
Abstract
Although the cofactors in the bacterial reaction centre of Rhodobacter sphaeroides wild type (WT) are arranged almost symmetrically in two branches, the light-induced electron transfer occurs selectively in one branch. As origin of this functional symmetry break, a hydrogen bond between the acetyl group of PL in the primary donor and His-L168 has been discussed. In this study, we investigate the existence and rigidity of this hydrogen bond with solid-state photo-CIDNP MAS NMR methods offering information on the local electronic structure due to highly sensitive and selective NMR experiments. On the time scale of the experiment, the hydrogen bond between PL and His-L168 appears to be stable and not to be affected by illumination confirming a structural asymmetry within the Special Pair.
Similar content being viewed by others

Introduction
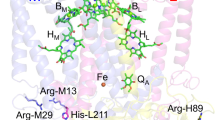
The reaction centre (RC) of the purple bacterium Rhodobacter (R.) sphaeroides is a membrane protein in which the primary charge separation, the first step of photosynthesis, is taking place. The availability of the x-ray structure of the RCs of this purple bacterium was a major break-through for the understanding of the early processes in photosynthesis1. The cofactors associated with the M- and L-subunits of bacterial RCs are arranged in two nearly symmetric branches spanning the membrane. Each branch consists of two bacteriochlorophylls a, a bacteriopheophytin a and a quinone. At the end of the branches, a non-heme iron is located (Fig. 1). Close to the B-branch and bound to the M subunit, a carotenoid molecule is present breaking the symmetry of the two branches.

Cofactor arrangement in the RC of R. sphaeroides WT: PM and PL ─ bacteriochlorophyll a dimer, forming the primary electron donor, the Special Pair P, BA/B ─ accessory bacteriochlorophyll a, ΦA/B ─ bacteriopheophytin a, QA/B ─ quinone A and terminal electron accepting quinone B, Fe2+ ─ non-heme iron, Car ─ carotenoid. The electron is transferred only via the A-branch (black arrow). For details, see text.
The primary electron-donor P, the so-called Special Pair, is formed by two overlapping bacteriochlorophylls a, PL and PM. Upon illumination, the Special Pair becomes electronically excited and transfers an electron to the ubiquinone QA via an accessory bacteriochlorophyll a (BA) and a bacteriopheophytin a (ΦA). In the final step, the electron is transferred to QB. Two photocycles coupled with the uptake of two protons reduce QB to QBH2 which diffuses out of the protein into the membrane-based quinone pool.
Although the two branches A and B are nearly symmetric, the electron transfer occurs selectively via the A-branch2,3. The directional electron transfer is reflected in the asymmetry of the electronic structure of the Special Pair in the various electronic states. In cation radical state P•+ reflecting the HOMO, techniques as EPR, ENDOR and solid-state photo-CIDNP NMR show more unpaired electron spin density on PL than on PM4,5,6,7. The localization of the LUMO mainly on cofactor PM, from which the electron transfer occurs, has been explored by the photo-CIDNP MAS NMR analysis of the donor triplet state 3P8. This asymmetry is already present in the electronic ground-state of the supermolecule P, as demonstrated by differences in chemical shifts9,10,11,12,13.
Mutagenesis as well as theoretical studies established that the orientation and coordination C-31-acetyl groups of PL and PM affect the electronic structure and the redox potential of the special pair14,15,16. As there is no hydrogen bonding partner available, the x-ray structures and Raman spectroscopy data of the acetyl group of PM also show no involvement in any coordination as the Mg-O distance is about 3.3 ± 0.3 Å, while this distance shrinks to 2.4 Å in QM/MM studies “essentially forming a sixth ligand to the metal”17,18,19,20. On the other hand, Raman spectroscopic and QM/MM studies on specifically mutated RC showed that the orientation of the acetyl group of PL depends on the protonation state of His-L168 as it is either involved in a hydrogen bond to His-L168 or, if no hydrogen bond is available, located very close to the magnesium ion of PM (Fig. 2)18,21,22,23. It was therefore suggested that a re-orientation of the acetyl group of PL acts as a valve to block the electron back-transfer upon cleavage of the hydrogen bond to His-L168 and thereby re-tuning of the electronic properties of the Special Pair16. The acetyl group might therefore be involved in the reorientation of protein polar groups that lead to electric polarization effects during the radical-pair formation24,25,26. So far, no experimental evidence on the cleavage or the dynamics of the hydrogen bond is known, since no appropriate method with enough sensitivity was available.

View on the Special Pair with the coordinating histidines. Depending on the protonation state of His-L168, the acetyl group of PL can either be involved in a hydrogen bond (white) or coordinating the magnesium ion of PM (orange). The coordination of the acetyl group might change upon electron transfer (orange arrow), tuning the electronic properties of the Special Pair during its photocycle. The acetyl group of PM is always coordinated to the magnesium ion of PL since no other coordination partner is available.
Nuclear magnetic resonance (NMR) spectroscopy can be a major technique to probe local dynamics. The lack of sensitivity usually related to this method, can be overcome by the solid-state photo-CIDNP effect allowing to study the photosynthetic cofactors in their native environment27,28. The solid-state photo-CIDNP effect induces a non-Boltzmann nuclear spin distribution after a photo-cycle in all natural photosynthetic RC as well as in some flavin proteins8,29,30,31,32,33,34,35,36. The enhancement is sufficiently strong to observe particular carbon positions on the cofactors forming the spin-correlated radical pair (SCRP), which is constituted by the donor and the acceptor cofactors, even in entire plants without any further isolation37. During the lifetime of the SCRP, multiple coherent mixing mechanisms take place leading to observable nuclear hyperpolarisation in the electronic ground state on the donor and the acceptor molecules. These mechanisms can be explained by level anti-crossings and are termed three-spin mixing (TSM) and differential decay (DD)38,39,40,41,42. In case of the quinone-blocked RC of R. sphaeroides WT, the Special Pair acts as the donor and the ΦA is the acceptor (Fig. 3).

Generation of nuclear hyperpolarisation via the solid-state photo-CIDNP mechanism on the primary donor, the Special Pair P (red and blue), as well as on the primary acceptor ΦA (yellow) in quinone depleted bacterial RC. The phytyl chains are omitted for sake of clarity. After light excitation of P (A), an electron is transferred to the ΦA forming a spin correlated radical pair (SCRP) (B). In this state, nuclear hyperpolarisation is generated which can be observed in the electronic ground-state after recombination (C) (for details, see main text). Hyperpolarisation of P and ΦA is accumulated by a series of photocycles and decays by nuclear T1 relaxation (A/D).
In this study, we apply photo-CIDNP MAS NMR experiments to investigate the hydrogen-bond interaction between the acetyl group of PL and His-L168 by measuring the chemical shift anisotropy (CSA) of C-31 of the acetyl group and comparing it to DFT calculations to test whether the assumed cleavage of the hydrogen bond between PL and His-L168 can be experimentally verified.
Methods
Sample preparation
Cultures of Rb. sphaeroides WT were grown anaerobically in the presence of 1.0 mM [3-13C]-δ-aminolevulinic acid • HCl (3-ALA) (Buchem B.V., Apeldoorn, The Netherlands) for selective 13C isotope labelling of BChl a and BPhe as described before [9]. The RCs were isolated and the quinones were removed as described earlier [12]. Approximately 10 mg of RC protein complex embedded in LDAO micelles was used for the NMR experiment.
Solid-state photo-CIDNP MAS NMR
All NMR experiments have been performed with a double-resonance MAS probe at a Bruker AVANCE III spectrometer (Bruker-Biospin, Karlsruhe, Germany) operating at a proton Larmor frequency of 400.15 MHz. The probe was equipped with a light fiber to illuminate the sample during the measurement as described in ref.27. As illumination source, a 488-nm continuous-wave laser (Genesis MX488–1000 STM OPS-Laser-Diode System, Coherent Europe B.V., The Netherlands) operating at 1 W was used. The sample was packed in a clear 4-mm sapphire rotor and frozen in the dark at a slow spinning frequency of 800 Hz to ensure a homogenous sample distribution43.
If not stated differently, NMR experiments were performed at a spinning frequency of 8 kHz with a recycle delay of 4 s and a temperature of 247 K. The \(\pi /2\) 13C pulses were applied at radio-frequency (rf) field strength of 72 kHz, while the rf field strength of the heteronuclear SWf-TPPM decoupling was set to 100 kHz44. For the 1D experiment, 1024 scans were recorded with an acquisition time of 20 ms. The spectral width was set to 30 kHz, with the offset placed in the centre of the spectrum, if not stated otherwise. For the 2D INADEQUATE experiments, the SR26 sequence with an rf field strength of 52 kHz was applied45. One full SR26 cycle was used for DQ excitation and reconversion each, resulting in a total mixing time of 4 ms. To ensure a large spectral width of 46 kHz, STiC phase shifts were used46. The carrier was placed at 100 ppm. A total of 640 scans were averaged per each of the 100 t1 increments collected. The t2 acquisition time was set to 20 ms. Heteronuclear SWf-TPPM decoupling was used during the t1 and t2 acquisition44. The SUPER experiment was performed at a spinning frequency of 6 kHz leading to a rf field of 72.72 kHz for CSA recoupling. 192 scans were averaged during each of the four γ-integral points used, leading to a total number of scans of 768. A total of 32 t1-increments were recorded. The spectral width was set to 32258.1 Hz taking into account the scaling factor of 0.15547. The carrier was placed at 192 ppm. The z-filter for the γ-integral was set to 100 µs. Heteronuclear SWf-TPPM decoupling was used during the t1 and t2 acquisition44. Frequency discrimination in all 2D experiments was achieved using the States-TPPI method48.
The simulation of the CSA line shapes were carried out with SIMPSON49. The script can be found in the supplementary information.
DFT calculations
Geometry optimization calculations were based on the crystal structure of Camara-Artigas et al. (PDB: 1M3X) and were carried out with the program ORCA 4.0.1.250,51. The resolution of identity approximation in combination with the corresponding auxiliary basis set was employed to speed up the calculation based on the BLYP functional in combination with a def2-SVP basis set52,53,54. The empirical dispersion correction of Grimme 3rd version (with Becke/Johnson) was employed to consider dispersion interactions55,56. The protein environment was considered by the conductor-like polarisable continuum model (CPCM) for which a dielectric constant of ε = 4 was selected57. The two investigated structural models are shown in the Supplementary Figs S1 and S2. Both models differ in the protonation pattern at His-L168 where solely model A forms a hydrogen bond between His-L168 and PL, see Fig. S1. The chemical shifts were calculated with the BLYP functional in ADF 2017 using good numerical quality and no frozen core. The empirical dispersion correction of Grimme 3rd version (with Becke/Johnson) was employed to consider dispersion interactions. For protons a single-zeta basis set without polarization was applied. For the carbon atoms a double-zeta singly polarized Slater-type basis set (DZP) was used. Application of a triple-zeta singly polarized Slater-type basis set (TZP) lowered the obtained agreement in isotropic chemical shifts.
Results and Discussion
So far, the assignment of the resonances of the cofactor signals has been performed by comparison of the resonances with the relevant chlorophyll in solution state or by homonuclear DARR or RFDR experiments9,10,12,58,59,60,61. Comparison to model molecules can lead to wrong assignments due to the drastic effect of the protein matrix on the electronic structure of the Special Pair. Two-dimensional homonuclear experiments on samples with several tetrapyrrole macrocycles might struggle from signal overlap. We therefore apply INADEQUATE experiments to unambiguously assign the resonances as it has already been performed on the 5-ALA labelling pattern62. Since the distances are significantly larger in the 3-ALA labelling pattern (Fig. 4A), we used the SR26 sequences which recouples weak dipolar interactions efficiently45.

(A) Labelling pattern in bacteriochlorophyll a achieved by feeding 3-δ-aminolevulinic acid (3-ALA). The atom numeration is according to IUPAC. (B) 1D 13C spectra of 3-ALA labeled RC of R. sphaeroides WT. (C,D) Detailed views on the low (C) and high (D) field regions of the INADEQUATE spectrum of 3-ALA labeled RC of R. sphaeroides WT. The double-quantum peak of C-12 PM correlated to C-131 of PM (marked with an asterisk) was in the range of noise.
Figure 4B shows the 1D spectrum as well as a detailed view on the 2D INADEQUATE spectrum (Fig. 4C,D). As can be seen, a clear correlation between neighbouring labeled carbons up to two bonds apart can be established. This connectivity, the fact that PL carries more electron density leading to more shielding, as well as the already known assignments from the DARR spectra allow to assign all resonances unambiguously as shown in Table 1. In course of this, due to the observed correlation signal with C-7 of PL at 64 ppm, we assign the resonance at 19 ppm to C-81 of PL which has been erroneously denoted as C-71 in ref.12. We do not observe the resonances of C-81 of PL at 32.1 ppm, C-12 of PM at 128.8 ppm and C-18 of PM at 50.9 ppm as it was reported earlier12. This might be due to the strong field dependence of the solid-state photo CIDNP effect and the different magnetic fields used for both experiments63,64. Nevertheless, the SR26 sequence shows a good performance and allows for recoupling over about 2.6 Å (i.e., two bonds) making it suitable for even sparsely labeled samples as they are used in photo-CIDNP MAS NMR.
Hence, the two C-31-acetyl carbons of PM and PL have different isotropic chemical shifts, occurring at 194.5 (PL) and 196.3 ppm (PM), pointing towards a different chemical environment as, for example, that the acetyl group of PL has hydrogen bond interaction with His-L168, while the acetyl group of PM coordinates the magnesium of PL. To obtain further insight into the chemical environment, we investigated two different DFT models in which His-L168 is protonated at either the \(\tau \) or the \(\pi \) position (Fig. 5). Depending on the protonation state of His-L168, the acetyl group of PL is either involved in a hydrogen bond to His-L168 (model A) or it coordinates to the magnesium ion of PM (model B).

Detailed view on the acetyl group of PM (blue) and PL (pink) in the two DFT models. On the left-hand side (model A), His-L168 is protonated in the τ-position forming a hydrogen bond of 1.6 Å to the acetyl group of PL suggesting a moderate hydrogen bonding interaction. The acetyl group of PM does not have a hydrogen bonding partner and is therefore coordinated to the magnesium ion of PL. On the right-hand side (model B), His-L168 is protonated in the π-position. Lacking a partner for hydrogen bonding, the acetyl group of PL coordinates to the magnesium ion of PM. The acetyl group of PM is always coordinated to the magnesium ion of PL. The extended presentations of the two models are shown in the Supplementary Information (Supplementary Figs S1 and S2).
To verify the existence of the hydrogen bond and to explore possible dynamics, we measured the chemical shift anisotropy (CSA) pattern of both groups via the SUPER technique47. In highly enriched samples, SUPER reintroduces homonuclear dipolar interactions which in conjunction with J-coupling leads to broadening of the CSA patterns at slow spinning speeds47,65. In our case, moderately fast spinning, only very few labels, weak dipolar interactions (~390 Hz) and the absence of J-couplings should not lead to significant broadening of the CSA pattern as it was verified by SIMPSON simulations (Supplementary Figure S4).
Figure 6 shows the experimental powder patterns of C-31 in the acetyl groups of PL and PM as well as their simulations which matched the experimental data best. Table 2 shows the experimentally obtained isotropic and anisotropic chemical shift values of PL and PM compared to the theoretically obtained isotropic and anisotropic chemical shift values obtained from the DFT calculations where His-L168 is protonated either in τ- or π-position (model A or B). The difference in the principal values of the CSA (δ11-δ33) of the two molecules show an unequal coordination mode of the two acetyl groups suggesting a hydrogen bond between the acetyl group of PL and His-L168.

Cross section from the experimental 2D SUPER spectra (top) and the best fit (bottom) patterns of the C-31-carbon in the acetyl groups of PM and PL. The principal values of the best fit are summarized in Tab. 2 in the “Experiment PL” and “Experiment PM” column.
The observed experimental anisotropy values \({\delta }_{aniso}\) of both powder patterns (\(12\,\mathrm{kHz}\,\cong 120\,\mathrm{ppm}\) (PL) and \(11.3\,\mathrm{kHz}\,\cong 112\,\mathrm{ppm}\) (PM)) are larger than the anisotropy values of the carboxylate group in glycine (\({\delta }_{aniso}\cong 7.5\,{\rm{kHz}}\)) providing strong evidence for the high rigidity of the system47. Motions with a correlation time \({{\rm{\tau }}}_{{\rm{c}}}\ll 85\,\mu s\,\,\) would lead to an averaging of the CSA, which is in the time scale of multiple photocycles in RC of R. sphaeroides WT66. Since the sample is under continuous illumination and therefore passes multiple photocycles during each scan, the size of the anisotropy implies that the acetyl group, if there is any structural change related to this group, does not remain changed on the timescale of nanoseconds or longer. If the acetyl group of PL would be changing its orientation, the movement in both directions need to be on a ps time scale that is not observable with this experiment.
The calculated isotropic chemical shifts of C-31 of PL show a reasonable agreement for model A. If His-L168 is protonated in the π-position (model B), the calculated isotropic chemical shift is off by about 10 ppm as C-31 of PL is coordinated to the magnesium ion of PM. The isotropic chemical shift as well as the principal values of the chemical shift anisotropy of C-31 of PM are independent of the protonation state of His-L168. We note, however, that the calculated values are off by about 5 ppm which is within the expected error of the employed approach67. Unfortunately, we are limited to GGA calculations due to the size of the system. Since the principal CSA values are caused by the electronic environment of the observed nucleus, the differences in the principal CSA values might therefore also be caused by differences in geometry of the model compared to the experimental case which is assumed to be close to the crystal structure. In this case, the high accuracy of the NMR data might be used to recalculate the orientation of the acetyl group and therefore for refinement of the arrangement of the cofactor in the protein pocket.
The findings are also in agreement with the observations of Li and Hong stating that the π-tautomer of histidine is only formed as an anionic tautomer at high pH and is metastable in the presence of water suggesting a short lifetime68. Hence, a stabilization of the π-tautomer of His-L168 can only be achieved by further metal-ion coordination or H-bonding, which is implausible in this case68,69. Therefore, we assume that His-L168 is protonated in the τ-position.
Conclusion
We used the selectivity and the strong enhancement of solid-state photo-CIDNP MAS NMR to identify and probe the dynamics of the two acetyl groups in the Special Pair of R. sphaeroides WT by measuring the isotropic chemical shifts and principal values of their CSA tensors. In conjunction with DFT calculations, we showed that a rigid hydrogen bond between His-L168 and the acetyl group of PL is present. The high values of the reduced anisotropy of the CSA of both acetyl groups imply that they are not changing their orientation on the time scale of ns to µs. This suggests that if the acetyl-group of PL is flipping to act as a valve preventing fast charge-recombination, the flip has to happen on the ps time scale after the light-induced electron transfer.
References
Deisenhofer, J. & Michel, H. In Molecular Mechanisms in Bioenergetics (ed. Ernster, L.) 103–120 (Elsevier Science Publishing, 1992).
Holzwarth, A. R. & Müller, M. G. Energetics and Kinetics of Radical Pairs in Reaction Centers from Rhodobacter sphaeroides. A Femtosecond Transient Absorption Study. Biochemistry 35, 11820–11831 (1996).
Céspedes-Camacho, I. F. & Matysik, J. In The Biophysics of Photosynthesis 141–170, https://doi.org/10.1007/978-1-4939-1148-6_5 (Springer New York, 2014).
Allen, J. P. et al. EPR, ENDOR, and Special TRIPLE measurements of P•+ in wild type and modified reaction centers from Rb. sphaeroides. Photosynth. Res. 99, 1–10 (2009).
Lendzian, F. et al. The electronic structure of the primary donor cation radical in Rhodobacter sphaeroides R-26: ENDOR and TRIPLE resonance studies in single crystals of reaction centers. BBA - Bioenerg. 1183, 139–160 (1993).
Rautter, J., Lendzian, F., Lubitz, W., Wang, S. & Allen, J. P. Comparative Study of Reaction Centers from Photosynthetic Purple Bacteria: Electron Paramagnetic Resonance and Electron Nuclear Double Resonance Spectroscopy. Biochemistry 33, 12077–12084 (1994).
Lubitz, W., Lendzian, F. & Bittl, R. Radicals, radical pairs and triplet states in photosynthesis. Acc. Chem. Res. 35, 313–320 (2002).
Thamarath, S. S. et al. Electron spin density distribution in the special pair triplet of rhodobacter sphaeroides R26 revealed by magnetic field dependence of the solid-state photo-CIDNP effect. J. Am. Chem. Soc. 134, 5921–5930 (2012).
Schulten, E. A. M. et al. 13C MAS NMR and Photo-CIDNP Reveal a Pronounced Asymmetry in the Electronic Ground State of the Special Pair of Rhodobacter sphaeroides Reaction Centers. Biochemistry 41, 8708–8717 (2002).
Prakash, S. et al. 13C Chemical Shift Map of the Active Cofactors in Photosynthetic Reaction Centers of Rhodobacter sphaeroides Revealed by Photo-CIDNP MAS NMR. Biochemistry 46, 8953–8960 (2007).
Daviso, E. et al. The electronic structure of the primary electron donor of reaction centers of purple bacteria at atomic resolution as observed by photo-CIDNP 13 C NMR. Proc. Natl. Acad. Sci. 106, 22281–22286 (2009).
Sai Sankar Gupta, K. B., Alia, A., de Groot, H. J. M. & Matysik, J. Symmetry break of special pair: Photochemically induced dynamic nuclear polarization NMR confirms control by nonaromatic substituents. J. Am. Chem. Soc. 135, 10382–10387 (2013).
Sai Sankar Gupta, K. B. et al. Spectral editing through laser-flash excitation in two-dimensional photo-CIDNP MAS NMR experiments. J. Magn. Reson. 246, 9–17 (2014).
Muegge, I. et al. Shift of the Special Pair Redox Potential: Electrostatic Energy Computations of Mutants of the Reaction Center from Rhodobacter sphaeroides †. Biochemistry 35, 8359–8370 (1996).
Apostolakis, J., Muegge, I., Ermler, U., Fritzsch, G. & Knapp, E. W. Free Energy Computations on the Shift of the Special Pair Redox Potential: Mutants of the Reaction Center of Rhodobacter sphaeroides. J. Am. Chem. Soc. 118, 3743–3752 (1996).
Wawrzyniak, P. K., Beerepoot, M. T. P., de Groot, H. J. M. & Buda, F. Acetyl group orientation modulates the electronic ground-state asymmetry of the special pair in purple bacterial reaction centers. Phys. Chem. Chem. Phys. 13, 10270–10279 (2011).
Ermler, U., Fritzsch, G., Buchanan, S. K. & Michel, H. Structure of the photosynthetic reaction centre from Rhodobacter sphaeroides at 2.65 å resolution: cofactors and protein-cofactor interactions. Structure 2, 925–936 (1994).
Hutter, M. C., Hughes, J. M., Reimers, J. R. & Hush, N. S. Modeling the bacterial photosynthetic reaction center. 2. A combined quantum mechanical molecular mechanical study of the structure of the cofactors in the reaction centers of purple bacteria. J. Phys. Chem. B 103, 4906–4915 (1999).
Wadsten, P. et al. Lipidic Sponge Phase Crystallization of Membrane Proteins. J. Mol. Biol. 364, 44–53 (2006).
Sebban, P. et al. pH Modulates the Quinone Position in the Photosynthetic Reaction Center from Rhodobacter sphaeroides in the Neutral and Charge Separated States. J. Mol. Biol. 371, 396–409 (2007).
Mattioli, T. A., Williams, J. A. C., Allen, J. P. & Robert, B. Changes in Primary Donor Hydrogen-Bonding Interactions in Mutant Reaction Centers from Rhodobacter sphaeroides: Identification of the Vibrational Frequencies of All the Conjugated Carbonyl Groups. Biochemistry 33, 1636–1643 (1994).
Mattioli, T. A., Lin, X., Allen, J. P. & Williams, J. C. Correlation between Multiple Hydrogen Bonding and Alteration of the Oxidation Potential of the Bacteriochlorophyll Dimer of Reaction Centers from Rhodobacter sphaeroides. Biochemistry 34, 6142–6152 (1995).
Hughes, J. M., Hutter, M. C., Reimers, J. R. & Hush, N. S. Modeling the Bacterial Photosynthetic Reaction Center. 4. The Structural, Electrochemical, and Hydrogen-Bonding Properties of 22 Mutants of Rhodobacter sphaeroides. J. Am. Chem. Soc. 123, 8550–8563 (2001).
Rubin, A. B. et al. Polarization effects in photosynthetic membranes. Int. J. Quantum Chem. 17, 587–593 (1980).
Trissl, H.-W., Bernhardt, K. & Lapin, M. Evidence for Protein Dielectric Relaxations in Reaction Centers Associated with the Primary Charge Separation Detected from Rhodospirillum rubrum Chromatophores by Combined Photovoltage and Absorption Measurements in the 1−15 ns Time Range †. Biochemistry 40, 5290–5298 (2001).
Sham, Y. Y., Muegge, I. & Warshel, A. The Effect of Protein Relaxation on Charge-Charge Interactions and Dielectric Constants of Proteins. Biophys. J. 74, 1744–1753 (1998).
Daviso, E., Jeschke, G. & Matysik, J. In Biophysical Techniques in Photosynthesis. Advances in Photosynthesis and Respiration (eds Aartsma, T. J. & Matysik, J.) 385–399, https://doi.org/10.1007/978-1-4020-8250-4_19 (Springer, Dordrecht, 2008).
Bode, B. E. et al. In Hyperpolarization Methods in NMR Spectroscopy (ed. Kuhn, L. T.) 1, 105–121 (Springer-Verlag Berlin Heidelberg, 2013).
Zysmilich, M. G. & McDermott, A. E. Photochemically Induced Dynamic Nuclear Polarization in the Solid-State 15N Spectra of Reaction Centers from Photosynthetic Bacteria Rhodobacter sphaeroides R-26. J. Am. Chem. Soc. 116, 8362–8363 (1994).
Zysmilich, M. G. & McDermott, A. E. Natural abundance solid-state carbon NMR studies of photosynthetic reaction centers with photoinduced polarization. Proc Natl Acad Sci USA 93, 6857–6860 (1996).
Matysik, J. et al. A set-up to study photochemically induced dynamic nuclear polarization in photosynthetic reaction centres by solid-state NMR. Indian J. Biochem. Biophys. 37, 418–23 (2000).
Alia et al. Photochemically induced dynamic nuclear polarization in photosystem I of plants observed by 13C magic-angle spinning NMR. J. Am. Chem. Soc. 126, 12819–12826 (2004).
Thamarath, S. S., Heberle, J., Hore, P. J., Kottke, T. & Matysik, J. Solid-state photo-CIDNP effect observed in phototropin LOV1-C57S by 13C magic-angle spinning NMR spectroscopy. J. Am. Chem. Soc. 132, 15542–15543 (2010).
Zill, J. C. et al. Photo-CIDNP in the Reaction Center of the Diatom Cyclotella meneghiniana Observed by13C MAS NMR the solid-state photo-CIDNP effect in a new kingdom of the tree of life. Zeitschrift fur Phys. Chemie 231, 347–367 (2017).
Zill, J. C. et al. 15N photo-CIDNP MAS NMR on both photosystems and magnetic field-dependent13C photo-CIDNP MAS NMR in photosystem II of the diatom Phaeodactylum tricornutum. Photosynth. Res. 1–21, https://doi.org/10.1007/s11120-018-0578-9 (2018).
Zill, J. C. et al. 15N photo-CIDNP MAS NMR analysis of reaction centers of Chloracidobacterium thermophilum. Photosynth. Res. 137, 295–305 (2018).
Janssen, G. J. et al. Photochemically induced dynamic nuclear polarization NMR on photosystem II: donor cofactor observed in entire plant. Sci. Rep. 8, 17853 (2018).
Jeschke, G. A new mechanism for chemically induced dynamic nuclear polarization in the solid state. J. Am. Chem. Soc. 120, 4425–4429 (1998).
Polenova, T. & McDermott, A. E. A Coherent Mixing Mechanism Explains the Photoinduced Nuclear Polarization in Photosynthetic Reaction Centers. J. Phys. Chem. B 103, 535–548 (1999).
Jeschke, G. & Matysik, J. A reassessment of the origin of photochemically induced dynamic nuclear polarization effects in solids. Chem. Phys. 294, 239–255 (2003).
Sosnovsky, D. V., Jeschke, G., Matysik, J., Vieth, H. M. & Ivanov, K. L. Level crossing analysis of chemically induced dynamic nuclear polarization: Towards a common description of liquid-state and solid-state cases. J. Chem. Phys. 144, 144202 (2016).
Sosnovsky, D. V. et al. Magnetic field and orientation dependence of solid-state CIDNP. J. Chem. Phys. https://doi.org/10.1063/1.5077078 (2019).
Fischer, M. R. et al. Carbon-13 magic angle spinning NMR study of the light-induced and temperature-dependent changes in Rhodobacter sphaeroides R26 reaction centers enriched in [4′-13C]tyrosine. Biochemistry 31, 11038–11049 (1992).
Thakur, R. S., Kurur, N. D. & Madhu, P. K. Swept-frequency two-pulse phase modulation for heteronuclear dipolar decoupling in solid-state NMR. Chem. Phys. Lett. 426, 459–463 (2006).
Kristiansen, P. E., Carravetta, M., Lai, W. C. & Levitt, M. H. A robust pulse sequence for the determination of small homonuclear dipolar couplings in magic-angle spinning NMR. Chem. Phys. Lett. 390, 1–7 (2004).
Märker, K., Hediger, S. & De Paëpe, G. Efficient 2D double-quantum solid-state NMR spectroscopy with large spectral widths. Chem. Commun. 53, 9155–9158 (2017).
Liu, S.-F., Mao, J.-D. & Schmidt-Rohr, K. A Robust Technique for Two-Dimensional Separation of Undistorted Chemical-Shift Anisotropy Powder Patterns in Magic-Angle-Spinning NMR. J. Magn. Reson. 155, 15–28 (2002).
Marion, D., Ikura, M., Tschudin, R. & Bax, A. Rapid recording of 2D NMR spectra without phase cycling. Application to the study of hydrogen exchange in proteins. J. Magn. Reson. 85, 393–399 (1989).
Bak, M., Rasmussen, J. T. & Nielsen, N. C. SIMPSON: A General Simulation Program for Solid-State NMR Spectroscopy. J. Magn. Reson. 147, (296–330 (2000).
Camara-Artigas, A., Brune, D. & Allen, J. P. Interactions between lipids and bacterial reaction centers determined by protein crystallography. Proc. Natl. Acad. Sci. 99, 11055–11060 (2002).
Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2, 73–78 (2012).
Lee, C., Yang, W. & Parr, R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789 (1988).
Weigend, F. & Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 7, 3297 (2005).
Weigend, F. Accurate Coulomb-fitting basis sets for H to Rn. Phys. Chem. Chem. Phys. 8, 1057 (2006).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (2010).
Grimme, S., Ehrlich, S. & Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 32, 1456–1465 (2011).
Takano, Y. & Houk, K. N. Benchmarking the Conductor-like Polarizable Continuum Model (CPCM) for Aqueous Solvation Free Energies of Neutral and Ionic Organic Molecules. J. Chem. Theory Comput. 1, 70–77 (2005).
Daviso, E. et al. Electron−Nuclear Spin Dynamics in a Bacterial Photosynthetic Reaction Center. J. Phys. Chem. C 113, 10269–10278 (2009).
Roy, E. et al. Characterization of the Primary Radical Pair in Reaction Centers of Heliobacillus mobilis by 13C Photo-CIDNP MAS NMR. Biochemistry 47, 4629–4635 (2008).
Janssen, G. J. et al. Observation of the solid-state photo-CIDNP effect in entire cells of cyanobacteria Synechocystis. Photosynth. Res. 104, 275–282 (2010).
Thamarath, S. S. et al. Whole Cell Nuclear Magnetic Resonance Characterization of Two Photochemically Active States of the Photosynthetic Reaction Center in Heliobacteria. Biochemistry 51, 5763–5773 (2012).
Najdanova, M., Gräsing, D., Alia, A. & Matysik, J. Analysis of the Electronic Structure of the Special Pair of a Bacterial Photosynthetic Reaction Center by 13C Photochemically Induced Dynamic Nuclear Polarization Magic-Angle Spinning NMR Using a Double-Quantum Axis. Photochem. Photobiol. 94, 69–80 (2018).
Prakash, S. et al. Magnetic field dependence of photo-CIDNP MAS NMR on photosynthetic reaction centers of Rhodobacter sphaeroides WT. J. Am. Chem. Soc. 127, 14290–14298 (2005).
Gräsing, D. et al. Field-cycling NMR with high-resolution detection under magic-angle spinning: determination of field-window for nuclear hyperpolarization in a photosynthetic reaction center. Sci. Rep. 7, 12111 (2017).
Hong, M. & Yao, X. Homonuclear decoupled chemical shift anisotropy in doubly labeled peptides by selective-pulse solid-state NMR. J. Magn. Reson. 160, 114–119 (2003).
Hoff, A. J. & Deisenhofer, J. Photophysics of photosynthesis. Structure and spectroscopy of reaction centers of purple bacteria. Phys. Rep. 287, 1–247 (1997).
Reiter, K., Mack, F. & Weigend, F. Calculation of Magnetic Shielding Constants with meta-GGA Functionals Employing the Multipole-Accelerated Resolution of the Identity: Implementation and Assessment of Accuracy and Efficiency. J. Chem. Theory Comput. 14, 191–197 (2018).
Li, S. & Hong, M. Protonation, tautomerization, and rotameric structure of histidine: A comprehensive study by magic-angle-spinning solid-state NMR. J. Am. Chem. Soc. 133, 1534–1544 (2011).
Haddad, K. C., Sudmeier, J. L., Bachovchin, D. A. & Bachovchin, W. W. alpha-Lytic protease can exist in two separately stable conformations with different His57 mobilities and catalytic activities. Proc. Natl. Acad. Sci. 102, 1006–1011 (2005).
Acknowledgements
D.G. would like to thank the Fonds der Chemischen Industrie for the granted scholarship. Generous support by the Deutsche Forschungsgemeinschaft is acknowledged. Deutsche Forschungsgemeinschaft (Grants MA4972-2/1 and 11/1). We thank ZIH Dresden for providing computational resources.
Author information
Authors and Affiliations
Contributions
Daniel Gräsing: performed experiments, wrote the paper. Katarzyna M. Dziubińska-Kühn: performed the DFT calculations. Stefan Zahn: designed the research, performed the DFT calculations, wrote the paper. A. Alia: prepared the sample, wrote the paper. Jörg Matysik: designed the research, wrote the paper.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gräsing, D., Dziubińska-Kühn, K.M., Zahn, S. et al. Studying hydrogen bonding and dynamics of the acetylate groups of the Special Pair of Rhodobacter sphaeroides WT. Sci Rep 9, 10528 (2019). https://doi.org/10.1038/s41598-019-46903-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-46903-4
This article is cited by
-
Photo-CIDNP in Solid State
Applied Magnetic Resonance (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
