
Abstract
Infection with CagA+ Helicobacter pylori strains is linked to an increased risk for gastric diseases, including gastric cancer. Recent evidence indicates that dynamic expansion and contraction of cagA copy number may serve as a novel mechanism to enhance disease development. Herein, comparative genomic analysis divided hpEurope into two groups: hpEurope/type-A and type-B. Only hpEurope/type-B displayed the multi-cagA genotype. Further analysis showed that cagPAI appears to have been independently introduced into two different H. pylori types, termed pre-type-A and pre-type-B, which consequently evolved to cagPAI type-A and type-B, respectively; importantly, all multi-cagA genotype strains displayed cagPAI type-B. Two direct cagA-flanking repeats of a genetic element termed CHA-ud were essential for the multi-cagA genotype in strain PMSS1 (hpEurope/type-B and cagPAI type-B). Furthermore, introduction of this genetic element into strain G27 (hpEurope/type-A and cagPAI type-A) was sufficient to generate the multi-cagA genotype. The critical steps in the evolution of the multi-cagA genotype involved creation of CHA-ud at cagA upstream in cagPAI type-B strains followed by its duplication to cagA downstream. En masse, elucidation of the mechanism by which H. pylori evolved to carry multiple copies of cagA helps to provide a better understanding of how this ancient pathogen interacts with its host.
Similar content being viewed by others


Introduction
Helicobacter pylori is a Gram-negative pathogen that inhabits the gastric mucosa of a significant portion of the human population and causes gastric diseases with severities that range from gastritis to gastric adenocarcinoma and mucosa-associated lymphoid tissue (MALT) lymphoma1,2. Once colonized, H. pylori infection chronically persists without treatment. Gastric cancer occurs at a prevalence of 1–2% in H. pylori infected subjects3. Given the association of H. pylori with gastric cancer and the fact that more than half of the world’s population is infected with this microbe, gastric carcinoma is the third leading cause of cancer-related death worldwide4. Accordingly, H. pylori is classified as a class I carcinogen by the World Health Organization5.
Modern humans were infected by H. pylori long before their first successful migration out of Africa 60 thousand years ago6,7. Since then, H. pylori has coevolved with human beings such that strains from different geographic regions have differentiated into H. pylori populations that are generally tightly associated with the human populations found in those areas. As a result, genetic differences seen among various H. pylori strains elicits interests in terms of evolution and virulence. Indeed, Multi Locus Sequence Typing (MLST) has been used to classify H. pylori into seven large population types: hpAfrica1, hpAfrica2, hpNEAfrica, hpEurope, hpEastAsia, hpAsia2 and hpSahul6,7. While the super-lineage hpAfrica2 was derived from the most ancestral H. pylori strains that migrated toward South Africa, the other populations descended from other super-lineages that migrated toward east and west Africa, and subsequently out of Africa. Migration of H. pylori with human beings out of Africa led to the establishment of two genetically distant populations, hpEurope and hpEastAsia, in which the strains were mainly isolated from Western and East Asian countries, respectively. In addition, before high-throughput sequencing methods became popular, prior studies considered gene arrangement in the H. pylori chromosome; McGee et al. speculated that strains 26695 and 43504 have different genome organizations, and Blomstergren et al. reported a strain that carried a large insertion in the cag Pathogenicity Island (cagPAI) region8,9. With the advent of modern sequencing technology and comparative genome analysis, Farnbacher et al. identified chromosome inversion pattern of strain B8, and further showed that B8 has a different gene arrangement in the cagPAI region as compared to other H. pylori strains10. Thus, due to the ability of H. pylori to rearrange its genome, different H. pylori cagPAI arrangements have been appreciated.
Among the various virulence factors of H. pylori, the oncoprotein cytotoxin-associated gene A (CagA) is thought to play a central role as a scaffolding protein in the development of gastric cancer11. The CagA effector is injected into host cells by a syringe-like structure termed the type IV secretion system (T4SS). Both CagA and the T4SS are encoded on a 40-kb DNA segment known as the cagPAI, which generally consists of 27 genes12. The cagPAI region is thought to have been introduced into the H. pylori genome via horizontal transfer from an unknown source. Once translocated to host cells, CagA undergoes phosphorylation by host cell kinases at a conserved tyrosine residue found within the EPIYA (Glu-Pro-Ile-Tyr-Ala) motif. CagA is known to interact with a wide variety of host cell signaling molecules and to perturb normal host cell signaling13,14. For example, phosphorylated CagA binds to a SH2-domain-containing protein tyrosine phosphatase (SHP2), and thus deregulates the phosphatase activity of SHP215. This in turn leads to hyperstimulation of Ras-Erk signaling.
In a recent study, we identified H. pylori strains that harbor multiple copies of the cagA gene, and showed that dynamic modulation of cagA copy number may impact development of gastric disease16. The multi-cagA genotype was only found in a subset of strains those were part of a collection of clinical isolates from the United States; this genotype was not seen in a large clinical collection from South Korea. Given this, we reasoned that the distribution of the multi-cagA genotype in the Western H. pylori population required further investigation. Furthermore, the mechanism by which H. pylori acquired the ability to modulate cagA copy number during coevolution with the human host remains to be elucidated. Thus herein, we performed PacBio whole genome sequencing of all of the clinical multi-cagA isolates we identified. The sequences were used as the foundation for comparative genomic analyses of a larger number of H. pylori strains that represent isolates derived from different worldwide geographic origins. We found that the multi-cagA genotype was only seen in a subset of hpEurope, termed hpEurope/type-B. Furthermore, we defined an essential cagA flanking genetic element that is required for generation of the multi-cagA genotype. Finally, we propose an evolutionary mechanism by which H. pylori developed the multi-cagA genotype. Overall, these results aid our understanding of H. pylori pathogenesis and its coevolution with the human host.
Results
General genomic features of all multi-cagA H. pylori clinical isolates
To begin to understand the process by which H. pylori developed the multi-cagA genotype, we sought to define the complete genome sequence of all identified H. pylori clinical isolates shown to carry multiple copies of cagA. Thus, we performed whole genome PacBio sequencing of six H. pylori strains containing the multi-cagA genotype (B125A, B128, B130A, B140, B147 and 7.13) along with two H. pylori strains containing a single-cagA (B136A and J182) (Table 1). Sequencing of each of the 8 strains resulted in a single circularized chromosome. Overall, these chromosomes showed a typical H. pylori genome GC content of 38.71–38.90% with a chromosomal length that ranged from approximately 1,646 kbp to 1,727 kbp. Of note, the genome sequences of three additional multi-cagA strains, PMSS1 (along with its mouse passaged derivative SS1) and J166, were released by other research groups17,18; our subsequent analyses included these whole genome sequences.
All multi-cagA H. pylori strains belong to hpEurope
Our prior analysis showed that the H. pylori multi-cagA genotype was found in American isolates but not in South Korean isolates16; thus, suggesting that the multi-cagA genotype is geographically-associated. To further categorize the multi-cagA strains, we performed phylogenetic analysis of the nine strains displaying this genotype (including the SS1 strain), with an additional 43 other H. pylori strains for which there were whole genome sequences available (Supplemental Table S1); this was accomplished via analysis of core genome pairwise distance (Fig. 1). The Neighbor-joining tree clearly classified the 52 H. pylori strains into hpAfrica1, hpAfrica2, hpEurope and hpEastAsia as previously described6,7. Of note, all strains carrying multiple copies of cagA belonged to hpEurope; this finding further suggests that the multi-cagA strains underwent a unique region-associated evolutionary history.

Neighbor-joining tree of the 52 H. pylori strains based on core genome pairwise distance. The Neighbor-joining tree was calculated from concatenated nucleic acid sequences of 927 orthologous genes (length 855,682 bp). Each H. pylori population and strains within it are indicated. The 9 multi-cagA genotype H. pylori strains grouped with hpEurope strains. The cagPAI type-B strains are defined in a latter section.
The multi-cagA genotype is associated with hpEurope/type-B
Genome rearrangement is an important factor that shapes genome structure and likely impacts gene expression19,20,21. This is especially true in H. pylori where many virulence gene polymorphisms are suggested to be the result of genome rearrangement22. Genomic rearrangement, as inferred by chromosomal reversal, is also commonly used to study bacterial phylogeny23,24,25,26. Thus, we analyzed the phylogenetic relationship inferred by chromosomal reversal distance using 46 of the 52 H. pylori strains in Fig. 1 (Supplemental Fig. S1); genome sequences of 6 H. pylori strains (N6, H-11, 59, NAD1, UM045 and NQ4216) were not completed and were excluded for this analysis. Importantly, most of the multi-cagA H. pylori strains were clustered together while the NY40 and ELS37 hpEurope stains clustered close to hpAfrica1 (Supplemental Fig. S1). Since all of the multi-cagA H. pylori strains belong to hpEurope, we further analyzed the phylogenetic relationship inferred by chromosomal reversal distance using just the 27 H. pylori strains that were assigned to hpEurope by core genome pairwise distance (Fig. 2); the whole-genome sequences of these 27 hpEurope strains were assembled as single circular contigs and thus provided full information on the genome structure. Analysis of these strains revealed that they further divided into two types, which we named hpEurope/type-A and type-B. Strikingly, the 9 multi-cagA strains were all classified as hpEurope/type-B along with SJM180, NY40, ELS37, B136A, J182, UM037, HUP-B14 and B8; the remaining 10 strains grouped into hpEurope/type-A.

Phylogenetic relationship of the 27 hpEurope strains for which a complete genome was available. A total of 80 local colinear blocks from each of the 27 genomes were converted to a signed permutation. The signed permutation was further used to infer their phylogenetic relationship based on chromosomal reversals with a unichromosomal circular model. The tree was mid-rooted as shown in the resulting dendrogram. The two clades into which hpEurope segregated were named hpEurope/type-A and type-B and are indicated by colors as described in the figure inset. The multi-cagA genotype strains all segregated into hpEurope/type-B. Scale bar indicates length of one rearrangement. The number of rearrangement is denoted on each branch.
The multi-cagA genotype co-occurs with a specific cagPAI rearrangement called type-B
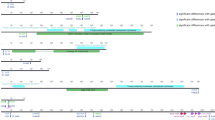
Our prior study showed that H. pylori PMSS1 dynamically alters cagA gene copy number. Furthermore, we previously suggested that the process by which this occurs likely involves homologous recombination at the repeated cagA homologous area (CHA)-ud sequences those flank cagA; the presence, pattern and number of copies of the CHA-ud sequence varies among different strains16. Since the multi-cagA genotype appears to play an important role in the development of gastric disease16, we sought to define the evolutionary mechanism behind development of the multi-cagA genotype. In cagPAI positive H. pylori strains, cagPAI is located within a chromosomal region delimited by the dapB and glr genes, designated as the dg region (Fig. 3). We first compared the dg regions in a total of 243 H. pylori strains (Supplemental Table S2). Among these strains, the overall gene arrangements of the dg region grouped into four types; pre-type-A, pre-type-B, type-A and type-B, which were represented by strain BM013A, SouthAfrica7, J99 and J166, respectively (Fig. 3). Most of the analyzed strains were cagPAI positive (73%, 178 out of 243) while about 27% (65 out of 243) were cagPAI negative (Table 2). Among the cagPAI negative strains, the majority of the strains (88%, 57 out of 65) were pre-type-A and the remaining (12%, 8 out of 65) were pre-type-B. Among the cagPAI positive strains, the majority (87%, 155 out of 178) were type-A and the remaining (13%, 23 out of 178) were type-B. Specifically, besides the 9 multi-cagA strains, which were type-B, the other 14 type-B strains were also shown to be part of hpEurope (Fig. 1).

Four types of gene arrangements within the chromosomal region delimited by dapB and glr (dg region). Representatives of each type of gene arrangements are shown and strain names are indicated in parentheses. Genes are drawn with filled arrows and DNA homology across the strain types are indicated by the same color. The dg region and cagPAI are indicated.
The pre-type-A strains (see BM013A in Fig. 3) carried a cluster of approximately nine genes within the dg region; interestingly, in the pre-type-B strains (see SouthAfrica7 in Fig. 3) seven genes of this cluster were inverted and, more importantly, an additional putative AAA family ATPase was inserted near the front of this cluster. The type-A strains (see J99 in Fig. 3) harbored an intact cagPAI upstream of the glr gene, while the type-B strains (see J166 in Fig. 3) harbored two separated cagPAI regions; the majority of the cagPAI (28 genes) was inverted downstream of the dapB gene and the cagA gene was separately located upstream of the glr gene.
Using the PacBio genomic sequencing and targeted Sanger dideoxy sequencing results, the dg regions of all of the multi-cagA strains were compared in Fig. 4. Notably, all of the multi-cagA strains belonged to cagPAI type-B. Furthermore, CHA-ud sequences were always found flanking cagA in these strains. Since all the multi-cagA strains harbored cagPAI type-B, we further asked a reciprocal question; do all of the type-B strains contain multiple copies of cagA? To assess this, we screened 234 clinical isolates obtained from South Korea and 80 clinical isolates obtained from the United States. We designed two sets of PCR primers (dapB-F/glnA-R and dapB-F/cagD-R) to enable us to identify the cagPAI type according to the position and orientation of the cagPAI in the dg region (Supplemental Fig. S2, Supplemental Table S3). As a result, we identified two more type-B strains (B136A and J182) from the United States (Table 1, Fig. 4). However, the multi-cagA genotype was not detected in these two strains. For further detailed analysis, we also performed whole genome sequencing of these two strains using PacBio technology (Table 1). Comparison of the dg region revealed that even though these two strains harbor a cagPAI type-B, only one CHA-ud sequence was present upstream of cagA (Fig. 4). Consistent with our prior results, these two strains, as well as some other cagPAI type-B strains (SJM180, NY40, ELS37, UM037, HUP-B14 and B8) also belonged to hpEurope/type-B. Therefore, all hpEurope/type-B strains analyzed in this study were cagPAI type-B strains (Fig. 2).

Gene arrangement within the dg region of strains with the multi-cagA genotype. All the strains carrying multiple copies of cagA harbored the type-B dg. Genes are drawn with filled arrows and DNA homology across the strain types is indicated by the same color. For comparison, the cagA copy number in strain J166, B125A, B128, 7.13, B130A, B140 and B147 is drawn as two copies. The first CHA-ud sequence upstream of cagA appears to have arisen from part of a AAA family ATPase gene. In strain B147, a ~39 kb insertion existed between CV730_03420 and CV730_03570. This insertion was delimited by two integrase genes indicated by empty arrows. The cagPAI region and CHA-ud are indicated.
To further characterize the number and location of the CHA-ud sequence, we searched for the CHA-ud sequence among the 178 cagPAI positive strains (Fig. 5). As a result, we discovered a relationship between CHA-ud and the cagPAI type. The CHA-ud only occurred within the dg region. Strains with cagPAI type-A possessed 0 or 1 CHA-ud; in these CHA-ud positive strains, CHA-ud always only appeared downstream of cagA. In comparison, strains with cagPAI type-B possessed 1 or 2 CHA-ud. In strains with one CHA-ud, the sequence always appeared upstream of cagA. Conversely, in strains with two CHA-ud, the sequences always flanked cagA. Given our prior supposition that cagA-flanking CHA-ud sequences were required for the homologous recombination that likely generates the multi-cagA genotype16, these data suggest that only cagPAI type-B strains containing two CHA-ud sequences have the potential to possess multiple cagA genes. Importantly, regardless of the cagA copy number, all of the 23 cagPAI type-B strains were part of the hpEurope clade (Figs 1 and 5). Specifically, the 17 strains with complete genome sequences were hpEurope/type-B (Fig. 2). Due to the incompleteness of the genome sequences of the remaining 6 cagPAI type-B strains, their groups in the hpEurope/type-A or type-B remain unclear. These data suggest that hpEurope/type-B strains underwent chromosomal rearrangements that resulted in the potential for the multi-cagA genotype.

CHA-ud sequence status in cagA-positive strains. The representative CHA-ud status within the dg regions of the 178 cagA-positive strains is shown. Genes are drawn with filled arrows and DNA homology across the strain types is indicated by the same color. The CHA-ud position is indicated. The number of strains showing each type of CHA-ud status is indicated in the box.
Genetic element required for generation of the multi-cagA genotype
Despite the previous work with H. pylori PMSS1 that suggested that two CHA-ud repeats are important for generation of the multiple cagA genotype and for deletion of cagA16, the mechanism behind the cagA duplication and deletion had not been explored experimentally. Therefore, we asked whether two direct CHA-ud repeats that flank cagA were essential for generation of the multiple cagA genotype using PMSS1 isogenic mutant strains, PMSS1/cagA-SF-1 and -SL-2 strains16. The PMSS1/cagA-SF-1 mutant strain contains a single CHA-ud upstream of cagA and the PMSS1/cagA-SL-2 mutant strain contains a single CHA-ud downstream of cagA (Fig. 6a). Using two previously described PCR strategies16, we screened 200 individual colonies derived from each PMSS1/cagA-SF-1 and -SL-2 strains for cagA duplication as well as deletion; we reasoned that recombination between CHA-ud elements could result in both possible genotypes. Representative PCR data were shown in Supplemental Fig. S3A. No duplication or deletion events of cagA were detected, further suggesting that two direct CHA-ud repeats that flank cagA are essential for evolution of the multiple cagA genotype.

Illustration of cagA copy number in PMSS1 and G27 isogenic mutant strains. The cagA gene copy number status in PMSS1 and G27 isogenic mutant strains were detected using colony PCR; and the cagA regions were confirmed by Sanger sequencing as described in Methods. Both PMSS1 isogenic mutant strains and the G27/revertant showed no changes in the cagA region; conversely, G27/2CHA-ud and G27/2CHA-d were able to delete and duplicate the cagA gene. Primer annealing sites for both colony PCR and sequencing as well as length of PCR amplicons are indicated. Recombination events in the cagA region are shown.
Given this, we next asked if introduction of two direct 449-bp CHA-ud repeats flanking cagA would be sufficient to generate the multi-cagA genotype in G27 H. pylori. G27 possesses cagPAI type-A with only one CHA-ud downstream of cagA (Fig. 3, Type-A). A markerless gene editing method using a kan-sacB cassette (ksc) was utilized to insert one additional 449-bp CHA-ud or one extra 179-bp sequence (designated as CHA-d) upstream of cagA (Supplemental Fig. S4). Four constructs (A-D) were designed; construct A was used to add ksc upstream of cagA, resulting in G27/ksc; construct B was used to remove ksc from G27/ksc, resulting in G27/revertant; construct C was used to replace ksc with CHA-ud, resulting in G27/2CHA-ud, and construct D was used to replace ksc with CHA-d, resulting in G27/2CHA-d. The resulting G27/2CHA-ud and G27/2CHA-d strains contained two direct repeats of CHA-ud and CHA-d flanking cagA, respectively. Two different colony PCR strategies using primer sets of G27dF2/dR2 and G27cagA1.1upF/cagAdownR2, were used to screen for cagA duplication and deletion in the G27/revertant, G27/2CHA-ud and G27/2CHA-d (Fig. 6b); representative PCR data were shown in Supplemental Fig. S3B. While the G27/2CHA-ud and G27/2CHA-d strains were both able to undergo recombination that resulted in cagA duplication and deletion, the G27/revertant did not show any recombination at this region. The cagA duplication and deletion seen in the G27/2CHA-ud and G27/2CHA-d strains were confirmed by Sanger dideoxy sequencing. Thus, introduction of two direct CHA-ud repeats or CHA-d repeats such that they flank cagA was sufficient to generate the multi-cagA genotype in G27 H. pylori.
Levels of CagA expression in five representative H. pylori single colony isolates of the G27/revertant, G27/2CHA-ud and G27/2CHA-d were measured by Western blot (Supplemental Fig. S5). As expected, the G27/revertant strains showed similar CagA expression levels to that of wild-type G27. Interestingly, CagA expression levels in the G27/2CHA-ud and G27/2CHA-d strains were also similar to that of the G27 wild-type and G27/revertant strains. In addition, wild-type G27, G27/revertant, G27/2CHA-ud and G27/CHA-d strains were all observed to translocate and phosphorylate CagA in the human gastric adenocarcinoma (AGS) cell line (Supplemental Fig. S5).
Evolutionary mechanism leading to the multi-cagA genotype
The above data allowed us to develop a model by which H. pylori could evolve to possess multiple copies of the cagA gene (Fig. 7). As mentioned previously, the cagPAI in H. pylori is believed to have been obtained from an unknown source via horizontal recombination27. Our phylogenetic analysis based on chromosomal reversal indicated that the strains in hpEurope present at least two distinct groups in terms of chromosomal structure. We also observed that among the total 243 strains analyzed, the ratio (6.3) of pre-type-A to pre-type-B strains was comparable to the ratio (6.7) of type-A to type-B strains. Therefore, we assume that prior to acquisition of the cagPAI, H. pylori evolved into two ancestral branches that are represented by pre-type-A and pre-type-B. These two ancestral branches then obtained cagPAI and became type-A and type-B, respectively. As represented by the majority of the cagPAI positive strains (63.8%, 155/243), one-step insertion of an intact cagPAI immediately upstream of glr in pre-type-A resulted in the formation of type-A (Fig. 7a). However, two steps were necessary for the evolution of type-B from pre-type-B. First, an intact cagPAI inserted upstream of glr, which is the same locus utilized in type-A. Second, the region delimited by the gene encoding a putative AAA family ATPase and cagB underwent inversion, resulting in the type-B strains (Fig. 7b). This inversion resulted in translocation of the ATPase gene to the region of upstream of cagA. Indeed, CHA-ud is in fact the 3′ terminal portion of this ATPase gene; thus, this inversion would explain the fact that all of the type-B strains carried CHA-ud upstream of cagA. Finally, a latter event duplicated CHA-ud such that it also appeared downstream of cagA; this may have involved activity of an insertion sequence (IS) since an IS is found in the cagA homologous area. Together, the two direct CHA-ud sequences then facilitated alteration of cagA copy number by homologous recombination.

Model for the evolutionary mechanism utilized to create the multi-cagA genotype. The dg region is shown. Genes are drawn with filled arrows and DNA homology across the strain types is indicated by the same color. (a) Formation of the type-A dg by insertion of the cagPAI into pre-typeA dg. (b) Formation of the multi-cagA genotype in type-B dg strains by three steps. IS* indicates an insertion sequence with a transposase in PMSS1. (c) Homologous recombination introduced CHA-ud into type-A strains.
In comparison, we note that in type-A strains, the CHA-ud found downstream of cagA likely had a different evolution history. This assertion is because the intact putative AAA family ATPase gene that appears to be the origin of CHA-ud, was neither found in pre-type-A nor in type-A genomes. Thus, intra-genomic recombination could not be the source of CHA-ud. Instead, since regions of cagA downstream are highly homologous in type-A and type-B strains, the CHA-ud likely came from inter-genomic recombination with a type-B strain (Fig. 7c). This fact may also explain why CHA-ud never appeared upstream of cagA in type-A strains; the upstream regions of cagA in type-A and type-B strains show little homology. The notion that the CHA-ud in type-A strains came from transformation and horizontal recombination of genetic material from type-B strains is supported by the fact that recombination among strains during mixed infections with multiple H. pylori strains are common28,29,30.
Discussion
H. pylori strain PMSS1 was previously shown to represent a heterogeneous population in terms of cagA copy number, harboring zero to four copies of cagA16. Moreover, cagA copy number was dynamic as it was able to expand and contract within individual isolates. A higher cagA copy number was correlated with a strain’s virulence potential; strains with more copies of cagA produced more CagA and induced higher cell elongation and more IL-8 expression. In addition, the multi-cagA strains were only found among American clinical isolates but not in Korean clinical isolates.
Herein we utilized phylogenetic analysis based on core genome pairwise distance to confirm that the multi-cagA strains genetically belong to hpEurope (Fig. 1). For phylogenetic analysis based on chromosomal reversal, although most of the multi-cagA H. pylori strains clustered together, the NY40 and ELS37 hpEurope stains clustered close to hpAfrica1; this may suggest that the NY40 and ELS37 strains harbor genome structures similar to hpAfrica1 strains (Supplemental Fig. S1). Furthermore, clustering based on chromosomal arrangement of the 27 hpEurope strains revealed two distinct chromosomal structures in hpEurope: hpEurope/type-A and type-B. More importantly, the multi-cagA strains were only found in hpEurope/type-B (Fig. 2). Previous studies implied that hpEurope originated from two distinct ancestral populations: northeastern Africa and Central Asia6,31. However, efforts to define subpopulations of hpEurope were unsuccessful, suggesting a complex evolutionary history and extensive horizontal recombination events in hpEurope. Indeed, H. pylori is a highly recombinant bacterium and that signal of clonal inheritance was likely rapidly lost. In the current study, phylogeny inference based on chromosomal rearrangement was used to avoid this issue; this technique was powerful in that it was able to distinguish subsets of hpEurope. Additionally, completion and assembly of more complete genomes will help to confirm and extend our findings.
We previously identified strains carrying multiple copies of cagA using a PCR strategy that detects adjacent multiple cagA genes; our results were confirmed by Sanger sequencing and Southern blot analysis16. To investigate these strains in greater genomic detail, we chose PacBio technology for whole-genome sequencing of these multi-cagA genotype strains. We reasoned that because PacBio sequencing has a longer read length (~20 kb), it should be better for identification of the adjacent repeat sequences32. However, we were unable to assemble multiple cagA copies in the genomes of the strains known to carry multiple cagA copies (B125A, B128, B130A, B140, B147 and 7.13).
Since prior targeted Sanger sequencing showed that the cagA copy number in B125A, B128, B130A, B140, B147, 7.13 and J166 were at least two copies16, the difference between the genome assemblies and the targeted sequencing approaches may indicate limitations of the assembly programs or indicate that the dynamic expansion and contraction of cagA copy number occurs at a population level and consists of heterogenous population in terms of cagA gene number. Support of the later possibility can be found in our prior finding that H. pylori strain 7.13 is estimated to harbor an average cagA number of approximately 1.416. Thus, genomic DNA for sequencing would have been isolated from heterogeneous populations of cells containing zero, one or two cagA copies; this issue could lead to issues with the downstream assembly process. Overall, these results suggest that many previous genome sequences may not reflect multiple copies of cagA that are actually present. Interestingly, among the analyzed sequences of 170 cagPAI positive strains available in the GenBank database, only the assemblies for strain PMSS1 and its derivative strain SS1 harbor more than one copy of the cagA gene. Further study will be required to examine the cagA gene numbers in other published type-B cagPAI strains since most of the next generation sequencing methods are not able to directly identify multiple cagA copies.
Previous analysis of heterogeneous population of strain PMSS1 strongly suggested that H. pylori can manipulate cagA number via homologous recombination within the CHA-ud repeat sequence16. In the current study, genome sequencing showed that the cagA genes in the multi-cagA strains were all flanked by the CHA-ud sequence. In addition, when either of the CHA-ud sequences that flank cagA were removed, PMSS1 lost the capability to generate the multi-cagA genotype. Previously expansion to multiple repeats was not detected in each single transformant of PMSS1/cagA-SF-1 and -SL-2 mutant strains16. In the current study, the prior observation of cagA duplication was confirmed by expanding the analysis to 200 individual colonies derived from each PMSS1 derivative (PMSS1/cagA-SF-1 and -SL-2). Furthermore, an additional PCR method was utilized to detect cagA deletion. Moreover, when G27 was engineered to contain CHA-ud or CHA-d sequences that flank cagA as direct repeats, G27 gained the ability to generate the multi-cagA genotype. This suggests that the CHA-ud genetic element flanking cagA is essential and sufficient for generation of the multi-cagA genotype; however, the CHA-ud sequence may not be unique for this capability; in theory any direct repeat that flanks cagA could result in a similar homologous recombination event. Indeed, introduction of flanking CHA-d sequences also resulted in the ability of G27 to duplicate cagA (Fig. 6). It is worth noting that G27/2CHA-ud and G27/2CHA-d strains carry approximately 1.1 and 1.3 copies of cagA, respectively (data not shown), while PMSS1 carries on average 3.7 copies of cagA16. The similar cagA copy number of G27/2CHA-ud and G27/2CHA-d strains to wild-type G27 might explain why there was no significant variation in the overall level of CagA expression in representative G27/revertant, G27/2CHA-ud and G27/2CHA-d strains as compared to that of wild-type G27 (Supplemental Fig. S5). Conversely, PMSS1 strains exhibited higher level of CagA expression than that of PMSS1/cagA-SF-1 and -SL-2 strains16.
The described genomic analyses lead us to propose a model whereby chromosomal rearrangements in the cagPAI region resulted in translocation and subsequently duplication of CHA-ud to flank cagA; this in turn leads to duplication of cagA (Fig. 7). In modern H. pylori, the cagPAI was thought to be derived from the most ancient H. pylori. However, our characterization of the dg region suggests that the cagPAI type-A and type-B strains evolved from two origins: pre-type-A and pre-type-B, respectively. The evolution of cagPAI type-A was similarly described by Fischer, but his model for cagPAI type-B evolution is different from our current model33. Data presented in Fig. 3 suggested that pre-type-B evolved separately from pre-type-A. Furthermore, our analysis (Fig. 5) suggests a role for the 3′ terminal region of a putative nonessential AAA family ATPase gene in pre-type-B strains in the origin of CHA-ud. Rearrangement within the dg region brought the putative ATPase gene to lie upstream of cagA, which successively became CHA-ud in all type-B strains. Next, CHA-ud duplicated to downstream of cagA probably through the activity of an IS element; IS elements were frequently present in this region34. It is worth noting that the phylogenetic analysis of chromosomal rearrangements using whole-genome sequences is based on the pattern of these rearrangements of co-linear blocks only for DNA regions that are shared by all of the strains. Thus, since some strains are cagPAI negative, the two types in hpEurope (Fig. 2) would not result from these two particular rearrangements in the cagPAI type-B strains. Together with the phylogenetic analyses, this model explains the distribution of multi-cagA strains only in Western hpEurope/type-B, but not in Western hpEurope/type-A and East Asian type H. pylori. From the observed data, it is likely that the evolution of the ATPase gene to become CHA-ud infrequently happened by a chromosomal reversal. However, since CagA plays an important role in bacterial pathogenesis, the multi-cagA genotype was retained. This strongly suggests that the multi-cagA genotype provided some evolutionary benefit to hpEurope/type-B and may do the same to other populations in the future.
Materials and Methods
Bacterial strains and culture
The H. pylori strains B125A, B128, B130A, B136A, B140, B147, J182, J166, 7.13, PMSS1, PMSS1/cagA-SF-1, PMSS1/cagA-SL-2, G27, and G27 isogenic mutant strains were cultured and stored as previously described16,35. Briefly, all H. pylori strains were grown on antibiotic supplemented Columbia blood agar plates under microaerobic conditions generated using an Anaeropack-Microaero gas-generating system (Mitsubishi Gas Chemical, Tokyo, Japan).
Genome sequencing, assembly and annotation
Single colonies were isolated from B125A, B128, B130A, B136A, B140, B147, J182, 7.13. One single colony from each strain was further cultured for whole genome sequencing and preparation of a frozen stock. Bacterial genomic DNA was extracted using the Wizard® Genomic DNA Purification Kit (Promega, USA) and then Chunlab Inc. Korea processed the subsequent PacBio genome sequencing. Briefly, qualified genomic DNA was prepared as 20 kb SMRTbell templates using the SMRTbell Template Prep Kit (Pacific Biosciences, USA) according to the manufacturer’s protocol. The sequencing was performed using the PacBio RSII sequencing platform (Pacific Biosciences, USA) with the default condition for RSII P6C4 chemistry. The raw data were filtered and assembled with the SMRT Analysis Software v2.3.0 with the HGAP2 protocol and default parameters. The assembled genomes were processed for subsequent annotation by the NCBI Prokaryotic Genome Annotation Pipeline version 4.336.
Phylogenetic analysis
Phylogenetic analysis based on core genome pairwise distance was performed using Efficient Database framework for comparative Genome Analyses using BLAST score Ratios (EDGAR 2.0)37. Briefly, 927 orthologous genes that were shared among a total of 52 H. pylori genomes (Supplemental Table S1) were identified by bidirectional BLASTn38. Each of the orthologous genes was aligned with MUSCLE39. The alignments were then trimmed and concatenated to a single 855,682-bp alignment. The concatenated alignment was further used to compute a pairwise genome distance matrix; a phylogenetic tree was then constructed based on this distance matrix using a Neighbor-joining method as implemented in PHYLIP40. The Neighbor-joining tree was visualized using Mega 7.041.
Phylogenetic analysis based on chromosomal inversion was computed using progressiveMauve and MGR42,43; all plasmids were first removed from the genome sequences. Briefly, the completed chromosomal sequences of the 46 general strains and 27 hpEurope strains were aligned with progressiveMauve using the default parameters; these resulted in 107 and 80 conserved local colinear blocks (LCBs), respectively. The relative position and orientation of the LCBs were output as signed permutations. Further, the permutations were used as input to MGR to compute phylogeny trees based on reversals with the unichromosomal circular mode. MGR generates a tree such that the sum of the rearrangements is minimized over all of the edges of the tree. Finally, the tree was midpoint-rooted and the tree topology was visualized using Mega 7.0.
Generation of G27 isogenic mutant strains
Four constructs targeting the G27 region upstream of cagA were generated (Supplemental Fig. S4). Construct A was made as follows. Using the G27 wild type as a template, amplicons of front and rear wings with XhoI and SmaI sites located at the adjacent ends were generated using primer sets of T1cagAup1kbF and T1cagAup500RXS and G27ctrl5 and T1cagA0R, respectively. Both amplicons were fused by splicing-by-overlap-extension (SOE) PCR so that the fused amplicon contained XhoI and SmaI sites in the overlap-joining region44,45. This product was cloned into the pGEM®-T Easy vector system (Promega, Madison, WI, USA) by TA cloning. A ksc, liberated from plasmid pKSF-II by double-digestion with XhoI and SmaI, was subsequently ligated with the XhoI-SmaI double-digested plasmid that contained the fused amplicon46,47. Construct B was made using the primer set of T1cagAup1kbF and T1cagA0R, and the amplicon was directly cloned into the pGEM®-T Easy vector system. The three amplicons (starting from left to right) of construct C were made using the primer set of T1cagAup1kbF and CnDF2F1R, CnDF1F2F and T1cagAdown600R, and T1cagAup500F and T1cagA0R, respectively. The first two amplicons were fused together by SOE PCR, followed by the fusion of the third amplicon (rear wing) for making the complete insert; the resulting product was cloned into the pGEM®-T Easy vector system. Construct D was generated using the same process and primer sets of T1cagAup1kbF and CnEChaDF1R, CnEF1ChaDF and CnEF3ChaDR, and CnEChaDF3F and T1cagA0R. Three marker-less G27 isogenic mutant strains were generated in two sequential steps. At stage I, the construct A was introduced into G27 by natural transformation48. The successful transformants (G27/ksc) were isolated using kanamycin selection via growth on horse blood agar plates supplemented with 25 µg/mL kanamycin. The double homologous recombination of the construct, resulting in kanamycin-resistant H. pylori single colony isolates, were further verified by three PCRs (Aa, Ba, Ca, see Supplemental Fig. S4) and DNA sequencing. At stage II, constructs B, C and D were transformed into the previously selected G27/ksc strain for generation of the G27/revertant, G27/2CHA-ud and G27/2CHA-d respectively. The unmarked mutant strains derived from the intended double homologous recombination were isolated via growth on 5% sucrose supplemented horse blood agar; resulting colonies were verified by PCR screening (Ab, Bb, Cb; Ac, Bc, Cc; Ad, Bd, Cd) and DNA sequencing. The primers used for the construction and verification of the G27 mutant strains are listed in Supplemental Table S3.
Genotyping of cagA copy number in H. pylori single colonies by colony PCR
Colony PCR was performed as previously described16. Briefly, a culture of PMSS1/cagA-SF-1, PMSS1/cagA-SL-2 or the G27 isogenic mutant strains was diluted in Brucella Broth and was plated onto horse blood agar plates. Plates were incubated for 4 days in microaerobic condition until single colonies appeared. Single colonies were picked and streaked onto a new blood agar plate for further culture. The cultures were used for isolation of genomic DNA and frozen stocks.
Colonies were transferred into 20 μL of distilled water and then were heated at 99 °C for 4 min. After centrifugation, the supernatant was used as the template for PCR to analyze cagA copy number in single colonies. A colony PCR was designed to identify none, single, or multiple cagA genes. The primers used for the cagA genotyping of the PMSS1 isogenic mutant strains and the G27 isogenic mutant strains are listed in Supplemental Table S3 and the alignment sites are illustrated in Fig. 6. The multiple cagA genotype was detected using the primer sets of dF2/dR2 for the PMSS1 isogenic mutants and G27dF2/dR2 for the G27 isogenic mutants. The cagA deletion was detected using the primer sets of cagAupF2/cagAdownR2 for the PMSS1 isogenic mutants and G27cagA1.1upF/cagAdownR2 for the G27 isogenic mutants. The presence of cagA was screened for using the F2 and G27R2 primer combination. Both cagA multiplication and deletion PCRs were performed as follows: 94 °C for 4 min; 35 cycles of 94 °C for 30 s, 54 °C for 15 s, and 72 °C for 90 s; a final step at 72 °C for 5 min. For the other PCR screening, the same PCR parameters, except a 30 s extension time, were used.
Analysis of H. pylori chromosomal region between dapB and glr gene (dg region)
The annotated sequences were visualized using CLgenomics version 1.55 software with the format provided by Chunlab Inc., Korea. The strains used for the analysis of the dg region were downloaded from EzBioCloud (https://www.ezbiocloud.net/) with the accession numbers listed in Supplemental Table S2. Sequence homology was searched for using BLAST implemented in CLgenomics version 1.55. For strain B147, our prior Sanger dideoxy sequencing was unable to resolve the region upstream of cagA16; PacBio genomic sequencing revealed that a 39 kb insertion that was flanked by two integrases was responsible for the failed Sanger dideoxy sequencing (Fig. 4).
DNA sequencing
Sanger dideoxy DNA sequencing was performed at Cosmo Genetech Co., Ltd (Seoul, Korea). The DNA sequencing primers are listed Supplemental Table S3.
Accession numbers
All of the genome sequences were deposited to the NCBI GenBank Prokaryotic Genomes (https://www.ncbi.nlm.nih.gov/nucleotide/) under Bioproject: PRJNA419269. Sanger dideoxy sequencing results were deposited to the NCBI GenBank with accession numbers from MK089812 to MK089815.
Immunoblot assay
To prepare lysates of infected cells, AGS cells were seeded onto 6-well cell culture plates at a density of 4 × 105 cells per well and were then incubated for 2 days at 37 °C. At 2 h prior to infection, cells were washed with PBS and the medium was changed to 2 ml of RPMI 1640. Liquid cultures of H. pylori were resuspended in RPMI 1640, and AGS cells were infected at a MOI of 100 for 5 h. Subsequently, cells were washed with PBS and then lysed with 150 µl of cell lysis buffer supplemented with protease inhibitor cocktail. Protein concentrations of infected cell lysates were measured using Pierce bicinchoninic acid (BCA) protein assay reagent (Thermo Fisher Scientific, Waltham, MA, USA). 20 µg of each respective protein sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and then transferred to a polyvinylidene fluoride membrane (Merck Millipore, Darmstadt, Germany). To detect CagA, phosphorylated CagA, UreA, and GAPDH, membranes were probed by the use of 1:10,000 dilution of rabbit polyclonal anti-CagA antibody b-300 (Santa Cruz Biotechnology), 1:10,000 dilution of mouse monoclonal anti-phosphotyrosine antibody pY99 (Santa Cruz Biotechnology, Dallas, TX, USA), 1:3,000 rabbit polyclonal anti-Urease α antibody b-234 (Santa Cruz Biotechnology), and 1:2,500 rabbit polyclonal anti-GAPDH antibody (AbFrontier, Seoul, South Korea), respectively. These membranes were detected with 1:5,000 goat anti-mouse IgG-horseradish peroxidase (IgGHRP) (AbFrontier) or goat anti-rabbit IgG-HRP (AbFrontier). The pY99 treated membranes were stripped with Re-Blot Plus Strong Solution (Merck Millipore), prior to detection of total CagA. Probed membranes were then developed using Western Bright enhanced chemiluminescence-HRP (ECL-HRP) substrate (Advansta, Menlo Park, CA, USA) on X-ray film (Agfa, Mortsel, Belgium). Relative CagA expression levels were measured as previously described16. A ratio of CagA to UreA was calculated for each of five representative samples, and each value was normalized to the value of wild type G27. Next, the mean CagA expression levels from each group were divided with the G27/revertant value to determine relative CagA protein levels and those values were plotted on a graph. Error bars represent standard deviations from each group (n = 5).
Data Availability
All data generated or analyzed during this study are included in this published article (and its Supplementary Information files) or are available from the corresponding author on reasonable request.
References
Marshall, B. J. & Warren, J. R. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1, 1311–1315 (1984).
Blaser, M. J. Helicobacter pylori and gastric diseases. BMJ 316, 1507–1510 (1998).
Noto, J. M. & Peek, R. M. Jr. Helicobacter pylori: an overview. Methods Mol Biol 921, 7–10, https://doi.org/10.1007/978-1-62703-005-2_2 (2012).
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68, 394–424, https://doi.org/10.3322/caac.21492 (2018).
Moller, H., Heseltine, E. & Vainio, H. Working group report on schistosomes, liver flukes and Helicobacter pylori. Int J Cancer 60, 587–589 (1995).
Falush, D. et al. Traces of human migrations in Helicobacter pylori populations. Science 299, 1582–1585, https://doi.org/10.1126/science.1080857 (2003).
Moodley, Y. et al. Age of the association between Helicobacter pylori and man. PLoS Pathog 8, e1002693, https://doi.org/10.1371/journal.ppat.1002693 (2012).
McGee, D. J., May, C. A., Garner, R. M., Himpsl, J. M. & Mobley, H. L. Isolation of Helicobacter pylori genes that modulate urease activity. J Bacteriol 181, 2477–2484 (1999).
Blomstergren, A., Lundin, A., Nilsson, C., Engstrand, L. & Lundeberg, J. Comparative analysis of the complete cag pathogenicity island sequence in four Helicobacter pylori isolates. Gene 328, 85–93, https://doi.org/10.1016/j.gene.2003.11.029 (2004).
Farnbacher, M. et al. Sequencing, annotation, and comparative genome analysis of the gerbil-adapted Helicobacter pylori strain B8. BMC Genomics 11, 335, https://doi.org/10.1186/1471-2164-11-335 (2010).
Hatakeyama, M. Helicobacter pylori CagA and gastric cancer: a paradigm for hit-and-run carcinogenesis. Cell Host Microbe 15, 306–316, https://doi.org/10.1016/j.chom.2014.02.008 (2014).
Odenbreit, S. et al. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287, 1497–1500 (2000).
Higashi, H. et al. Helicobacter pylori CagA induces Ras-independent morphogenetic response through SHP-2 recruitment and activation. J Biol Chem 279, 17205–17216, https://doi.org/10.1074/jbc.M309964200 (2004).
Suzuki, M. et al. Interaction of CagA with Crk plays an important role in Helicobacter pylori-induced loss of gastric epithelial cell adhesion. J Exp Med 202, 1235–1247, https://doi.org/10.1084/jem.20051027 (2005).
Higashi, H. et al. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 295, 683–686, https://doi.org/10.1126/science.1067147 (2002).
Jang, S. et al. Dynamic Expansion and Contraction of cagA Copy Number in Helicobacter pylori Impact Development of Gastric Disease. MBio 8, https://doi.org/10.1128/mBio.01779-16 (2017).
Draper, J. L. et al. Fallacy of the Unique Genome: Sequence Diversity within Single Helicobacter pylori Strains. MBio 8, https://doi.org/10.1128/mBio.02321-16 (2017).
Linz, B. et al. A mutation burst during the acute phase of Helicobacter pylori infection in humans and rhesus macaques. Nat Commun 5, 4165, https://doi.org/10.1038/ncomms5165 (2014).
Sousa, C., de Lorenzo, V. & Cebolla, A. Modulation of gene expression through chromosomal positioning in Escherichia coli. Microbiology 143 (Pt 6), 2071–2078,https://doi.org/10.1099/00221287-143-6-2071 (1997).
Couturier, E. & Rocha, E. P. Replication-associated gene dosage effects shape the genomes of fast-growing bacteria but only for transcription and translation genes. Mol Microbiol 59, 1506–1518, https://doi.org/10.1111/j.1365-2958.2006.05046.x (2006).
Darling, A. E., Miklos, I. & Ragan, M. A. Dynamics of genome rearrangement in bacterial populations. PLoS Genet 4, e1000128, https://doi.org/10.1371/journal.pgen.1000128 (2008).
Furuta, Y. et al. Birth and death of genes linked to chromosomal inversion. Proc Natl Acad Sci USA 108, 1501–1506, https://doi.org/10.1073/pnas.1012579108 (2011).
Boore, J. L. & Brown, W. M. Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Opin Genet Dev 8, 668–674 (1998).
Wang, L. S., Warnow, T., Moret, B. M., Jansen, R. K. & Raubeson, L. A. Distance-based genome rearrangement phylogeny. J Mol Evol 63, 473–483, https://doi.org/10.1007/s00239-005-0216-y (2006).
Herniou, E. A. et al. Use of whole genome sequence data to infer baculovirus phylogeny. J Virol 75, 8117–8126 (2001).
Sankoff, D. & Blanchette, M. Multiple genome rearrangement and breakpoint phylogeny. J Comput Biol 5, 555–570, https://doi.org/10.1089/cmb.1998.5.555 (1998).
Censini, S. et al. cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci USA 93, 14648–14653 (1996).
Patra, R. et al. Multiple infection and microdiversity among Helicobacter pylori isolates in a single host in India. PLoS One 7, e43370, https://doi.org/10.1371/journal.pone.0043370 (2012).
Cao, Q. et al. Progressive genomic convergence of two Helicobacter pylori strains during mixed infection of a patient with chronic gastritis. Gut 64, 554–561, https://doi.org/10.1136/gutjnl-2014-307345 (2015).
Morales-Espinosa, R. et al. Colonization of Mexican patients by multiple Helicobacter pylori strains with different vacA and cagA genotypes. J Clin Microbiol 37, 3001–3004 (1999).
Linz, B. et al. An African origin for the intimate association between humans and Helicobacter pylori. Nature 445, 915–918, https://doi.org/10.1038/nature05562 (2007).
Krsticevic, F. J., Schrago, C. G. & Carvalho, A. B. Long-Read Single Molecule Sequencing to Resolve Tandem Gene Copies: The Mst77Y Region on the Drosophila melanogaster Y Chromosome. G3 (Bethesda) 5, 1145–1150, https://doi.org/10.1534/g3.115.017277 (2015).
Fischer, W. Assembly and molecular mode of action of the Helicobacter pylori Cag type IV secretion apparatus. FEBS J 278, 1203–1212, https://doi.org/10.1111/j.1742-4658.2011.08036.x (2011).
Kersulyte, D. et al. Differences in genotypes of Helicobacter pylori from different human populations. J Bacteriol 182, 3210–3218 (2000).
Carpenter, B. M. et al. Expanding the Helicobacter pylori genetic toolbox: modification of an endogenous plasmid for use as a transcriptional reporter and complementation vector. Appl Environ Microbiol 73, 7506–7514, https://doi.org/10.1128/AEM.01084-07 (2007).
Haft, D. H. et al. RefSeq: an update on prokaryotic genome annotation and curation. Nucleic Acids Res 46, D851–D860, https://doi.org/10.1093/nar/gkx1068 (2018).
Blom, J. et al. EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res 44, W22–28, https://doi.org/10.1093/nar/gkw255 (2016).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402 (1997).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797, https://doi.org/10.1093/nar/gkh340 (2004).
Baum, B. R. PHYLIP: Phylogeny Inference Package. Version 3.2. Joel Felsenstein. The Quarterly Review of Biology 64, 539–541, https://doi.org/10.1086/416571 (1989).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 33, 1870–1874, https://doi.org/10.1093/molbev/msw054 (2016).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5, e11147, https://doi.org/10.1371/journal.pone.0011147 (2010).
Bourque, G. & Pevzner, P. A. Genome-scale evolution: reconstructing gene orders in the ancestral species. Genome Res 12, 26–36 (2002).
Horton, R. M. et al. Gene splicing by overlap extension. Methods Enzymol 217, 270–279 (1993).
Horton, R. M., Cai, Z., Ho, S. M. & Pease, L. R. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. BioTechniques 8(5):528-535 (November 1990). Biotechniques 54, 129–133, https://doi.org/10.2144/000114017 (2013).
Copass, M., Grandi, G. & Rappuoli, R. Introduction of unmarked mutations in the Helicobacter pylori vacA gene with a sucrose sensitivity marker. Infect Immun 65, 1949–1952 (1997).
Mehta, N., Olson, J. W. & Maier, R. J. Characterization of Helicobacter pylori nickel metabolism accessory proteins needed for maturation of both urease and hydrogenase. J Bacteriol 185, 726–734 (2003).
Israel, D. A., Lou, A. S. & Blaser, M. J. Characteristics of Helicobacter pylori natural transformation. FEMS Microbiol Lett 186, 275–280, https://doi.org/10.1111/j.1574-6968.2000.tb09117.x (2000).
Acknowledgements
The authors thank Dr. Jochen Blom at Justus-Liebig-University Gießen for uploading data to the EDGAR platform. The authors also acknowledge the EDGAR platform, which is financially supported by the BMBF grant FKZ 031A533 within the de.NBI network. This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT and Future Planning (2017R1A2B4008960), and funded by the Ministry of Education (2018R1D1A1B07043204). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
H.S., L.G. and J.-H.C. designed the experiments, interpreted the data, and wrote the manuscript. H.S. and K.T. performed all experiments. S.J. constructed the G27 isogenic strains and PCR analysis. A.K. and Y.H.C. performed genome sequencing. Y.-J.C., M.L. and N.G. performed genome data analysis. D.S.M. interpreted the data and wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Su, H., Tissera, K., Jang, S. et al. Evolutionary mechanism leading to the multi-cagA genotype in Helicobacter pylori. Sci Rep 9, 11203 (2019). https://doi.org/10.1038/s41598-019-47240-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-47240-2
This article is cited by
-
Prevalence of Helicobacter pylori Virulence Genes and Their Association with Chronic Gastritis in Beijing, China
Current Microbiology (2023)
-
Characterization of East-Asian Helicobacter pylori encoding Western EPIYA-ABC CagA
Journal of Microbiology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
