).
In silico analyses
In silico analyses of the variants found were performed with the following bioinformatics tools: SIFT, PolyPhen-2, F-SNP, Mutation Taster, Predict SNP-2, SILVA, MotifbreakR, HSF, MethPrimer and miRBase. Characteristcs of each tool used is described in Supplementary Table S1.
Pathogenicity score
In order to evaluate the pathogenic potential of the variants found, which could increase the susceptibility to TE, we set up a score for each variant based in the statistics analyses (difference of allelic frequencies between TE group and genomic databases), haplotype block formation and in silico predictions of pathogenicity. If the variant affected some of these features it was scored and at the end we added the points.
Differential gene expression (DGE) analysis
The raw data of the transcriptome GSE63935 available on the Gene Expression Omnibus (GEO) database27 was reassessed here. In the aforementioned study the RNA of human pluripotent stem cell-derived neural constructs (mix of neural, endothelial, mesenchymal and macrophage precursor) was extracted 2 and 6 days after exposure to toxicants. Here we compared the expression data of ESCO2, SALL4 and TBX5 genes in both periods after thalidomide exposure and compared with control cells (exposed to saline solution). Secondary data analysis was performed through edgeR package in RStudio v.1.0.136. P-values were adjusted by false discovery rate correction, considering an Adjusted P-value of <0.05 significant.
Interaction networks and gene ontology analysis
We performed protein-protein interaction network analysis and gene ontology enrichment analysis in the STRING database42.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Vargesson, N. Thalidomide-induced teratogenesis: history and mechanisms. Birth Defects Res C Embryo Today 105, 140–156 (2015).
Sampaio, E. P., Sarno, E. N., Galilly, R., Cohn, Z. A. & Kaplan, G. Thalidomide selectively inhibits tumor necrosis factor alpha production by stimulated human monocytes. J Exp Med 173, 699–703 (1991).
D’Amato, R. J., Loughnan, M. S., Flynn, E. & Folkman, J. Thalidomide is an inhibitor of angiogenesis. Proc Natl Acad Sci USA 91, 4082–4085 (1994).
Paumgartten, F. J. & de Souza, N. R. Clinical use and control of the dispensing of thalidomide in Brasilia-Federal District, Brazil, from 2001 to 2012. Cien Saude Cole 18, 3401–3048 (2013).
Sales Luiz Vianna, F. et al. Pharmacoepidemiology and thalidomide embryopathy surveillance in Brazil. Reprod Toxicol 53, 63–67 (2015).
Sales Luiz, V. F., Kowalski, T. W., Fraga, L. R., Sanseverino, M. T. & Schuler-Faccini, L. The impact of thalidomide use in birth defects in Brazil. Eur J Med Genet 60, 12–15 (2017).
Vianna, F. S. et al. Recognition of the phenotype of thalidomide embryopathy in countries endemic for leprosy: new cases and review of the main dysmorphological findings. Clin Dysmorphol 22, 59–63 (2013).
Newman, C. G. The thalidomide syndrome: risks of exposure and spectrum of malformations. Clin Perinatol 13, 555–573 (1986).
Vianna, F. S. et al. Epidemiological surveillance of birth defects compatible with thalidomide embryopathy in Brazil. PLoS One 6, 21735 (2011).
Cassina, M., Salviati, L., Di Gianantonio, E. & Clementi, M. Genetic susceptibility to teratogens: state of the art. Reprod Toxicol 34, 186–191 (2012).
Kowalski, T. W. et al. New Findings in eNOS gene and Thalidomide Embryopathy Suggest pre-transcriptional effect variants as susceptibility factors. Sci Rep 6, 23404 (2016).
Vianna, F. S. et al. Genomic and in silico analyses of CRBN gene and thalidomide embryopathy in humans. Reprod Toxicol 66, 99–106 (2016).
Lenz, W. Phenocopies. J Med Genet 10, 34–49 (1973).
Schüle, B., Oviedo, A., Johnston, K., Pai, S. & Francke, U. Inactivating mutations in ESCO2 cause SC phocomelia and Roberts syndrome: no phenotype-genotype correlation. Am J Hum Genet 77, 1117–1128 (2005).
Vega, H. et al. Roberts syndrome is caused by mutations in ESCO2, a human homolog of yeast ECO1 that is essential for the establishment of sister chromatid cohesion. Nat Genet 37, 468–470 (2005).
Li, Q. Y. et al. Holt-Oram syndrome is caused by mutations in TBX5, a member of the Brachyury (T) gene family. Nat Genet 15, 21–29 (1997).
Al-Baradie, R. et al. Duane radial ray syndrome (Okihiro syndrome) maps to 20q13 and results from mutations in SALL4, a new member of the SAL family. Am J Hum Genet 71, 1195–1199 (2002).
Harvey, M. P. & Logan, O. Sall4 acts downstream of tbx5 and is required for pectoral fin outgrowth. Development 133, 1165–1173 (2006).
Koshiba-Takeuchi, K. et al. Cooperative and antagonistic interactions between Sall4 and Tbx5 pattern the mouse limb and heart. Nat Genet 38, 175–183 (2006).
Cassina, M., Cagnoli, G. A., Zuccarello, D., Di Gianantonio, E. & Clementi, M. Human teratogens and genetic phenocopies: Understanding pathogenesis through human genes mutation. Eur J Med Genet 60, 22–31 (2017).
Knobloch, J. & Rüther, U. Shedding light on an old mystery: thalidomide suppresses survival pathways to induce limb defects. Cell Cycle 7, 1121–1127 (2008).
Khalil, A. et al. A HAND to TBX5 Explains the Link Between Thalidomide and Cardiac Diseases. Sci Rep 7, 1416 (2017).
Donovan, K. A. et al. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray Syndrome. Elife 7, e38430 (2018).
Matyskiela, M. E. et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat Chem Biol 14, 981–987 (2018).
Landrum, M. J. et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42, 980–D985 (2014).
Richards, S. et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424 (2015).
Schwartz, M. P. et al. Human pluripotent stem cell-derived neural constructs for predicting neural toxicity. Proc Natl Acad Sci USA 112, 12516–12521 (2015).
Knight, J. C. Regulatory polymorphisms underlying complex disease traits. J Mol Med (Berl) 83, 97–109 (2005).
Lupo, P. J. et al. A GCH1 haplotype and risk of neural tube defects in the National Birth Defects Prevention Study. Mol Genet Metab 107, 592–595 (2012).
Morin, R. D. et al. Application of massively parallel sequencing to microRNA profiling and discovery in human embryonic stem cells. Genome Res 18, 610–621 (2008).
Ito, T. et al. Identification of a primary target of thalidomide teratogenicity. Science 327, 1345–1350 (2010).
Miller, M. T. & Strömland, K. Teratogen update: Thalidomide: A review, with a focus on ocular findings and new potential uses. Teratology 60, 306–321 (1999).
Steimle, J. D. & Moskowitz, I. P. TBX5: A Key Regulator of Heart Development. Curr Top Dev Biol 122, 195–221 (2017).
Holm, H. et al. Several common variants modulate heart rate, PR interval and QRS duration. Nat Genet 42, 117–122 (2010).
Szot, J. O. et al. A Screening Approach to Identify Clinically Actionable Variants Causing Congenital Heart Disease in Exome. Data. Circ Genom Precis Med 11, e001978 (2010).
Pazoki, R. et al. SNPs Identified as Modulators of ECG Traits in the General Population Do Not Markedly Affect ECG Traits during Acute Myocardial Infarction nor Ventricular Fibrillation Risk in This Condition. PLoS One 8, e57216 (2013).
Bonachea, E. M. et al. Use of a targeted, combinatorial next-generation sequencing approach for the study of bicuspid aortic valve. BMC Medical Genomic 7, 56 (2014).
Wang, F. et al. A TBX5 3′UTR variant increases the risk of congenital heart disease in the Han Chinese population. Cell Discov 3, 17026 (2017).
Yamada, Y. et al. Identification of TNFSF13, SPATC1L, SLC22A25 and SALL4 as novel susceptibility loci for atrial fibrillation by an exome-wide association study. Mol Med Rep 16, 5823–5832 (2017).
Hatcher, C. J., Goldstein, M. M., Mah, C. S., Delia, C. S. & Basson, C. T. Identification and localization of TBX5 transcription factor during human cardiac morphogenesis. Dev Dyn 219, 90–5 (2000).
Tatetsu, H. et al. SALL4, the missing link between stem cells, development and cancer. Gene 584, 111–9 (2016).
von Mering, C. et al. STRING: a database of predicted functional association between proteins. Nucleic Acids Res 31, 258–261 (2003).
Hansen, J. M. & Harris, C. A novel hypothesis for thalidomide-induced limb teratogenesis: redox misregulation of the NF-kappaB pathway. Antioxid Redox Signal 6, 1–14 (2004).
Kowalski, T. W., Sanseverino, M. T., Schuler-Faccini, L. & Vianna, F. S. Thalidomide embryopathy: Follow-up of cases born between 1959 and 2010. Birth Defects Res A Clin Mol Teratol 103, 794–803 (2015).
Gordillo, M., Vega, H. & Jabs, E. W. Roberts Syndrome in GeneReviews (R). Seattle, WA: in press (2013).
Kohlhase, J. SALL4-Related Disorders in GeneReviews (R). Seattle, WA: in press (2015).
McDermott, D. A., Fong, J. C. & Basson, C. T. Holt-Oram Syndrome in GeneReviews (R). Seattle, WA: in press (2015).
Akiyama, R. et al. Sall4-Gli3 system in early limb progenitors is essential for the development of limb skeletal elements. Proc Natl Acad Sci USA 112, 5075–5080 (2015).
Sheeba, C. J. & Logan, M. P. The Roles of T-Box Genes in Vertebrate Limb Development. Curr Top Dev Biol 122, 355–381 (2017).
Acknowledgements
The authors acknowledge the Brazilian Association of People Affected by Thalidomide Syndrome (ABPST), National Institute of Population Medical Genetics (INAGEMP) (grant CNPq 573993/2008-4, CNPq 465549/2014 and FAPERGS 17/2551.0000521-0), Research and Events Incentive Fund of Hospital de Clínicas de Porto Alegre (FIPE/HCPA) (Grant 10-0244) and Universal Project Fund (Grant CNPq 423249/2016-9).
Author information
Authors and Affiliations
Contributions
J.A.G., Conception and design, Acquisition of data, Analysis and interpretation of data, Wrote the manuscrip; T.W.K., Conception and design, Analysis and interpretation of data, Article Review; L.R.F., Analysis and interpretation of data, Article Review; G.S.M., Analysis and interpretation of data, Article Review; M.T.V.S., Conception and design; L.S.F., Conception and design, Article Review; F.S.L.V., Conception and design, Analysis and interpretation of data, Article Review.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gomes, J.d.A., Kowalski, T.W., Fraga, L.R. et al. The role of ESCO2, SALL4 and TBX5 genes in the susceptibility to thalidomide teratogenesis. Sci Rep 9, 11413 (2019). https://doi.org/10.1038/s41598-019-47739-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47739-8
This article is cited by
-
Possible New Candidates Involved to Thalidomide-Related Limbs and Cardiac Defects: A Systems Biology Approach
Biochemical Genetics (2024)
-
CRL4-Cereblon complex in Thalidomide Embryopathy: a translational investigation
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
