
Abstract
Tibetan pig is native to the Qinghai-Tibet Plateau and has adapted to the high-altitude environmental condition such as hypoxia. However, its origin and genetic mechanisms underlying high-altitude adaptation still remain controversial and enigmatic. Herein, we analyze 229 genomes of wild and domestic pigs from Eurasia, including 63 Tibetan pigs, and detect 49.6 million high-quality variants. Phylogenomic and structure analyses show that Tibetan pigs have a close relationship with low-land domestic pigs in China, implying a common domestication origin. Positively selected genes in Tibetan pigs involved in high-altitude physiology, such as hypoxia, cardiovascular systems, UV damage, DNA repair. Three of loci with strong signals of selection are associated with EPAS1, CYP4F2, and THSD7A genes, related to hypoxia and circulation. We validated four non-coding mutations nearby EPAS1 and CYP4F2 showing reduced transcriptional activity in Tibetan pigs. A high-frequency missense mutation is found in THSD7A (Lys561Arg) in Tibetan pigs. The selective sweeps in Tibetan pigs was found in association with selection against non-coding variants, indicating an important role of regulatory mutations in Tibetan pig evolution. This study is important in understanding the evolution of Tibetan pigs and advancing our knowledge on animal adaptation to high-altitude environments.
Similar content being viewed by others


Introduction
The Qinghai-Tibet Plateau is a hotspot for high-altitude adaptation studies in diverse native organisms, including humans1,2,3,4, domestic animals5,6,7, and wild life8,9. Tibetan pig is the indigenous pig (Sus scrofa domesticus) breed native to the Qinghai-Tibet Plateau, providing Tibetans with stable source of meat. Earliest records from the book of Tang show that Tibetans raised domestic pig in the 7th century10. Tibetan pig adapts well to harsh plateau environments and extensive feeding condition, mainly in search of food by themselves. Physiological studies show that the Tibetan pig have evolved physiological adaptations to the high-altitude hypoxia, such as a thicker alveolar septum with more developed capillaries11, larger and strong heart12.
The origin of Tibetan pigs is still under debate. Earlier studies have proposed different origin models. The earliest study based on phylogenomic analysis of mitochondrial DNA (mtDNA) sequence variations in 567 domestic pigs (including 29 Tibetan pigs) and 155 wild boars across Asia conducted by Wu et al. (2007) showed that majority of Tibetan pigs shared haplogroups with domestic pigs from Yangtze River and northern China13. Later, Yang et al. (2011) suggested a local origin of Tibetan pigs from Tibetan highlands by analyzing mtDNA variants in more pig samples from Asia14. In a recent nuclear genome research, Li et al.(2013) considered them as wild boars that have evolved without artificial selection12. However, Ai et al. (2014) defined the Tibetan pig as a domestic breed and found the essential role of admixture with neighboring Chinese domestic pigs during their breeding15.
Clarifying the relationship between Tibetan and other Chinese wild and domestic pigs will provide important information to guide the choice of research approach used to reveal genetic mechanism underlying high-altitude adaptation in Tibetan pigs. Whole genome nuclear variants would provide more comprehensive information for studying the origin of Tibetan pigs than nuclear chip study and a complementary perspective to mtDNA evidence. In this study, we conducted whole genome analysis of 229 pigs, including Tibetan as well as other pig populations across Eurasia. First, we focused on the origin of Tibetan pigs by conducting phylogenomic and population structure analysis. Then, we compared the genomes of Tibetan pigs with those of low-land pigs that showed the closest relationship with Tibetan pigs in the phylogenomic analysis. Finally, we screened the signatures in Tibetan pig genomes that experienced selection since their arrival in Tibet. This study will provide useful information in resolving the origin and mechanism underlying high-altitude adaptation in Tibetan pigs and give signals on the importance of clear origin history before conducting evolutionary adaptation analysis of special population or species in the future study.
Results
Whole genome resequencing and identification of sequence variants
In this study, we sampled 48 domestic pigs and wild boars (Tibetan pig: 11, lowland pigs: 28 samples from 11 breeds, wild boar: 9) across China for whole genome resequencing (Supplementary Table s1). A total of 601 Gb of raw paired-end reads were generated. In order to produce more comprehensive and more reliable results, 181 genomes of Eurasian wild boars and domestic pigs and four other outgroup species from SRA database (http://www.ncbi.nlm.nih.gov/Traces/sra/) were also incorporated in our analysis (Supplementary Table s1).
The combined dataset contained a total of 3.2Tb of sequences that were mapped to the pig reference genome (Sus scrofa 10.2) after trimming low quality regions with QcReads16. The average sequencing depths for the different breeds ranged from 2.35× to 22.29× (Supplementary Tables s1 and s2). Over 49.6 million SNPs were identified among the 229 Eurasian pig samples.
Genetic structure analysis
First, we studied the genetic structure of Tibetan pigs and their relationship with other Chinese wild and domestic pigs. The relationship between the pig populations might have been affected by recent gene flows. It is common to observe the introduction of European commercial pig breeds into China in order to improve the performance of local pigs17. To exclude the effect of recently intercontinental gene flows, we examined the population structure18 of all the samples to estimate the influence of European commercial pigs on Chinese pigs. Unexpectedly, we observed that over 30% of the 183 Asian individuals had European genetic components, with a proportion ranging from 10% to 99% (Supplementary Fig. s1 and Table s3). To reduce the effects from undisclosed gene flows, only 98 Asian pig samples (26 Tibetan pigs, 20 Chinese wild boars, 52 Chinese domestic pigs from 13 breeds) (Fig. 1A) that showed European genetic component fraction less than 5% were included in the subsequent analysis (Supplementary Table s3).

Geographic distribution and population genetic analysis of the Tibetan pigs and other Chinese pigs. (A) Sites origin for the different Chinese breeds and wild boars. Sampling sites of the wild boar only include newly sequenced samples. (B) Whole-genome Neighbor-Joining tree of pigs in this study. The branch length of OG artificially shortened and is shown as a dashed line. (C) Principal component analysis. PC1 is the first principal component. PC2 is the second principal component. (D) Population structure analysis with K from 2 to 5. Abbreviations (B,D) defined as follow. OG: outgroup (Sumatra wild boar).
Three genetic methods were employed to aid in interpreting the evolutionary history of Tibetan pigs. Wild boars from Sumatra in Indonesia, proposed site for wild boar origin19, were used as outgroup, as they had more genetic similarity with pigs as compared to Sus cebifrons, Sus celebensis, Sus verrucosus and Sus barbatus (Supplementary Fig. s1 and Table s3). The rooted Neighbor-Joining (NJ) phylogenetic tree of the pig genomes across China was constructed20 and the wild boars were located near the root of the tree (Fig. 1B). All domestic pigs diverged from the Chinese wild boars and outgroup (wild boars from Sumatra), and formed two different clades separated by Nanling Mountains (Fig. 1A), namely northern group and southern group (Fig. 1B). The principal component analysis (PCA) demonstrated that the domestic pigs from southern group separated from the wild boars and other domestic pigs from northern group in PC1 (8.1%), while Chinese wild boars and domestic pigs from northern group separated in PC2 (4.7%) (Fig. 1C). This result suggests that the genetic structure of the southern group differs greatly from the wild boar and domestic pigs from northern group in China. Tibetan pigs were interspersed among domestic pigs from northern group. We also employed STRUCTURE18 to analyze the population structure among the samples with different values for K (from 2 to 5). The southern group genetic component was the first to separate from wild boar and other domestic pigs from the northern group (K = 3, Fig. 1D). The Tibetan pigs shared similar genetic components with other Chinese domestic pigs from northern group and differed from the Chinese wild boar (CWB) (K = 4 to 5, Fig. 1D). The STRUCTURE results showed a similar genetic pattern with PCA analysis.
Analysis of selective signatures in the Tibetan pig genomes
The above analysis indicated a close relationship between Tibetan pigs and low-land domestic pigs from northern group in China. To investigate on the genetic adaptation to high altitude in Tibetan pigs, we only compared the genomes of Tibetan pigs with those of low-land domestic pigs from the northern China. Furthermore, Chinese pigs with more than 5% European genetic component were also removed to avoid the influence from recent gene flows between Eurasia populations. Finally, our dataset included 26 Tibetan pigs (average altitude >3,000 m) and 29 low-land pigs from northern group (average altitude of no more than 800 m) (Supplementary Table s1) that were used as the control population.
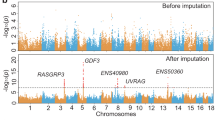
Genomic regions that have experienced selection show specific signatures, such as diverging allele frequencies between populations21 and extended haplotype homozygosity22. We scanned for selective sweeps using two comparative genomic methods, FST (fixation index) and XP-EHH23, in 10 kb siding windows. A total of 33,432,165 autosomal SNPs were identified within the genomic sequences of Tibetan and control populations. In differentiation analysis, high FST values with elevated derived allele frequency was used to detect genomic regions in the Tibetan pigs highly differentiated from other low-land pigs. Candidate sweeps were identified as a clustering of at least three consecutive (except for undetermined genomic gaps) 10-kb sliding windows with genome-wide top 1% FST or XP-EHH values. A total of 4.68 Mb (the longest sweep: 270 kb, average length: 45 kb) and 14.07 Mb (the longest sweep: 220 kb, average length: 54 kb) of genomic sequence were defined as selective sweeps in genome of Tibetan pig by FST and XP-EHH analyses, respectively (Fig. 2, Supplementary Tables s4 and s5). Within the sequences identified by FST and XP-EHH, 70 and 211 potentially positively selected genes (PSGs) were identified, respectively, with eight genes identified by both approaches (Supplementary Table s6). Majority of our 273 candidate PSGs were found for the first time, only these four are in common (SERGEF, RAPGEF2, LEF1, HIF1A) with the 215 candidate PSGs reported by Li et al.12, only six genes (THSD7A, SEC. 63, OSBPL1A, MFSD2A, FAM149A, DPPA4) were in common when compared with the 489 genes identified by Ai et al.’s15.

Selection signals on the autosomes of the Tibetan pigs. (A) Genome-wide FST values in the Tibetan pigs. The black dotted line represents top 99% threshold of FST at whole-genome level. (B) XP-EHH values of the autosomes in the Tibetan pigs. Each dot represents the average value of a10-kb sliding window. The red dotted line represents top 99% threshold of XP-EHH at whole-genome level.
Literature mining of the 273 PSGs biological function indicated that many PSGs participated in physiological processes, such as response to hypoxia, cardiovascular system, lung and gas exchange, mitochondria or respiratory chain, DNA damage repair, spermatogenesis, embryo development, tumor/cancer, neural development, immunity and apoptosis (Supplementary Table s7).
Six PSGs (EPAS1, HIF1A, RNF4, TNFSF10, PDE1A, PDE3) were related to “response to hypoxia”. EPAS1, encoding hypoxia-inducible factor 2-alpha subunit, is well known for its role in adaptation to hypoxia in humans and animals native to high-altitude levels24,25,26. EPAS1 is the only gene located within a 70 kb sweep region on chromosome 3 in Tibetan pigs (chr3: 100, 170, 001–100, 240, 000) (Fig. 2A). To analyze the haplotypes, the 40 most differentiated SNPs (FST > = 0.4, red box in Fig. 3A) across the entire gene region of EPAS1 (39.7 kb) were phased in all of the Eurasian samples. Intriguingly, we found that the Tibetan pigs contained a medium-frequency haplotype (haplotype XXV), that was extremely differentiated from the other Eurasian haplotypes (Fig. 3B,C), this differentiated pattern was similar to previous observation of EPAS1 between Tibetans and Han Chinese27.

Selective signals and haplotypes of EPAS1. (A) FST values of each SNP between the Tibetan and control populations. The x axis is the physical position on chromosome 3 (Sus scrofa 10.2 build). The region between the two green dashed lines is the candidate sweep. The red box defines the SNPs with the largest genetic differentiation, used to analyze haplotypes. (B) Haplotype pattern of highly differentiated SNPs between the Tibetan and control pig populations using 227 pigs from East Asia and Europe. Each column is a polymorphic genomic location and each row is a phased haplotype. The blue cell represents the ancestor allele and the red cell represents the derived allele. OG: outgroup, TP: Tibetan pigs, CDP: Chinese domestic pig, CWB: Chinese wild boar, EDP: European domestic pig, EWB: European wild boar. (C) The Median-Joining network of haplotypes within EPAS1. Only haplotypes with frequency more than 1 were used to draw the network.
Genes involved in circulatory and respiratory systems were also detected under positive selection in Tibetan pigs. 34 candidate PSGs (Supplementary Table s7) were detected in association with the cardiovascular system and five genes (CYSLTR2, PHF14, RNF150, TIMELESS, SCAP) in association with lung and gas exchange. BCR (Breakpoint Cluster Region), which was found within a 180 kb sweep region (Chr14: 52, 720, 001–52, 900, 000) (Fig. 2B) formed the fusion protein BCR-ABL with ABL that affects hypoxia-induced pulmonary hypertension28 and the expression of vascular endothelial growth factor29. THSD7A, across two adjacent discontinuous sweeps (Fig. 2A and Supplementary Fig. s2A), is a conserved gene in vertebrates that is known to be involved in endothelial cell migration and embryonic angiogenesis30,31. A missense mutation (Lys561Arg) in THSD7A was observed with elevated derived allele frequency (DAF) in the Tibetan pigs (Tibetan pigs: DAF = 0.79, low-land pigs: DAF = 0.25), at a site that is highly conserved among vertebrates (Supplementary Fig. s3). This missense mutation site and 21 additional most differentiated SNPs between the Tibetan and control populations (FST > = 0.4, red box in Supplementary Fig. 2A) were used to perform haplotype analysis. We discovered that Haplotype III (containing the Lys561Arg mutation) was observed only in domestic pigs and showed an increased frequency (frequency = 0.4) in the Tibetan pigs (Supplementary Fig. s2B,C). The variants of THSD7A might have assisted Tibetan pigs to overcome the effects of hypoxia during pregnancy32.
Many of the genes related to spermatogenesis (CLGN, RFX4, MORC1, TXNDC8, GGN, CATSPERG, DPPA4, DHX36, PPP1R2, GALNT3, NRG3, DKK3) were also identified in sweep regions of Tibetan pig genomes, that might have aided counteracting deleteriously hypoxic effects in reproduction process33,34. CLGN (Calmegin) and RFX4 (Regulatory factor X, 4) are involved in the spermatogenesis process35,36, and both were the only genes found in a long sweep regions (CLGN: Chr8: 92,110,001–92,230,000, 120 kb; RFX4: Chr5: 13,450,001–13,540,000, 90 kb) with very strong FST and XP-EHH signals (Fig. 2A,B, Supplementary Table s4 and s5).
The hypoxia-inducible factor (HIF), including HIF1 and HIF2, signaling pathway plays an essential role during the response to hypoxia in many organisms37,38. We found that multiple genes from the HIF pathways (including 12 candidate PSGs: EPAS1, HIF1A, PRMT1, PIK3R2, FRAP1, RNF7, RNF4, EIF4E, TXN, SPSB2, CUL5, and NOX4) were under selection in the Tibetan pigs (Fig. 2A,B, Supplementary Fig. s4). HIFs are heterodimers transcriptional factors, composed of a distinct oxygen-sensitive α subunit and a constitutive β subunit39. While HIF-α proteins (encoded by HIF1A or HIF2A) are constitutively synthesized and degraded under normoxic conditions, hypoxia inhibits α protein degradation by various genes regulation40,41,42. The 12 candidate PSGs (observed in our findings) mainly regulated the stability of HIF-α under different steps (gene names in red in Supplementary Fig. s4). Finally, HIFs activates the transcription of a series of genes, to increase oxygen delivery and reduce oxygen consumption, by recognizing and binding to hypoxic response elements (HREs) in the promoters of these genes43,44.
Enrichment distribution of variants with different divergence levels
In our observation, no genetic differentiated (FST > = 0.05) missense variant was identified from 91 of the total 104 selective sweeps (FST analysis) in the Tibetan pig genomes, indicating that coding variants might have not accounted for majority of selective sweeps. Previous reports have implied an role of regulatory elements in pig domestication45,46,47. This raises an interesting question on the role of noncoding variants from the selective sweeps in Tibetan pig genomes. To analyze the role of coding and noncoding variants during the evolution of Tibetan pig genomes, we performed enrichment analysis of variants with different divergence levels between Tibetan and lowland pigs at the whole genome and within-sweep levels.
We first analyzed the enrichment pattern of 13,461,622 autosomal SNPs in Tibetan pig genomes. SNPs were classified into coding and different noncoding categories for enrichment analysis and linear regression analysis was used to measure the relationship between enrichment ratio and FST value (see Materials and Methods section for details). We found that enrichment ratios for SNPs from all functional regions decreased along with increasing differentiation levels (Fig. 4A), indicative of evolution under purifying selection. SNPs from “UTR”, “Conserved”, “Histone”, “FAIRE”, “DHS” and “TFBS” showed statistically significant negative correlation with FST order (Supplementary Table s8). However, the enrichment ratios for intergenic and intronic SNPs were not related to FST value and remained at a near constant level that centered at 1 (Fig. 4A and Supplementary Table s8), indicating a process of neutral evolution. The pattern of enrichment ratio decrease with increasing FST order for variants from functional regions implied that functional variants at the whole genome level have evolved under strong functional constraints and have experienced purifying selections during the evolution of Tibetan pigs.

Enrichment of variants under different divergence levels and transcription activity assay of alleles in predicted motifs near EPAS1. (A) Enrichment pattern of SNPs at whole genome level. SNPs were divided to different bins by FST between Tibetan and lowland pigs. (B) Enrichment pattern of SNPs from selected sweep regions in Tibetan pigs. (C) Selective signals of EPAS1. The black vertical bar in downstream of EPAS1 indicated differentiated SNP positions between Tibetan and lowland pigs. The red dotted line represents top 99% threshold of FST at whole-genome level. (D) Predicted motifs with differentiated SNPs in downstream of EPAS1. (E) Transcription activity assay of different alleles within predicted motifs near EPAS1 in pig lung fibroblast cell. AA names of different pGLS3 vectors means ancestral allele and DA means derived allele. (F) Transcription activity assay of different alleles within predicted motifs near EPAS1 in BEAS-2B cell. The two-tailed t test was used for statistical assessment of transcription activity change.
To compare the enrichment pattern of variants from selective sweeps and the whole genome, we conducted the enrichment ratio analysis on SNPs from selective sweeps in Tibetan pig genomes. In total, we obtained 16,140 SNPs from 4.68 Mb sweep regions in FST outlier windows. Interestingly, the related pattern between enrichment ratio and FST value for SNPs in selective sweeps is contrary to the pattern observed in whole genome level (Fig. 4B). We found that the enrichment ratios showed positive correlation with FST order for all SNPs but SNPs from Intron. Here, only enrichment ratio for SNPs from “Motif” show statistically significant positive correlation with FST order and a high coefficient (P = 0.03; coefficient = 2.44) (Supplementary Table s8). Furthermore, the enrichment ratio of “Coding” and “UTR” increase dramatically when FST increase from 0.4 to > = 0.5, implying an important role of SNPs from “Coding” and “UTR” among the highly differentiated SNPs in Tibetan pig. The enrichment of highly differentiated nocoding-regulatory SNPs in selective sweeps indicated that the evolution of Tibetan pigs was related to a selection against regulatory SNPs, especially for SNPs from transcription factor (TF) recognizing motifs (“Motif”).
Functional analysis of regulatory variants in selective sweeps of Tibetan pigs
The enrichment of highly differentiated mutations in transcription factor (TF) recognizing motifs from sweep regions may imply their important role during high-altitude adaptation in Tibetan pigs. TF recognizing motifs are important for transcriptional regulation48. Variants in these TF recognizing motifs from selective sweep regions in Tibetan pigs might have affected the binding affinity of transcription factors and altered gene expressional regulation. In total, 78 mutations from 97 TF recognizing motifs were detected in 4.68 Mb FST selected sweep regions in Tibetan pigs, and about 44 TF recognizing motifs contain SNPs showing high level of differentiation (FST > 0.15) between high-altitude Tibetan pigs and lowland pig populations (Supplementary Table s9). To examine the role of highly differentiated mutations in the TF recognizing motifs, we presented two cases by analyzing their genotypes and compared transcriptional activity difference between ancestral and derived alleles in the gene expressional regulation.
At first, we focused on the EPAS1 locus since previous researches had revealed that selection on this gene is associated with high altitude adaptation in a variety of animal species49,50 and has also experienced strong selection in Tibetan pigs from our observation (Fig. 4C). In our analysis, no differentiated (FST > 0.05) missense mutation was observed in the EPAS1 locus in Tibetan pigs. By aligning to the human orthologous sequence, we detected a clustering of three predicted TF recognizing motifs with five highly differentiated SNPs (FST > = 0.27) in 465 bp noncoding DNA fragment (chr3: 100, 231, 640–100, 232, 104) at 18.3 kb downstream of the EPAS1 locus (Fig. 4D). The TF recognizing motif cluster was located in the EPAS1 selective sweep region. The three TF recognizing motifs were predicted as the binding sites for transcription factors hepatocyte nuclear factor 4 (HNF4), V-maf musculoaponeurotic fibrosarcoma oncogene homolog (v-Maf), PR domain containing 1 (PRDM1), respectively (Fig. 4D). HNF4 is a hypoxia-responding transcription factor interacting with HIF-1 (hypoxia-inducible factor 1) to regulate the expression of erythropoietin (Epo) to compensate for reduced oxygen supply to organs by modulating erythropoiesis under hypoxia51. PRDM1 is a plasma cell-specific transcription factor and its expression decreased under hypoxia condition52. To reveal the linkage disequilibrium in EPAS1 sweep, we combined the five differentiated SNPs from TF recognizing motifs and 40 most differentiated SNPs from EPAS1 to analyze the haplotype structure in all Eurasia samples. We discovered that the haplotype XXV of EPAS1 was linked to the derived alleles of all the five SNPs and this haplotype was only presented in Tibetan pigs (Supplementary Fig. s5). We further analyzed the derived allele frequency of these SNPs in all the 227 Eurasia pigs. We found that the derived alleles of the three SNPs among recognizing motifs of HNF4 and PRDM1 were observed only in Asian pig population and showed increased frequency in Tibetan pig (Fig. 4D), suggesting an important regulatory role for high-altitude adaptation of Tibetan pig.
To investigate regulatory effects on HNF4 and PRDM1 recognizing motifs in downstream of EPAS1, we clone ancestral and derived HNF4 and PRDM1 motifs (Supplementary Table s9) into luciferase reporter plasmids to test their transcriptional regulatory activity under different level of oxygen concentration (21% and 2%). After transfection into the pig lung fibroblast cell line (21% oxygen), both ancestral-type vectors (HNF4-AA and PRDM1-AA) show statistically significant increase in luciferase expression when compared with the empty vector (pGL3-promoter) implying enhancer activity of the two predicted TF recognizing motifs (Fig. 4E). However, a reduction of transcriptional activity was observed for both derived-type HNF4-DA and PRDM1-DA motifs when compared to their ancestral-types (Fig. 4E). We also investigated the luciferase activity of differentiated SNPs in HNF4-DA and PRDM1-DA reporter vectors after hypoxia incubation (2% oxygen) in 48 h. We found that the two ancestral reporter vectors showed increased luciferase expression (Fig. 4E). Interestingly, both derived-type HNF4-DA and PRDM1-DA motifs also showed decreased luciferase expression as compared to corresponding ancestral-types in hypoxia experimental replicates. Similar results were also observed in human bronchial epithelial cell line (BEAS-2B) (Fig. 4F). In a recent report, a down-regulated expression of EPAS1 was detected in association with high altitude adaptation in Tibetans, implying that the adaption could be due to selection for a change in EPAS1 expressional level47. Our experimental data indicated that the mutations in the EPAS1 downstream noncoding regulatory sequence could have affected gene expression and have been putatively involved in the high altitude adaptation in Tibetan pigs.
We also detected another cluster of three TF recognizing motif cluster with three highly differentiated SNPs (FST > = 0.4) within a 408 bp fragment (chr2: 61,358,862 –61,359,269) in the immediate upstream (123 bp) of CYP4F2 transcription start site (Supplementary Fig. s6A,B and Table s9). This gene encodes an omega-hydroxylase and synthesizes 20-hydroxyeicosatetraenoic acid (20-HETE) which plays an important role in blood pressure control53,54. Three TF recognizing motifs were predicted as the binding sites for transcription factors forkhead box protein A (FOXA), specificity protein 1 (SP1), nuclear factor erythroid 2 (NFE2), respectively. NFE2 is essential for regulating erythroid and megakaryocytic maturation and differentiation54,55, and mutation of NFE2 was associated with polycythemia56. The functional convergence of NFE2 and CYP4F2, and the observation of NFE2 binding site in the CYP4F2 promoter region implied an expressional regulation of CYP4F2 by NFE2. The derived alleles of three SNPs were also observed in Chinese wild boars with high frequency, implying their origin from wild boar (Supplementary Fig. s6B).
To test the mutation effect on the NFE2 recognizing motif in CYP4F2 promoter, we conducted an experimental assay to test the transcriptional activity of NFE2 recoginizing motifs (Supplementary Table s9). We compared the luciferase activity of ancestral and derived NFE2 motifs under normoxia (21%) and hypoxia (2%) conditions. pGL3-Basic vector with ancestral/derived NFE2 motif fragments inserted in the promoter region were transfected into pig lung fibroblast and BEAS-2B cells. Anestral-type (NFE2-AA) showed statistically significant increase (P < 0.01) in luciferase expression when compared to the empty vector (pGL3-basic) in all cell lines and oxygen condition (Supplementary Fig. s6C,D), implying that the NFE2 motif is a promoter sequence for the CYP4F2 gene. Furthermore, we found that the activity of derived-type vectors (NFE2-) decreased compared to ancestral-type in all conditions (Supplementary Fig. s6C,D). This result implied that the mutation in the NFE2 motif may have altered the promoter activity of CYP4F2 resulting in different CYP4F2 expression, which might have played a role in high-altitude adaptation in Tibetan pigs.
Discussion
Clarifying history of the origin of Tibetan pigs is critical in guiding investigation of the genetic mechanism underlying their high-altitude adaptation and resolving previous controversy about their origin. In this study, we analyzed the genomic variants in 229 pig genomes across Eurasia through phylogenomic and population structure approach. We discovered that Tibetan pigs had a similar genetic structure with domestic pigs from northern group. Genome comparison between Tibetan pigs and low-land domestic pigs from northern group, unveiled signatures associated with high-altitude adaptation in Tibetan pig genomes. Furthermore, we also revealed the important role of noncoding regulatory SNPs in high-altitude adaptation of Tibetan pigs.
Our results suggests Tibetan pigs possibly share a common ancestor with other domestic pigs from low-land regions in north of Nanling Mountains. Phylogenomic and population structure analyses of whole genome variants revealed that Tibetan pigs showed close phylogenetic relationship and similar genetic background with other domestic pigs from low-land areas in north of Nanling Mountains, all of which diverged from wild boars from different regions of China and domestic pigs in south of Nanling Mountains. The genetic structure of Tibetan pigs observed from nuclear genomic analyses in our study is in accordance with the mtDNA pattern previously observed in Wu et al.’s report13. The Tibetan pigs phylogenetically clustered with domestic pigs from northern group, rather than with East Asian wild boars, indicating the less possibility of Tibetan pigs beings wild as previously reported by Li et al.12, and the less likelihood to be domesticated from Tibetan wild boars, based on evidences from partial mtDNA sequences, as an event paralleling to the domestication in low-land regions14. We inferred that the inconsistency between the whole genome and mtDNA analysis may possibly be due to partial maternal introgression of wild boars into Tibetan pigs that were included in Yang et al.’s. study, since the partial mtDNA sequence could provide only limited information. Our analysis confirmed close relationship between Tibetan and neighboring low-land pig populations from the chip data analysis15.
Tibetan pigs may have moved to Qinghai-Tibetan Plateau from middle Yellow River basin. Recent study based on ancient mtDNA found pig remains from middle Yellow River region (including the earliest archaeological sits of pigs, about 10,500–7,575 before present) contained the mtDNA haplotypes (haplotypes H2, H3, H4 and H10) that are dominant in both younger archaeological and modern populations and was thought as one of the centers for early Chinese pig domestication57. By further analyzing this report, we found that mtDNA haplotypes H2, H3, H4 and H10 also accounted for 296 of the 348 sequences in modern Tibetan pigs. Furthermore, domesticating or breeding pigs only might have happened after the shift in life style from hunter-gatherer to sustained settlement and development of agriculture. Pigs are not so skilled at migrating but usually captive fed, and relies upon humans for food. The earliest archaeology records of dometic plants and pigs in middle Yellow River region (Nanzhuangtou sit, about 10,500–9,700 years ago)58 is mucher earlier than that in Tibet (Karuo site, about 4,300–4,700 years ago)59. Tibetan began to plant millets and settle down about 5,200 years ago60. Thus, combining evidences from both our genetic analysis and previous archaeological reports, we hypothesized that Tibetan pigs were transported to the Qinghai-Tibetan Plateau from middle Yellow River region less than 5,200 years ago.
The PSGs identified (n = 273) in this analysis showed only a few overlaps with previous researches by Li et al.12 (Li et al., 2013) and Ai et al.15. We discovered that many missense mutations in the positively selected genes reported by Li et al. (2013) showed similar allele frequency in Tibetan pigs and low-land pigs in China (data not shown), indicating that these missense mutations were not specifically related to the evolution of Tibetan pigs. Furthermore, our results also have little common PSGs with Ai’s candidate gene list due to low SNP density and design bias of illumina porcine SNP60 chip. We found that illumina porcine SNP60 chip only covered 75 of 93,789 SNPs located in selected sweep regions of Tibetan pigs in this study, as the illumina porcine SNP60 chip was designed by SNPs from European pig populations61.
Our study revealed the genetic mechanism underlying the high-altitude adaptation in Tibetan pigs. PSGs involved in different physiological functions might have collaborated in aiding Tibetan pigs to overcome different physiological pressure caused by high-altitude environment, such as hypoxia, UV damage and impaired reproduction62. 12 PSGs related to HIF pathways were identified under selection in Tibetan pigs, indicative of the important role of HIF pathways in hypoxia adaptation process of Tibetan pigs. Here, the most common gene, EPAS1, was also detected in a sweep region with strong selection signal in Tibetan pigs. We found a specific medium-frequency haplotype XXV of EPAS1 in Tibetan pigs. Further linkage disequilibrium analysis shows that haplotype XXV was linked to five derived-alleles in downstream of EPAS1, three of which has been proven to have repressed regulation activity. Recently, Peng et al. discovered an EPAS1 adaptive haplotype in Tibetans down-regulates EPAS1 transcription, which contributed to the genetic adaptation of Tibetans to high-altitude hypoxia50. Our results implied a similar adaptive molecular mechanism in EPAS1 between Tibetans and Tibetan pigs. Furthermore, we also discovered an elevated derived-missense mutation (Lys561Arg) on a very conserved site of THSD7A in Tibetan pigs. THSD7A is involved in endothelial cell migration and embryonic angiogenesis30,31 and this missense mutation might have assisted Tibetan pigs to overcome impaired reproduction caused by high-altitude hypoxia.
Our analysis revealed an important function of highly differentiated regulatory SNPs between high-altitude Tibetan pigs and lowland pig populations during the evolution of Tibetan pigs. From whole genomic level view, highly differentiated regulatory SNPs have evolved under strong evolutionary constraints (purifying selection), possibly due to having important biological regulatory function. Comparatively, the enrichment of highly differentiated regulatory SNPs in selective sweeps of Tibetan pigs indicated that the selective sweeps were possibly due to positive selection against these regulatory SNPs. Since much larger number of regulatory SNPs than protein coding SNPs were observed in the selective sweeps, the regulatory SNPs might have played more role during the high altitude adaptation in Tibetan pigs as compared to the protein coding SNPs. From our observation, the sweeps in EPAS1 and CYP4F2 could be associated with regulatory SNPs at downstream of EPAS1 and the promoter sequence of CYP4F2. Inferring from our observation, the regulatory variants might be associated with an important role during high-altitude adaptation in Tibetan pigs and more attention should be paid on it in future while studying evolution in other animals.
Material and Methods
All methods were performed in accordance with the guidelines approved by the Kunming Institute of Zoology, Chinese Academy of Sciences. All experimental protocols were approved by the Kunming Institute of Zoology, Chinese Academy of Sciences.
Sample collection and data download
Ear or muscle tissues from 48 samples (including wild boars and domestic pigs) from China (Supplementary Table s1) were collected and kept in 95% alcohol. Among this, 11 Tibetan pigs were sampled from the Qinghai-Tibet Plateau (average altitude >3,000 m, see Fig. 1A), and nine wild boars were collected from geographically isolated locations in China. The remaining 28 domestic pigs (average of altitude <600 m) comprises of 11 different pig breeds from China (Supplementary Table s1). We then downloaded genomes of 182 Eurasian pigs (including 138 Chinese samples and 44 European pigs) and six samples as outgroup (Supplementary Table s1) from the Sequence Read Archive (SRA) database (http://www.ncbi.nlm.nih.gov/Traces/sra/).
Whole genome Re-sequencing, read mapping and SNP calling
The 48 pigs were used for the whole genome resequencing. Genomic DNA was extracted using a routing phenol-chloroform method and precipitated by 75% alcohol. The resequencing libraries were constructed with 500-bp inserts according to the Illumina library construction protocols. 100-bp paired-end reads were generated with the HiSeq2000 platform (Illumina).
To obtain reliable alignment results, low-quality sequences (phred quality score <20) from all data sets were trimmed by QcReads (http://sourceforge.net/projects/qcreads/). The controlled genomic reads were then mapped to the Duroc reference genome (NCBI build Sscrofa 10.2) with the BWA program63 (http://sourceforge.net/projects/bio-bwa/). Before SNPs calling, SAMtools64 was used to sorting, merging and removing PCR duplicates generated during genomic library construction. To ensure reliability of the downstream analyses, we selected only sequences with mapping quality greater than 20 for SNP calling. To call high quality SNPs, a high consensus quality (≥20) or a high SNP quality (≥20) required if homozygous at a genomic site; and an intermediate criterion (consensus quality≥10 and SNP quality≥10) required if heterozygous. SNPs of each individual were called using SAMtools. The sequence reads are available at GSA (Genome Sequence Archive) under accession CRA001606.
Population genetic analysis
To exclude the effect of recently intercontinental gene flows between Eurasia pigs, we used STRUCTURE (2.3.4) to analyze the population structure of all the 233 samples (K set as 3) with 3 iterations. Finally, only 98 Chinese pig samples (26 Tibetan pigs, 20 Chinese wild boars, 52 Chinese domestic pigs from 13 breeds) showing European genetic component fraction less than 5% were included in the subsequent analysis (Supplementary Table s3).
Only SNPs with good sample coverage (above 89% individual) from autosomes were used for the analyses of the genetic structure of the 98 Chinese pigs. Furthermore, the physical distance between SNPs was greater than 100 kb to avoid any bias due to linkage disequilibrium (LD)65. Finally, 22,664 SNPs passed quality control test and were used for the population genetic analysis. Two wild boars from Sumatra, Indonesia, were used as the out-group. For constructing the Neighbor-Joining (NJ) phylogenetic tree, the miss ratio of the SNPs of each individual is less than 10% (80 out of the 98 Chinese pig qualified for this condition). MEGA620 was used to construct the NJ tree. STRUCTURE 2.3.418 was used to infer the population structure within the different Chinese pig populations with different K values (from 2 to 5). Principal component analysis (PCA) was accomplished with R language (http://www.r-project.org/).
Selective sweep analysis
The fixation index (FST)66 was used to measure the population differentiation between 26 Tibetan pigs and 29 Chinese low-altitude control pigs. Previous reports indicate that the pig ancestor emerged from South East Asia 5.3-3.5 Myr ago61. In addition, the genetic background of the wild boar from Sumatra is more similar to the Eurasian pigs than to Sus cebifrons, Sus celebensis, Sus verrucosus and Sus barbatus (Supplementary Fig. s1 and Table s3). Therefore, the wild boar from Sumatra in Indonesia is more suitable for defining the derived alleles within domesticated pigs. Only sites that were homozygous in two Sumatra wild boars were defined as the ancestral allele. First, 33,432,165 SNPs were called in the autosomes of 26 Tibetan pigs and 29 control pigs. Next, 28,283,678 SNPs were found to be homozygous in the two Sumatra wild boars. The allele frequencies within each population were calculated with a correction for sample size and sequencing depth at each SNP site (details described in supplementary note) and then used for the FST analysis. The range of FST is between 0 and 1. However, it is possible to get negative values, which have no biological interpretation; therefore, negative values were set to 0. Phased SNPs are needed for the XP-EHH analysis between 26 Tibetan pigs and 29 control pigs. 7,408,187 SNPs with good sample coverage (Tibetan pigs > = 24, control pigs > = 25) in the autosomes were phased by the program fastPHASE67 (http://stephenslab.uchicago.edu/software.html). Cross Population Extended Haplotype Homozogysity (XP-EHH) values for each SNP were calculated by the XP-EHH program (http://hgdp.uchicago.edu/Software/). Sliding windows were used for both the FST and the XP-EHH analyses to avoid the influence of genetic drift. As the extent of LD is no more than 10 kb in Chinese domestic pigs68, the window size was set as 10 kb. Average values were calculated for all SNPs within each sliding window. Regions with a clustering of at least three consecutive (except undetermined genomic gaps) sliding windows above the genome-wide top 1% of FST or XP-EHH values were defined as selected sweeps.
Enrichment analysis of differentiated variants in Tibetan pigs
The SNPs were classified into different categories, coding, UTR, intronic, and intergenic, to compare their differentiation patterns. We also classified the SNPs into different regulatory categories by annotation of homologous sequence to human in our other unpublished work, such as transcription factor binding sites (“TFBS”), transcription factor recognizing motifs (“Motif”), DNase I hypersensitive sites (“DHS”), Formaldehyde-Assisted Isolation of Regulatory Elements (“FAIRE”), and histone chemical modification sites (“Histone”). Furthermore, conserved sequence (phastCons score > = 0.2) among 100 vertebrate genomes from UCSC database (http://genome.ucsc.edu) were also included in our analysis. The SNPs were divided into different subsets by FST values. An enrichment ratio was developed to measure the distribution of different categories of SNPs in FST bins that were ordered by increasing levels of genetic differentiation. For a specific group of SNPs in a FST bin, the enrichment ratio was denoted as the ratio of the number of observed SNPs (Pobserved) to the number of SNPs expected under random (Pexpected). A Pobserved/Pexpected ratio greater than 1 indicates an enrichment of SNPs in a FST bin, while a ratio less than 1 indicates a deficiency of SNPs in the FST bin.
Cell culture
Pig lung fibroblast cell line was purchased from the Kunming Cell Bank, Kunming Institute of Zoology, Chinese Academy of Sciences (Kunming, China). Human bronchial epithelial cell line (BEAS-2B) was obtained from the Yunnan University (Kunming, China). Cells were cultured in DMEM (Gibco, New York, USA) supplemented with 10% fetal bovine serum (Gibco, New York, USA), 100 U/mL penicillin, 100 μg/mL streptomycin (Beyotime Biotechnology, Hangzhou, China), and incubated at 37°C and 5% CO2.
Plasmids construction and luciferase assays
DNA fragments (50 bp: chr2: 61,359,235-61,359,284; 49 bp: chr2: 61,368,465-61,368,513; 49 bp: chr3: 58,229,710-58,229,758; 50 bp: chr3: 100,231,621-100,231,670; 50 bp: chr3: 100,232,077-100,232,126) containing ancestral-type or derived-type Motif were synthesized and cloned into pGL3-basic or pGL3-promoter vector (Promega) in BamHI and SalI digestion sites. Each construct was confirmed by sequencing.
For luciferase assays, cells were seeded onto 24-well plates corresponding to ∼80% confluency. Cells were transfected with various reporter construct along with pRL-TK Renilla luciferase plasmid (Promega) by using Lipofectamine 3000 Reagent (Invitrogen) according to the manufacturer’s instructions. For hypoxic assays, cells were incubated in an atmosphere of 2% O2, 93% N2 and 5% CO2 after transfection. After forty-eight hours, cells were collected for luciferase activity assays using the Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer’s instructions. Luminescence signals were captured in a Varioskan Flash Multimode Reader (Thermo Scientific). Firefly signals were normalized with the Renilla luciferase internal control. Six independent transfection and assays were performed. P-values between ancestral-type and derived-type Motif were calculated using the two-tailed t-test.
References
Lorenzo, F. R. et al. A genetic mechanism for Tibetan high-altitude adaptation. Nat. Genet. 46, 951–956 (2014).
Peng, Y. et al. Genetic variations in Tibetan populations and high-altitude adaptation at the Himalayas. Mol. Biol. Evol. 28, 1075–1081 (2011).
Xu, S. et al. A genome-wide search for signals of high-altitude adaptation in Tibetans. Mol. Biol. Evol. 28, 1003–1011 (2011).
Yi, X. et al. Sequencing of 50 human exomes reveals adaptation to high altitude. Science 329, 75–78 (2010).
Li, Y. et al. Population variation revealed high-altitude adaptation of Tibetan mastiffs. Mol. Biol. Evol. 31, 1200–1205 (2014).
Qiu, Q. et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 44, 946–949 (2012).
Wang, M. S. et al. Genomic analyses reveal potential independent adaptation to high altitude in Tibetan chickens. Mol. Biol. Evol. 32, 1880–1889 (2015).
Cai, Q. et al. The genome sequence of the ground tit Pseudopodoces humilis provides insights into its adaptation to high altitude. Genome Biol. 14, R29 (2013).
Ge, R. L. et al. Draft genome sequence of the Tibetan antelope. Nat. Commun. 4, 1858 (2013).
(Later Jin) Liu Xu, Old Book of Tang, (National Library of China Publishing House, Beijing, 2014).
Baima, Y. Z. et al. Preliminary study on pulmonary tissue and hypoxia adaptation to plateau for Tibetan pigs. Hubei Agricultural Sciences 51, 2776–27779 (2012).
Li, M. et al. Genomic analyses identify distinct patterns of selection in domesticated pigs and Tibetan wild boars. Nat. Genet. 45, 1431–1438 (2013).
Wu, G. S. et al. Population phylogenomic analysis of mitochondrial DNA in wild boars and domestic pigs revealed multiple domestication events in East Asia. Genome Biol. 8, R245 (2007).
Yang, S. et al. The local origin of the Tibetan pig and additional insights into the origin of Asian pigs. PLoS One 6, e28215 (2011).
Ai, H. et al. Population history and genomic signatures for high-altitude adaptation in Tibetan pigs. BMC Genom. 15, 834 (2014).
Ma, Y., Xie, H., Han, X., Irwin, D. M. & Zhang, Y. P. QcReads: an adapter and quality trimming tool for next-generation sequencing reads. J. Genet. Genomics 40, 639–642 (2013).
Yang, B. et al. Genome-wide SNP data unveils the globalization of domesticated pigs. Genet. Sel. Evol. 49, 71 (2017).
Hubisz, M. J., Falush, D., Stephens, M. & Pritchard, J. K. Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 9, 1322–1332 (2009).
Larson, G. et al. Worldwide phylogeography of wild boar reveals multiple centers of pig domestication. Science 307, 1618–1621 (2005).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Sabeti, P. C. et al. Positive natural selection in the human lineage. Science 312, 1614–1620 (2006).
Sabeti, P. C. et al. Detecting recent positive selection in the human genome from haplotype structure. Nature 419, 832–837 (2002).
Sabeti, P. C. et al. Genome-wide detection and characterization of positive selection in human populations. Nature 449, 913–918 (2007).
Gao, W. X., Wu, G. & Gao, Y. Q. Pathophysiological changes in mitochondria of mammalian exposed to hypoxia at high altitude. Chinese Journal of Applied Physiology 30, 502–505 (2014).
Simonson, T. S. et al. Genetic evidence for high-altitude adaptation in Tibet. Science 329, 72–75 (2010).
Wu, X. Y. et al. Novel SNP of EPAS1 gene associated with higher hemoglobin concentration revealed the hypoxia adaptation of yak (Bos grunniens). J. Integr. Agr. 14, 741–748 (2015).
Huerta-Sanchez, E. et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512, 194–197 (2014).
Yu, M. et al. Lack of bcr and abr promotes hypoxia-induced pulmonary hypertension in mice. PLoS One 7, e49756 (2012).
Mayerhofer, M., Valent, P., Sperr, W. R., Griffin, J. D. & Sillaber, C. BCR/ABL induces expression of vascular endothelial growth factor and its transcriptional activator, hypoxia inducible factor-1alpha, through a pathway involving phosphoinositide 3-kinase and the mammalian target of rapamycin. Blood 100, 3767–3775 (2002).
Kuo, M. W., Wang, C. H., Wu, H. C., Chang, S. J. & Chuang, Y. J. Soluble THSD7A is an N-glycoprotein that promotes endothelial cell migration and tube formation in angiogenesis. PLoS One 6, e29000 (2011).
Wang, C. H. et al. Zebrafish Thsd7a is a neural protein required for angiogenic patterning during development. Dev. Dyn. 240, 1412–1421 (2011).
Moore, L. G., Charles, S. M. & Julian, C. G. Humans at high altitude: Hypoxia and fetal growth. Respir. Physiol. Neurobiol. 178, 181–190 (2011).
Dunwoodie, S. L. The role of hypoxia in development of the mammalian embryo. Dev. Cell 17, 755–773 (2009).
Liao, W. G. et al. Hypobaric hypoxia causes deleterious effects on spermatogenesis in rats. Reproduction 139, 1031–1308 (2010).
Ikawa, M. et al. The putative chaperone calmegin is required for sperm fertility. Nature 387, 607–611 (1997).
Wolfe, S. A., vanWert, J. M. & Grimes, S. R. Transcription factor RFX4 binding to the testis-specific histone H1t promoter in spermatocytes may be important for regulation of H1t gene transcription during spermatogenesis. J. Cell. Biochem. 105, 61–69 (2008).
Hu, C. J., Wang, L. Y., Chodosh, L. A., Keith, B. & Simon, M. C. Differential roles of hypoxia-inducible factor 1 alpha (HIF-1 alpha) and HIF-2 alpha in hypoxic gene regulation. Mol. Cell. Biol. 23, 9361–9374 (2003).
Semenza, G. L. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci. STKE 2007, cm8 (2007).
Wang, G. L., Jiang, B. H., Rue, E. A. & Semenza, G. L. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. USA 92, 5510–5514 (1995).
Bruick, R. K. & McKnight, S. L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337–1340 (2001).
Epstein, A. C. et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107, 43–54 (2001).
Maxwell, P. H. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275 (1999).
Majmundar, A. J., Wong, W. H. J. & Simon, M. C. Hypoxia-Inducible Factors and the Response to Hypoxic Stress. Mol. Cell 40, 294–309 (2010).
Wenger, R. H., Stiehl, D. P. & Camenisch, G. Integration of oxygen signaling at the consensus HRE. Sci. STKE 2005, re12 (2005).
Lu, M. D. et al. Genetic variations associated with six-white-point coat pigmentation in Diannan small-ear pigs. Sci. Rep. 6, 27534 (2016).
Yang, Y., Adeola, A. C., Xie, H. B. & Zhang, Y. P. Genomic and transcriptomic analyses reveal selection of genes for puberty in Bama Xiang pigs. Zool. Res. 39, 424–430 (2018).
Yang, Y. et al. Artificial selection drives differential gene expression during pig domestication. J. Genet. Genomics, (In Press), (2019).
Xue, G. P. An AP2 domain transcription factor HvCBF1 activates expression of cold-responsive genes in barley through interaction with a (G/a)(C/t)CGAC motif. Biochim. Biophys. Acta 1577, 63–72 (2002).
Gou, X. et al. Whole-genome sequencing of six dog breeds from continuous altitudes reveals adaptation to high-altitude hypoxia. Genome Res. 24, 1308–1315 (2014).
Peng, Y. et al. Down-Regulation of EPAS1 Transcription and Genetic Adaptation of Tibetans to High-Altitude Hypoxia. Mol. Biol. Evol. 34, 818–830 (2017).
Zhang, W., Tsuchiya, T. & Yasukochi, Y. Transitional change in interaction between HIF-1 and HNF-4 in response to hypoxia. J. Hum. Genet. 44, 293–299 (1999).
Kawano, Y. et al. Hypoxia reduces CD138 expression and induces an immature and stem cell-like transcriptional program in myeloma cells. Int. J. Oncol. 43, 1809–1816 (2013).
Fava, C. et al. The V433M variant of the CYP4F2 is associated with ischemic stroke in male Swedes beyond its effect on blood pressure. Hypertension 52, 373–380 (2008).
Lasker, J. M. et al. Formation of 20-hydroxyeicosatetraenoic acid, a vasoactive and natriuretic eicosanoid, in human kidney. Role of Cyp4F2 and Cyp4A11. J. Biol. Chem. 275, 4118–4126 (2000).
Sayer, M. S. et al. Ectopic expression of transcription factor NF-E2 alters the phenotype of erythroid and monoblastoid cells. J. Biol. Chem. 275, 25292–25298 (2000).
Yigit, N. et al. Nuclear factor-erythroid 2, nerve growth factor receptor, and CD34-microvessel density are differentially expressed in primary myelofibrosis, polycythemia vera, and essential thrombocythemia. Hum. Pathol. 46, 1217–1225 (2015).
Xiang, H. et al. Origin and dispersal of early domestic pigs in northern China. Sci. Rep. 7, 5602 (2017).
Xu, H. S., Jin, J. G. & Yang, Y. H. Test excavation to Nanzhuangtou site in Xushui County, Hebei Province. Archaeology 11, 961–970 (1992).
Huang, W. Note on holocene gazo site of Changdu, XiZang. Vertebrata PalAsiatica 18, 163–168 (1980).
Chen, F. H. et al. Agriculture facilitated permanent human occupation of the Tibetan Plateau after 3600 BP. Science 347, 248–250 (2015).
Groenen, M. A. et al. Analyses of pig genomes provide insight into porcine demography and evolution. Nature 491, 393–398 (2012).
Vitzthum, V. J. Fifty fertile years: anthropologists’ studies of reproduction in high altitude natives. Am. J. Hum. Biol. 25, 179–189 (2013).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Lee, T. H., Guo, H., Wang, X., Kim, C. & Paterson, A. H. SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data. BMC Genom. 15, 162 (2014).
Akey, J. M., Zhang, G., Zhang, K., Jin, L. & Shriver, M. D. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 12, 1805–1814 (2002).
Scheet, P. & Stephens, M. A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am. J. Hum. Genet. 78, 629–644 (2006).
Amaral, A. J., Megens, H. J., Crooijmans, R. P., Heuven, H. C. & Groenen, M. A. Linkage disequilibrium decay and haplotype block structure in the pig. Genetics 179, 569–579 (2008).
Acknowledgements
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA2004010301), the National Natural Science Foundation of China (31472000), the State Key Laboratory of Genetic Resources and Evolution (GREKF13-01), and the Animal Branch of the Germplasm Bank of Wild Species, Chinese Academy of Sciences (the Large Research Infrastructure Funding).
Author information
Authors and Affiliations
Contributions
Y.P.Z., H.B.X. and Y.F.M. designed the research. Y.F.M., X.M.H., C.P.H. and L.Z. performed the experiments. Y.F.M., X.M.H. and H.B.X. analyzed the data and wrote the paper; A.C.A. and D.M.I. revised and edited the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ma, YF., Han, XM., Huang, CP. et al. Population Genomics Analysis Revealed Origin and High-altitude Adaptation of Tibetan Pigs. Sci Rep 9, 11463 (2019). https://doi.org/10.1038/s41598-019-47711-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47711-6
This article is cited by
-
Structural variant landscapes reveal convergent signatures of evolution in sheep and goats
Genome Biology (2024)
-
Landscape genomics reveals regions associated with adaptive phenotypic and genetic variation in Ethiopian indigenous chickens
BMC Genomics (2024)
-
Whole-genome analysis reveals distinct adaptation signatures to diverse environments in Chinese domestic pigs
Journal of Animal Science and Biotechnology (2024)
-
Genetic characterization of Tibetan pigs adapted to high altitude under natural selection based on a large whole-genome dataset
Scientific Reports (2024)
-
Ssc-miR-101-3p inhibits hypoxia-induced apoptosis and inflammatory response in alveolar type-II epithelial cells of Tibetan pigs via targeting FOXO3
Scientific Reports (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
