
Abstract
Viral diseases of crustaceans are increasingly recognised as challenges to shellfish farms and fisheries. Here we describe the first naturally-occurring virus reported in any clawed lobster species. Hypertrophied nuclei with emarginated chromatin, characteristic histopathological lesions of DNA virus infection, were observed within the hepatopancreatic epithelial cells of juvenile European lobsters (Homarus gammarus). Transmission electron microscopy revealed infection with a bacilliform virus containing a rod shaped nucleocapsid enveloped in an elliptical membrane. Assembly of PCR-free shotgun metagenomic sequencing produced a circular genome of 107,063 bp containing 97 open reading frames, the majority of which share sequence similarity with a virus infecting the black tiger shrimp: Penaeus monodon nudivirus (PmNV). Multiple phylogenetic analyses confirm the new virus to be a novel member of the Nudiviridae: Homarus gammarus nudivirus (HgNV). Evidence of occlusion body formation, characteristic of PmNV and its closest relatives, was not observed, questioning the horizontal transmission strategy of HgNV outside of the host. We discuss the potential impacts of HgNV on juvenile lobster growth and mortality and present HgNV-specific primers to serve as a diagnostic tool for monitoring the virus in wild and farmed lobster stocks.
Similar content being viewed by others


Introduction
Viral pathogens and the diseases that they impart are a particularly significant source of production loss in the cultivation of crustaceans1. Despite the preponderance of known viruses in a wide range of crustacean hosts and their ubiquity in the aquatic environment, with abundance estimates of ~108 viruses per ml of productive coastal waters2, there have been no reported examples of naturally-occurring viruses infecting any clawed-lobster species (Decapoda; Nephropidae).
The currently unclassified Panulirus argus virus 1 (PaV1) is so far the only virus described from lobsters, in this case infecting mesodermal cells of the Caribbean spiny lobster (Panulirus argus) in Florida and throughout the Caribbean3. The infection, characterised by a milky colouration of the haemolymph and lethargy of the host, initially infects fixed phagocytes in the hepatopancreas (HP), prior to spreading to cells of the connective tissues. Since its initial discovery in juvenile life stages in the United States, PaV1 has been found in wild and cultured host populations throughout the Caribbean4. Remarkably, healthy lobsters demonstrate avoidance behaviours towards those infected with the PaV1 virus5. As with several other invertebrate pathogens, mortality rate is higher in animals with a carapace length of less than 16 mm6. Until now, there have been no reports of PaV1-like viruses or any other viruses in any species within the clawed lobster genera of the family Nephropidae. In experimental conditions, White spot syndrome virus (WSSV), a double stranded DNA (dsDNA) virus of the family Nimaviridae, has been demonstrated to infect (and cause disease in) both American lobster (Homarus americanus)7 and the European lobster (H. gammarus)8 as well as numerous other decapod crustacean taxa9. However, WSSV has not been detected in wild or cultured Nephropidae.
Viral diseases have led to substantial bottlenecks to shrimp aquaculture production. Monodon baculovirus (MBV), the causative agent of spherical baculovirosis, was the first virus reported in penaeid shrimp10. Phylogenetic analyses and genomic reconstruction has since suggested that MBV be reclassified as Penaeus monodon nudivirus (PmNV) and be reassigned to the Nudiviridae11,12, a family of dsDNA viruses which to that point was exclusively comprised of viruses infecting insects. Although initially named to reflect a lack of occlusion body formation (large protein lattices which protect the bacilliform-shaped virions and facilitate transmission outside of the host), there are now multiple examples within the Nudiviridae where occlusion bodies have been observed, or where sequence and structural homologs of the polyhedrin gene have been found within the genome12,13,14. Seven fully sequenced virus species have been characterised as nudiviruses: Penaeus monodon nudivirus (PmNV)12; Gryllus bimaculatus nudivirus (GbNV), infecting the nymph and adult stages of several cricket species15; Heliothis zea nudivirus-1 (HzNV-1), a persistent pathogen of insect cell lines14; Helicoverpa (syn. Heliothis) zea nudivirus-2 (HzNV-2), the sexually transmitted corn earworm moth virus which can cause sterility in the host16; Oryctes rhinoceros nudivirus (OrNV), a biological control agent used to manage palm rhinoceros beetle populations17; Tipula oleracea nudivirus (ToNV) a causative agent of nucleopolydreosis in crane fly larvae13; and Drosophila innubila nudivirus (DiNV)18,19, which causes significant reductions to fecundity and lifespan18. Three further viruses isolated from metagenomic sequencing of Drosophila melanogaster (Kallithea virus20, Tomelloso virus and Esparto virus) have also been described as nudiviruses (Table 1). However, the genomes of these three Drosophila viruses have yet to be analysed with respect to their phylogenetic position. There is also evidence of ancestral nudivirus integration into the host genome (Nilaparvata lugens endogenous nudivirus (NleNV))21 and a sister group of the nudiviruses, the bracoviruses, associated with Braconid wasp hosts, where viral genes are also integrated into the host genome22. Finally, a large DNA virus infecting the hepatopancreas of the European brown shrimp, Crangon crangon has also been proposed as a putative member of the Nudiviridae albeit based upon limited genomic information23,24.
As part of a large UK-based lobster rearing study assessing the growth of hatchery-reared European lobsters in novel sea-based container culture (SBCC) systems (Lobster Grower, www.lobstergrower.co.uk), we conducted a histology-led health screening of a large cohort of individuals (n = 1,698), sampled at several time points throughout a multi-year production cycle. We observed a distinctive histopathology of the hepatopancreas of juvenile lobsters in both hatchery and sea container phases of production. Intranuclear inclusions appeared within the hepatopancreatocytes of affected individuals; later confirmed by transmission electron microscopy (TEM) as of viral aetiology. Genome assembly of PCR-free shotgun metagenomic sequences confirmed the presence of a novel member of the Nudiviridae; hereby named Homarus gammarus nudivirus, the first virus described infecting any clawed lobster genus. Here, we present the fully annotated genome of HgNV, comprising a single contiguous sequence, together with diagnostic primers and reference histology and ultrastructure to aid in future identification in natural and aquaculture settings. HgNV is now the second confirmed aquatic nudivirus.
Results
Histological sectioning reveals virus-associated pathology
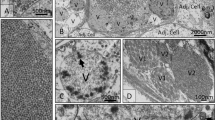
Lobsters did not appear to display any clinical signs of infection with HgNV. Histopathology of the virus infection was apparently limited to the tubule epithelial cells of the hepatopancreas (HP), observed in fibrilar (F) and reserve (R) cells. Infected cells contained hypertrophic nuclei occupied by a single, large eosinophilic inclusion. This inclusion displaced the host chromatin resulting in the latter’s emargination against the nuclear envelope (Fig. 1A,B). In some cases, this margination of the chromatin causes the formation of septa leading to the appearance of intranuclear compartmentalisation. Viral infection occurred either within the nuclei of isolated cells, within the closely opposing cells of a single tubule, within numerous cells of several closely opposed tubules, or generally throughout the tubules of the hepatopancreas. Often, epithelial cells containing virus-infected nuclei detached from the basement membrane of the tubule and were sloughed to the tubule lumen, presumably for excretion via the gut.

Homarus gammarus nudivirus (HgNV) infection within the hepatopancreas. (A) Section through the hepatopancreas, haemal sinus (HS) surrounds the tubules, cross section of the tubules shows a clear lumen (*). Infected nuclei within the epithelial cells of the tubules are enlarged, with emarginated chromatin and possess an eosinophilic inclusion body (white arrows). Infected cells (black arrow) may be sloughed into the lumen of the tubules. H&E Stain. Scale bar = 50 µm. (B) Infections can be seen within multiple epithelial cells, infected nuclei appearing larger than uninfected nuclei. Margination of the chromatin can form septa leading to the appearance of discrete intranuclear compartmentalisation (arrow). H&E Stain. Scale bar = 20 µm. (C,D) HgNV-specific DNA polymerase probe hybridised to infected nuclei (arrows) within epithelial cells of the hepatopancreas. In-situ hybridisation. Scale bar = 100 µm, 50 µm respectively. (E) Nucleus from a HgNV infected cell containing rod-shaped virions. Virions accumulate at the periphery of the nuclear membrane (arrow), TEM. Scale bar = 500 nm. (F) Longitudinal (white arrow) and transverse sections (black arrow) of HgNV virions within the nucleus. Virions possess an electron dense nucleocapsid surrounded by a trilaminar membrane (envelope). The rod shaped nucleocapsid appears to bend within the envelope forming a “u” or “v” shape in some cases (line arrows). TEM. Scale bar = 500 nm.
Infection prevalence in hatchery and sea-based juvenile lobsters
Intranuclear inclusions were observed in 12.72% of all samples processed for histology (145/1140) across the two-year sampling period. In sea-based lobsters the prevalence of intranuclear inclusions was highest at 39 weeks post deployment (17%) (Fig. 2). At this time point, the percentage of individuals displaying histological signs of viral aetiology was the same in both the hatchery and sea-based populations. However, whereas intranuclear inclusions were not evident in sea-based lobsters at 104 weeks (0%), prevalence had peaked in hatchery reared lobsters (53%) at this time (Fig. 2). Prevalence was generally observed to be higher in hatchery-based individuals compared to those retained in SBCC systems.

Prevalence of intranuclear inclusions in sea-based and hatchery lobsters over 104 weeks. Proportion of surviving lobsters displaying histopathological signs of viral infection. Green triangles: hatchery-based animals. Blue circles: sea-based animals. Sample size indicated at each point.
Three of the 150 (52 week) sea-based lobsters tested were PCR positive for HgNV, all of which were histologically positive. An additional four sea-based samples, displaying histopathological signs of intranuclear inclusions, did not amplify with HgNV-specific primers. Of the 12 hatchery-based animals tested, five were PCR positive for HgNV, three of which were histologically positive. An additional hatchery-based sample showed histological signs of nuclear infection but did not test positive with PCR. In-situ hybridisation of the HgNV-specific amplicon probe confirms detection and demonstrates localisation of the target HgNV DNA polymerase gene in infected tissues (Fig. 1C,D).
Transmission electron microscopy (TEM) confirms the presence of viral infection
Transmission electron microscopy revealed the presence of masses of enveloped virions accumulated at the nuclear membrane and surrounding the virogenic stroma (Fig. 1E,F). Virions exhibited an electron-dense nucleocapsid showing a bacilliform morphology and were contained within an elliptical membrane. In some cases, the rod-shaped nucleocapsids appeared “u” or “v” shaped within the envelope (Fig. 1F). The mean length of the enveloped virions was 180.43 ± 16.9 nm, with a mean diameter of 136.07 ± 11.28 nm (n = 20). The mean length of the nucleocapsids was 154 ± 20 nm, with a mean diameter of 36 ± 4 nm, mean envelope width was 5.2 ± 0.2 nm (n = 20).
Complete genome assembly of candidate virus
The alignment of multiple independent assemblies produced a full genome consensus sequence of 107,063 bp (Accession: MK439999). Reassembling the concatenated reads from all samples, after mapping to the candidate consensus sequence, increased coverage to an average of 400.50× (SD: 65.16). The assembled contig of 107,063 bp is concordant with the size of other known nudivirus genomes, as is the estimated GC content of 35.34% (Table 1). REAPR detected no errors or breaks in the assembled genome. PCR confirmation and sequencing of reduced coverage areas revealed the presence of repeating units, which sometimes varied in copy number between independent samples. Sanger reads sequenced from three separate samples confirmed correct assembled sequence.
Tandem repeats associated with viral replication
The HgNV genome does not contain any A/T-rich, palindromic, homologous regions (hrs) that are known to support the origin of replication in baculoviruses and play important roles in viral transcription25,26. However, seven direct repeats (drs), ranging from 58.8 to 188 bp were detected (Table 2), two of which fall within protein coding regions. EcoRI centres or significant palindromic regions, both typical of hrs, were not detected within these repeating regions. However, dr1-dr4 are clustered within 3.3% of the entire genome; a region of 3,531 bp (Fig. 3). A cluster of drs also appear within the PmNV genome12.

HgNV circular genome plot. Visual representation of HgNV layout scaled to the complete 107 063 bp contiguous sequence. Outermost track shows GC content (%) across complete sequence. Dark blue track displays gene predictions localised to the forward strand, whereas light blue displays those on the reverse. The innermost track depicts direct repeat regions. Links highlight genes involved in similar functions; yellow - DNA replication and repair, red - nucleotide metabolism, green – RNA transcription, pale blue – per os infectivity, pink – packaging and assembly, and grey – apoptosis inhibition.
Open reading frame (ORF) prediction and genome annotation
Prokka predicted 101 protein coding regions in the HgNV genome. FGenesV0 and GeneMarkerS predicted 103 and 89 ORFs, respectively. Ninety-seven ORFs were supported by two or more programs and were distributed evenly across both strands (Fig. 3, Table 3); 49 on the plus strand and 48 on the minus. The gene density of the HgNV genome was estimated to be 1.10 per kb and 69% of ORFs aligned most closely with predicted genes from the PmNV genome.
The exact number of genes conserved across all the nudiviruses is somewhat unclear. However, re-analyses of all sequenced nudivirus genomes revealed a set of 21 core genes conserved between baculoviruses and nudiviruses13. The core genes were typically grouped into one of five functional groups: DNA processing, RNA transcription, per os infectivity, package and assembly and conserved genes of unknown function. The HgNV genome contained 7 genes involved in DNA processing; DNA polymerase, helicase, two copies of helicase2, integrase, fen-1 and ligase. Gene predictions similar to three of the four thymidine kinase (tk) genes involved in nucleotide metabolism were also found. All five core baculovirus/nudivirus genes involved in RNA transcription were found; p47, lef-4 (late expression factor), lef-5, lef-8 and lef-9. As were 8 genes involved in per os infectivity: pif-0 (p74), pif-1, pif-2, pif-3, pif-4 (19 k/odv-e28), pif-5 (odv-e56) and pif-6 (ac68) and pif-8 (vp91/p95). The 11K-like gene was also found27. HgNV contained less than half of the genes encoding packaging, assembly and release processes conserved amongst the Baculoviridae. These include a 38 k gene, p6.9, two copies of vlf-1, vp39, p33 (ac92) and ac-81. Similar to PmNV, HgNV also possessed 2 copies of the Iap genes involved in apoptosis inhibition. Furthermore, HgNV encoded sequences similar to PmNVorf99 and PmNVorf62, reported to be common in nudiviruses (Fig. 4). Other genes typically common to baculoviruses, including methyltransferase and two neighbouring copies of odv-e66, were also found in the HgNV genome.

HgNV homologs to conserved nudivirus sequences. Colours as in Fig. 3. *Fused to a single gene. **Multiple copy number. Shaded cells of second column indicate ‘core nudivirus genes’ shared with the Baculoviridae. + Reported present.
Promotor regions preceding ORF predictions
Analysis of the 300 bp region upstream of each ORF start codon revealed the presence of promotor motifs in all but 12 of the predicted coding regions (Table 3). Early promotors defined by a TATA box with or without E1 motifs were predicted for 73 ORFs. No E2 motifs were detected. Late (L) promoters were predicted for 59 ORFs with HzNV-1 specific late promoters predicted for two coding regions; HgNV_ORF23, exhibiting a protein kinase structural domain and HgNV_ORF63 coding for the p74 gene. A combination of early and late promoters were predicted to precede 47 potential coding regions.
Phylogenetic characterisation of HgNV
Single gene phylogenies using the DNA polymerase and helicase genes showed contrasting positioning of the Nudivirus and Baculovirus clades, however both grouped HgNV with PmNV, together with ToNV, HzNV-1 and HzNV-2 (Fig. 5A,B). Multigene analyses of all shared genes involved in transcription (lef-4, lef -5, lef -8, lef -9 and p47) and per os infectivity (pif-0, pif-1, pif-2, pif-3, pif-4, pif-5, pif-6) within the nudiviruses very robustly supported this grouping, with Maximum Likelihood (ML) bootstrap values of 100% and 98% respectively (Fig. 5C,D).

Single and multigene phylogenies of known nudiviruses. Maximum Likelihood analyses of nudivirus phylogeny, including whispovirus and baculovirus outgroups. Node labels indicate bootstrap support (%). (A) – Single gene phylogeny of DNA polymerase. (B) – Single gene phylogeny of DNA helicase. (C) – Multigene phylogeny of late expression factors (lef-4, lef-5, lef-8, lef-9 and p47). (D) – Multigene phylogeny of per os infectivity genes (pif-0, pif-1, pif-2, pif-3, pif-4, pif-5 and pif-6). DiNV – Drosophila innubila nudivirus, KNV – Kallithea virus (D. melanogaster), ENV – Esparto virus (D. melanogaster), TNV – Tomelloso virus (D. melanogaster), OrNV – Oryctes rhinoceros nudivirus, NleNV – Nilaparvata lugens endogenous nudivirus, GbNV – Gryllus bimaculatus, HzNV-1 – Heliothis zea nudivirus 1, HzNV-2 – Heliocoverpa (syn. Heliothis) zea nudivirus 2, HgNV – Homarus gammarus nudivirus, PmNV – Penaeus monodon nudivirus, ToNV - Tipula oleracea nudivirus, NeleNPV – Neodipirion lecontei nucleopolyhedrovirus, NeseNPV – Neodipirion sertifer nucleopolyhedrovirus, AcMNPV – Autographa californica multiple nucleopolyhedrovirus, AgseGV – Agrotis segetum granulovirus, and WSSV – white spot syndrome virus. Coloured clade groupings refer to proposed genera: yellow – Alphanudiviruses, pink – Betanudiviruses, green – Gammanudiviruses, blue – Deltanudiviruses, red – Baculoviruses, cream – Whispovirus.
Discussion
Here we provide the first description of a naturally-occurring virus infection of nephropid lobsters. The virus, Homarus gammarus nudivirus (HgNV) is a new species within the family Nudiviridae; a group of dsDNA viruses that infect arthropod (mainly insect) hosts. Histopathology and ultrastructure of HgNV is similar to numerous other bacilliform viruses described to infect Crustacea, wherein viral replication within the host nucleus displaces host chromatin and results in aberrant, hypertrophied nuclei, visible in routine histological preparations. In many cases, infected epithelial cells are sloughed off the basement membrane of the tubule into the lumen, for excretion via the faeces. It is important to consider that intranuclear inclusions may also be indicative of other pathogens as this may explain discrepancies in prevalence when comparing PCR and histology data. Furthermore, digestive tissues are known to contain inhibitors which can impact PCR success28,29. However, in-situ hybridisation confirms that HgNV is inducing this pathology in infected cells (Fig. 1C,D).
Comprehensive genome analysis of infected lobsters revealed that HgNV is most closely related to PmNV, a virus infecting the black tiger shrimp, Penaeus monodon. However, despite a high degree of conservation in gene order, the percentage identity of HgNV gene predictions to known annotations was fairly low, averaging just over 38%. Our Maximum Likelihood phylogenies were concordant with previously published trees, which indicate that PmNV may belong to a separate genus within the Nudiviridae; the Gammanudiviruses12. Our phylogenetic analyses show that HgNV also belonged to this clade and, together with PmNV, could represent a radiation of nudiviruses infecting diverse aquatic crustacean taxa. Based on the long branch lengths of the neighbouring lineages, it is likely that ToNV and both HzNV-1 and HzNV-2 belong to separate genera; provisionally referred to as Deltanudivirus and Betanudivirus respectively (Fig. 5). We also show that the newly sequenced Drosophila nudiviruses belong to the Alphanudivirus clade and present the most substantial nudivirus phylogeny to date. The multigene phylogeny of the late expression factors provides bootstrap support of 100% in all but one node (98%) (Fig. 5C).
Nudiviruses contain a distinct repertoire of genes involved in DNA processing, compared to the baculoviruses. Two lef genes and an alk-exo gene are absent in HgNV. Lef-1 has been shown to be associated with DNA primase activity which aids in polymerisation, and lef-2 is thought to stabilize the binding of lef-1 to the DNA molecule30, amplifying replication31. HgNV contains two copies of the helicase-2 gene which are also found in the PmNV genome (HgNV_ORF66, HgNV_ORF69). Both genes are predicted to contain features characteristic of helicase activity. An integrase gene is also common to all sequenced nudiviruses and is represented by HgNV_ORF42, which contains the phase_integrase domain involved in the integration of viral DNA into the host genome. This is noted to facilitate persistent infection of HzNV-1 in its host32.
Of the five core genes involved in RNA transcription, P47 (HgNV_ORF08) encodes a viral transcription regulator, involved in late stage infections, reported to make up one of the four subunits of RNA polymerase, whereas the four remaining lef genes are thought to regulate late and very late gene expression, and are named to reflect their synthesis during infection. In contrast, early gene expression is instead initiated by host-derived RNA polymerase33.
Baculovirus life cycles are typically split between occlusion-derived virus (ODV) and budded virus (BV) stages. Per os infectivity genes, conserved within the HgNV genome, are required for the infectivity of ODVs that facilitate the transmission of viral particles from one host to the next, whereas BVs spread virions to neighbouring cells. Pif-0,1 and 2 encode envelope proteins vital for oral infection and are thought to bind virions to the midgut cells34. However, pif-3 does not affect midgut binding. Pif-3 is instead hypothesised to interact with the viral cytoskeleton and play a role in translocation of the capsid35. It is believed that pif-1, 2, 3 and 4 form a multimolecular protein complex that is vital for oral infectivity, with other pif genes associating with the core complex at a lower affinity36. Pif-7¸originally described as the ODV-envelope protein Ac110, also associates with the complex but was not found in the HgNV genome. However, Pif-8, previously described as the structural protein vp91/p95, was detected (HgNV_ORF05) and is predicted to contain chitin-binding peritrophin-A domains. The peritophic membrane surrounds the food bolus and lines the gut of most crustaceans and serves to separate large particulate matter from the epithelial cells and limits the penetration of microbes37. HgNV also encodes a homolog to an 11 K protein noted to enhance oral infection. These 11 K proteins typically contain binding motifs common to mucins, peritrophins and chitinases and could facilitate midgut binding, typically occurring after the alkaline dissolution of the occlusion lattice38.
The reduction in genes encoding packaging, assembly and release is likely a reflection of the lack of occlusion bodies in the transmission strategy adopted by the Nudiviridae. HgNV_ORF47 encodes a 38K-like gene which mediates the dephosphorylation of the C terminus of the p6.9 gene; a gene responsible for the encapsulation of the viral genome. Although not detected through BLAST alignment, likely a result of its highly repetitive sequence, HgNV_ORF40 was identified as the p6.9 gene after alignment with other annotated sequences. Furthermore, alignment of the hypothesised PmNV coding region of the p6.9 gene to the HgNV genome corresponds to a region within the HgNV_ORF40 predicted ORF. Similarly to PmNV, HgNV shares two separate sequence homologs to the vlf-1 gene (HgNV_ORF43, HgNV_ORF80), responsible for very late gene expression and proper formation of the nucleocapsid39. HgNV also encodes a second major capsid protein: p33/ac92 (HgNV_ORF04). HgNV_ORF04 reports an Erv1_Alr feature, belonging to a family of sulfhydryl oxidases. Prior analyses and purification of ac92 suggests it is a flavin adenine dinucleotide (FAD) containing sulfhydryl oxidase40. The major viral capsid protein vp39 is thought to be a core baculovirus/nudivirus gene and was reportedly mislabelled as a 31K-like structural protein in PmNV (ORF_022). HgNV_ORF15 shares 33% identity across 99% of the PmNV_022 sequence and 21% identity across 91% of HzNV-1 ORF89 and HzNv-2 ORF52, also annotated in GenBank as 31K-like proteins. However, the similarity of HgNV_ORF15 with the vp39 genes of other nudiviruses is much lower. Protein domains could not be predicted to aid in its clarification.
The PmNV_099-like coding region is also shared amongst the nudiviruses and HgNV_ORF88 indeed shares 40% sequence identity with PmNV_099, which is described as ‘microtubule-associated-like’12,13. This gene could play a role in the rearrangement of the host nucleus during viral replication, whereby host chromatin is translocated to the inner nuclear membrane, a process thought to be dependent on viral interaction with host tubulin35. Much like ac92, HgNV_ORF88 is also predicted to contain the ERV/ALR sulfhydryl oxidase feature which can play a role in virion assembly by catalysing disulphide bond formation between cysteine residues41. In regard to HgNV gene predictions found in the baculoviruses, HgNV_ORF02 shares 35.86% identity to a methyltransferase annotated in the PmNV genome, hypothesised to be involved in viral RNA capping42. As is the case with PmNV, HgNV encodes two neighbouring odv-e66 predictions responsible for the trafficking of viral proteins during infection43. Odv-e66 was also reported as the first chondroitin lyase44. Chondroitin is an extracellular matrix polysaccharide and its degradation by pathogenic bacteria facilitates access to the target cell44. Chondroitin AC/alginate lyase Interpro features were also identified in HgNV_ORF24 and HgNV_ORF25.
There are 37 core genes reported to be conserved amongst the baculoviruses45. Assuming the 31K-like gene is in fact a vp39 homolog, HgNV encodes all 21 core baculovirus genes proposed to be conserved across the nudiviruses (Fig. 4). Nearly half of HgNV predicted coding regions were preceded by both early and late promoter regions (Table 3), suggesting plasticity in the way HgNV can regulate gene expression. However, as stated by Bezier et al.13 gene expression chronology should not be generalised to promoter motifs alone. Transcriptome analysis of the baculovirus AcMNPV failed to associate reliable sequence motifs with gene expression patterns46.
We did not observe evidence of occlusion body formation within our histological or TEM analyses. Similarly, we did not detect sequence homologs of the poly/gran gene, which encodes the protein that forms the structural lattice. Ingestion of occlusion bodies allows passage to the digestive tract, where alkalinity of the gut causes the proteinous lattice to dissolve, releasing the virions within and initiating infection33. Much like the baculoviruses, the nudiviruses surrounding HgNV (HzNV-2, ToNV and PmNV) can rely on occlusion bodies to facilitate transmission outside of the host. As it would seem that HgNV does not form occlusion bodies, it begs the question of how viral particles remain viable during horizontal transmission. An alternative infection strategy would be that HgNV persists as a latent virus within the host and its evolution has favoured the maintenance of low virulence, which subsequently translates to an increase in transmission through longer lasting infections, as infection doesn’t incapacitate the host. Viruses infecting cells of the digestive tract sloughed out of the animal may remain viable until the degradation of the excreted cell. The ingestion of faeces may therefore serve as possible route of transmission for HgNV47. Latency within the host is a shared strategy true of several other shrimp viruses and supported by field data relating to the infection of the marine shrimp Crangon crangon by a putative nudivirus, where prevalence can reach 100% in wild populations23,48,49. Alternatively, HgNV may persist in reservoirs outside of its currently known host.
Due to the short life-cycle and seasonal development of their host, insect viruses, like the baculoviruses, are unable rely on either latency or reservoir strategies35. Therefore, resistant occlusion bodies would ensure viability outside of the host and facilitate transmission to the next. However, compared to penaeid shrimp, lobsters have very long life-cycles (decades). As such, a virus infecting these animals can rely on latency and is not required to survive long periods within the environment. In further support of this theory, occlusion-derived viruses infecting the insect midgut rely on occlusion body-associated enhancins, or similar factors, that digest the chitin lining of the midgut and facilitate entry35. However, the hepatopancreas of the lobster is not chitinous50. Therefore, HgNV would not depend on OB-associated proteases to gain entry into hepatopancreatic cells. Slack and Arif (2007) hypothesise that baculovirus ancestors were not occluded and instead relied on alkaline proteolytic activation during infection. It is hypothesised that contrasting ecological niches occupied by the insect host life cycle, limit baculoviruses infection to larval stages15. Therefore, occlusion body-facilitated horizontal transmission is vital for its longevity within the environment. The non-occluded nudiviruses, however, have demonstrated their ability to infect adult life stages. Therefore evolutionary maintenance of occlusion body transmission offers little benefit over vertical transmission or latency within the aging host15.
The expanding diversity of the Nudiviridae suggests that lack of occlusion alone is not a distinguishing characteristic of these viruses; several occlude prior to horizontal transmission whereas others do not. It is therefore likely that other characteristics of the genome underlie the separation of the group from the baculoviruses. Little is known about the nudivirus lifecycle and so this, and the means by which they gain entry into the host cell and cause infection, may also serve as discernible features of the proposed genus.
We did not observe any accompanying clinical signs in HgNV-infected individuals. Evidence suggests a persistent asymptomatic virus may even offer benefit to the individuals within an infected population. Invertebrates lack a typical adaptive immune system, however, host cells infected with latent Hz-1 virus (HzNV-1) are resistant to a more virulent infection of the same virus via homologous interference32. Nevertheless, despite widespread latency within the Nudiviridae, many cause delayed development and eventually death13. Whether HgNV has an effect on growth development or mortality of the European lobster remains to be shown. Furthermore environmental and/or physiological stimuli can result in massive viral amplification which give even low virulent viruses the potential to cause mass mortalities within a population49. This may be of particular importance as invertebrate aquaculture grows in popularity. The increased prevalence of HgNV in hatchery vs SBCC lobsters suggests either that conditions within SBCC are not conducive to high prevalence (e.g. lower transmission potential) or, that lobsters infected with HgNV have higher mortality during early deployment and thus are not present at later stage sampling points. However, in relation to the latter, given that early mortality in SBCC and hatchery populations did not differ (data not shown), HgNV as a driver of mortality in SBCCs appears unlikely. It should be noted that recirculating systems likely serve as drivers for increased prevalence in older hatchery-reared stocks (52–104 weeks post deployment controls) and juvenile lobsters are not typically on-grown in hatchery environments for such extended periods. Further work on the role of HgNV in early life stage growth and mortality is now required.
Materials and Methods
Experimental design and sample collection
Over the period of July 2016 to April 2017, 14,507 hatchery-reared juvenile lobsters were deployed in SBCCs anchored off the coast of Cornwall (St. Austell Bay 50° 18.956 N, 4°44.063 W). The majority of those deployments (10,987 animals), including those used in the current study, occurred in the summer of 2016. Routine sampling (3, 6, 28, 39, 52 and 104 weeks post deployment) was carried out to monitor the incidence of disease in SBCC populations. In total, 1,698 animals were sampled over the 2-year period. Another set of lobsters were retained within the National Lobster Hatchery, Padstow UK, and sampled at the same time points, over this period. Carapace length and survival were measured at each time point. Upon sampling, larger animals (39–104 weeks post deployment) were anaesthetised on ice for several minutes prior to bisection through the dorsal line. One half was fixed in Davidson’s Seawater fixative for histological processing, the other fixed in molecular grade ethanol for sequence analysis. A small piece of hepatopancreas was removed and fixed for transmission election microscopy. Smaller animals (0–28 weeks post deployment) were fixed whole and underwent separate analyses.
Six juvenile lobsters displaying pronounced histopathology associated with viral infection were selected from hatchery and SBCC settings, allowing for comparative molecular and transmission election microscopy analyses. Five of the six animals had spent one to two years growing in controlled hatchery raceways. The remaining individual had spent 52 weeks in SBCC in the open sea.
Histopathology
Bisected lobsters were placed in to histological cassettes and fixed in Davidson’s Seawater Fixative for 24–48 h before transfer to 70% industrial denatured alcohol (IDA). Cassettes were processed using a Leica Peloris Rapid Tissue Processor and subsequently embedded in paraffin wax. Histological sections were cut using a rotary microtome set at 3 µm thickness, adhered to glass slides and stained using a standard haematoxylin and eosin (H&E) protocol. Slides were examined using a Nikon Eclipse light microscope and NIS imaging software at the International Centre of Excellence for Aquatic Animal Health at the Cefas Laboratory, Weymouth, UK.
Transmission electron microscopy
Hepatopancreas samples were fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.4) and later rinsed in 0.1 M sodium cacodylate buffer prior to processing. Post-fixation was carried out in 1% osmium tetroxide/0.1 M sodium cacodylate buffer for 1 h. Tissues were washed in three changes of 0.01 M sodium cacodylate buffer and were subsequently dehydrated through a graded acetone series before embedding in Agar 100 epoxy resin (Agar Scientific, Agar 100 pre-mix kit medium). Embedded tissues were polymerised overnight at 60 °C. Semi-thin (1–2 µm) sections were cut and stained with Toluidine blue for viewing with a light microscope to identify suitable target areas. Ultra-thin sections (70–90 µM) of targeted areas were mounted on uncoated copper grids and stained with 2% aqueous uranyl acetate and Reynold’s lead citrate51. Grids were examined using a JEOL JEM1400 transmission electron microscope and digital images captured using an AMT XR80 camera and AMT V602 software.
DNA extraction and sequencing
DNA for genomic reconstruction was extracted using the CTAB/phenol:chloroform extraction protocol as described in Holt et al.52. DNA for HgNV screens of HP tissue was extracted using the EZ1 Advanced XL and DNA Tissue Kit (Qiagen). Extracted DNA was cleaned with polyethylene-glycol (PEG) precipitation and submitted to the sequencing service at the University of Exeter, UK for shotgun library preparation using the TruSeq DNA PCR-Free Library Prep Kit. Pooled libraries underwent high-throughput sequencing using an Illumina Miseq (2 × 300 bp).
Sequence analysis
The raw Illumina paired-end sequence reads generated were quality-checked using FastQC v0.11.453 and subsequently trimmed to remove adapter sequences and low-quality bases using Trimmomatic v0.3654. Sequence reads were error-corrected and digitally normalised using bbnorm (part of BBMap v38.22)55 and reads of each sample were assembled individually with Unicycler v0.4.7 (using default parameters and–no_correct)56. Quality-trimmed paired reads from individual samples were also assembled de novo using the A5-miseq assembly pipeline57. Contigs representing putative HgNV were aligned using progressiveMauve (build date Jun 26 2018)58 in order to obtain a consensus sequence. In order to identify viral contigs, Prokka59 was used to identify protein-coding regions spanning the assembled contigs and these were subsequently annotated using the BLASTp algorithm of Diamond v0.7.960 and the full NCBI non-redundant (nr) protein database (20170515). Sequences representing dsDNA viruses were identified by visualising the Diamond output in MEGAN6 Community Edition v6.5.561 and corresponding contig sequences were extracted. Paired reads from all samples were subsequently mapped to the candidate genome contigs using BWA-MEM 0.7.12-r103962 and visualised with the Integrative Genomics Viewer (IGV) v2.3.6863 Assembly quality and accuracy were assessed with QualiMap v2.064 and REAPR (version 1.0.18)65. Predicted open reading frames (ORFs) were identified using three different tools, including Prokka, FGenesV0 (softberry.com) and GeneMarkS66 (amino acid size of 50, circular genome). ORFs that were supported by two or more programs were analysed further. In cases where multiple ORFs were predicted to overlap, the largest sequence was chosen. Supported ORFs were annotated using NCBI BLASTp and the full NCBI nr protein sequence database (20180803).
Tandem repeats within the final assembled genome were identified using the tandem repeats finder using default parameters67. Repetitive regions with an alignment score of 100 or more were further analysed for palindromic sequences using the MEME program and a minimal size of 20 bp68. Promoter sequences were located within 300 nucleotides upstream of ORF start codon predictions using the Geneious software package v.11.1.469. Early promoters contain TATA[AT][AT][AT] sequences. TATA boxes may also associate with CA[TG]T (E1) or CGTGC (E2) 20–40 nucleotides downstream. The baculovirus late promotor (L) corresponding to [ATG]TAAG and the HzNV-1-specific late promoter (HL) were also queried using the sequence TTATAGTAT.
A circular map of the HgNV genome was plotted using shinyCircos70. The assembled HgNV genome and corresponding ORF predictions are deposited in GenBank under the genome accession number MK439999.
Molecular confirmation of genome assembly
To resolve ambiguous regions of the genome assembly, primers were designed that span areas of lower coverage and INDEL queries. PCR amplification was performed in 50 µL volumes using 10 µL of Promega 5X Green GoTaq Flexi Buffer, 5 µL of MgCl2, 0.5 µL of each primer (Final concentration; 1 µM), 0.5 µL of DNTPs, 0.25 µL of GoTaq DNA Polymerase, 30.75 µL of molecular grade water and 2.5 µL of template DNA. Initial denaturation was carried out at 94 °C for 2 min. This was followed by 30 PCR cycles of denaturation at 94 °C for 1 min, annealing at 60 °C for 1 min and extension at 72 °C for 1 min, followed by a final extension at 72 °C for 5 min. Sequenced amplicons were aligned to the candidate genome using the multiple sequence alignment program (MAFFT Version 7)71 and assembly was assessed across query regions.
Diagnostic primers were constructed from the alignment of DNA polymerase gene sequences. HgNV_DNAPol_F1: 5′ACTTGAAGCTGTGCGTGACT 3′ and HgNV_DNAPol_R1: 5′ TGTATGTCTTGCGGCCCATT 3′ produce an amplicon of 383 bp and only anneal to HgNV when queried with Primer-BLAST and the nr database. PCR conditions were as above. Amplicons were cleaned with the GeneJET PCR Purification Kit (Thermo, US) and sequenced via the Eurofins TubeSeq service. HgNV_DNAPol primers were tested on 150 SBCC and 12 hatchery lobsters (sampled at 52 weeks post-deployment). Shrimp tissues infected with WSSV and a putative nudivirus were tested as negative controls and did not amplify.
Phylogenetic tree construction
Homologous target genes were aligned using the multiple sequence alignment program MAFFT Version 771; and the E-INS-I iterative refinement method. Multigene alignments were constructed by concatenating gene sequences prior to alignment. A maximum likelihood phylogenetic tree inference was constructed using RAxML-HPC BlackBox version 872 on the CIPRES Science Gateway73 using a generalised time-reversible (GTR) model with CAT approximation (all parameters estimated from the data).
In-situ hybridisation
An extended HgNV-specific DNA polymerase probe which spanned and the HgNV_DNAPol amplicon sequence was designed to optimise the hybridisation protocol. HgNV_DNAPol_ISH_1838f: 5′ AGATTGAGCAGAGTGTAGCCC 3′ and HgNV_DNAPol_ISH_2799R 5′ ACCTTCCGATGATAGTTCTTCC 3′ produce an amplicon of 961 bp. In-situ hybridisation of the extended HgNV probe was carried out following the protocol described by Bojko et al. 201974 using a 2X washing buffer (20X SSC, 0.2% BSA, 6 M Urea). However, NBT/NCIP incubation was limited to 15 minutes and slides were instead counterstained with 0.1% Fast Green solution.
Data Availability
Sequences have been deposited in GenBank under the BioProject PRJNA516791.
References
Stentiford, G. D. et al. Disease will limit future food supply from the global crustacean fishery and aquaculture sectors. J. Invertebr. Pathol. 110, 141–157 (2012).
Suttle, C. A. Viruses in the sea. Nature 437, 356 (2005).
Shields, J. D. A new pathogenic virus in the Caribbean spiny lobster Panulirus argus from the Florida Keys. Dis. Aquat. Organ. 59, 109–118 (2004).
Shields, J. D. Diseases of spiny lobsters: A review. J. Invertebr. Pathol. 106, 79–91 (2011).
Behringer, D. C., Butler, I. M. & Shields, J. Avoidance of disease by social lobsters. Nature 441(441), 421 (2006).
Butler, M. J., Behringer, D. C. & Shields, J. D. Transmission of Panulirus argus virus 1 (PaV1) and its effect on the survival of juvenile Caribbean spiny lobster. Dis. Aquat. Organ. 79, 173–182 (2008).
Clark, K. F., Greenwood, S. J., Acorn, A. R. & Byrne, P. J. Molecular immune response of the American lobster (Homarus americanus) to the white spot syndrome virus. J. Invertebr. Pathol. 114, 298–308 (2013).
Bateman, K. S., Munro, J., Uglow, B., Small, H. J. & Stentiford, G. D. Susceptibility of juvenile European lobster Homarus gammarus to shrimp products infected with high and low doses of white spot syndrome virus. Dis. Aquat. Organ. 100, 169–184 (2012).
Pradeep, B. & Rai, P. Biology, host range, pathogenesis and diagnosis of White spot syndrome virus. India. J. Virol. 23, 161–174 (2012).
Lightner, D. & Redman, R. A baculovirus-caused disease of the penaeid shrimp, Penaeus monodon. J. Invertebr. Pathol. 38, 299–302 (1981).
Wang, Y. & Jehle, J. A. Nudiviruses and other large, double-stranded circular DNA viruses of invertebrates: New insights on an old topic. J. Invertebr. Pathol. 101, 187–193 (2009).
Yang, Y. T. et al. The genome and occlusion bodies of marine Penaeus monodon nudivirus (PmNV, also known as MBV and PemoNPV) suggest that it should be assigned to a new nudivirus genus that is distinct from the terrestrial nudiviruses. BMC Genomics 15 (2014).
Bézier, A. et al. The genome of the Nucleopolyhedrosis-causing virus from Tipula oleracea sheds new light on the Nudiviridae family. J. Virol. 89, 3008–3025 (2015).
Cheng, C. et al. Analysis of the complete genome sequence of the Hz-1 virus suggests that it is related to members of the Baculoviridae. J. Virol. 76, 9024–9034 (2002).
Wang, Y., Kleespies, R. G., Huger, A. M. & Jehle, J. A. The genome of Gryllus bimaculatus nudivirus indicates an ancient diversification of baculovirus-related nonoccluded nudiviruses of insects. J. Virol. 81, 5395–5406 (2007).
Burand, J. P. et al. Analysis of the genome of the sexually transmitted insect virus Helicoverpa zea nudivirus 2. Viruses 4, 28–61 (2012).
Wang, Y., Bininda-Emonds, O. R. P., van Oers, M. M., Vlak, J. M. & Jehle, J. A. The genome of Oryctes rhinoceros nudivirus provides novel insight into the evolution of nuclear arthropod-specific large circular double-stranded DNA viruses. Virus Genes 42, 444–456 (2011).
Unckless, R. L. A DNA virus of drosophila. PLoS One 6 (2011).
Hill, T. & Unckless, R. L. The dynamic evolution of Drosophila innubila Nudivirus. Infect. Genet. Evol. 57, 151–157 (2018).
Webster, C. L. et al. The Discovery, Distribution, and Evolution of Viruses Associated with Drosophila melanogaster. PLOS Biology. 13(7), e1002210 (2015).
Cheng, R.-L. et al. Brown planthopper nudivirus DNA integrated in its host genome. J. Virol. 88, 5310–5318 (2014).
Bézier, A. et al. Polydnaviruses of braconid wasps derive from an ancestral nudivirus. Science. 323, 926–931 (2009).
Van Eynde, B. et al. Development and application of a duplex PCR assay for detection of Crangon crangon bacilliform virus in populations of European brown shrimp (Crangon crangon). J. Invertebr. Pathol. 153, 195–202 (2018).
Bateman, K. S. & Stentiford, G. D. A taxonomic review of viruses infecting crustaceans with an emphasis on wild hosts. J. Invertebr. Pathol. 147, 86–110 (2017).
Pearson, M., Bjornson, R., Pearson, G. & Rohrmann, G. The Autographa californica baculovirus genome: Evidence for multiple replication origins. Science (80-.). 257, 1382–1384 (1992).
Guarino, L. A. & Summers, M. D. Interspersed homologous DNA of Autographa californica nuclear polyhedrosis virus enhances delayed-early gene expression. J. Virol. 60, 215–23 (1986).
Beperet, I., Simón, O., Williams, T., López-Ferber, M. & Caballero, P. The ‘11K’ gene family members sf68, sf95 and sf138 modulate transmissibility and insecticidal properties of Spodoptera frugiperda multiple nucleopolyhedrovirus. J. Invertebr. Pathol. 127, 101–109 (2015).
El-maklizi, M. A., Ouf, A., Ferreira, A., Hedar, S. & Cruz-rivera, E. A localized PCR inhibitor in a porcelain crab suggests a protective role. peerJ. 2, e689 (2014).
Schrader, C., Schielke, A., Ellerbroek, L. & Johne, R. PCR inhibitors – occurrence, properties and removal. J. Appl. Microbiol. 113, 1014–1026 (2012).
Mikhailov, V. S. & Rohrmann, G. F. Baculovirus replication factor LEF-1 is a DNA primase. J Virol 76, 2287–2297 (2002).
Wu, C. P. et al. Autographa californica multiple nucleopolyhedrovirus LEF-2 is a capsid protein required for amplification but not initiation of viral DNA replication. J. Virol. 84, 5015–24 (2010).
Lin, C.-L. et al. Persistent Hz-1 virus infection in insect cells: Evidence for insertion of viral DNA into host chromosomes and viral infection in a latent status. J. Virol. 73, 128–139 (1999).
Acharya, A. & Gopinathan, K. P. Characterization of late gene expression factors lef -9 and lef -8 from Bombyx mori nucleopolyhedrovirus. J. Gen. Virol. 83, 2015–2023 (2015).
Haas-Stapleton, E. J., Washburn, J. O. & Volkman, L. E. P74 mediates specific binding of Autographa californica M nucleopolyhedrovirus occlusion-derived virus to primary cellular targets in the midgut epithelia of Heliothis virescens larvae. J. Virol. 78, 6786–6791 (2004).
Slack, J. & Arif, B. M. The baculoviruses occlusion-derived virus: virion structure and function. Adv. Virus Res. 69, 99–165 (2006).
Boogaard, B., van Oers, M. M. & van Lent, J. W. M. An advanced view on baculovirus per os infectivity factors. Insects 9 (2018).
Martin, G. G., Simcox, R. & Nguyen, A. Peritrophic membrane of the penaeid shrimp Sicyonia ingentis: structure, formation, and permeability. Biol Bull. 275–285 (2006).
Lapointe, R. et al. Characterization of two Autographa californica nucleopolyhedrovirus proteins, Ac145 and Ac150, which affect oral infectivity in a host-dependent manner. J. Virol. 78, 6439–6448 (2004).
Vanarsdall, A. L., Okano, K., George, F. & Rohrmann, G. F. Characterization of the role of very late expression factor 1 in baculovirus capsid structure and dna processing characterization of the role of very late expression factor 1 in baculovirus capsid structure and DNA processing. J. Virol. 80, 1724–1733 (2006).
Long, C. M., Rohrmann, G. F. & Merrill, G. F. The conserved baculovirus protein p33 (Ac92) is a flavin adenine dinucleotide-linked sulfhydryl oxidase. Virology 388, 231–235 (2009).
Hakim, M., Mandelbaum, A. & Fass, D. Structure of a baculovirus sulfhydryl oxidase, a highly divergent member of the Erv flavoenzyme family. J. Virol. 85, 9406–9413 (2011).
Koonint, E. V. Computer-assisted identification of a putative methyltransferase domain in NS5 protein of flaviviruses and 22 protein of reovirus. J. Gen. Virol. 74, 733–740 (1993).
Braunagel, S. C. et al. Trafficking of ODV-E66 is mediated via a sorting motif and other viral proteins: Facilitated trafficking to the inner nuclear membrane. PNAS. 101, 8372–7 (2004).
Kawaguchi, Y., Sugiura, N., Kimata, K., Kimura, M. & Kakuta, Y. The crystal structure of novel chondroitin lyase ODV-E66, a baculovirus envelope protein. FEBS Lett. 587, 3943–3948 (2013).
Garavaglia, M. J., Belén, A., Iserte, A., Belaich, N. & Daniel, P. The ac53, ac78, ac101, and ac103 genes are newly discovered core genes in the family Baculoviridae. J. Virol. 86, 12069–12079 (2012).
Chen, Y. et al. The transcriptome of the baculovirus Autographa californica multiple nucleopolyhedrovirus in Trichoplusia ni cells. J. Virol. 87, 6391–6405 (2013).
Lavalli, K. L. & Barshaw, D. E. Post-larval American lobsters (Homarus americanus) living in burrows may be suspension feeding. Marine Behaviour and Physiology. 15, 255–264 (2009).
Stentiford, G. D., Bateman, K. & Feist, S. W. Pathology and ultrastructure of an intranuclear bacilliform virus (IBV) infecting brown shrimp Crangon crangon (Decapoda: Crangonidae). Dis. Aquat. Organ. 58, 89–97 (2004).
Walker, P. J. & Winton, J. R. Emerging viral diseases of fish and shrimp. Veterinary Research 41 (2010).
Barker, P. L. & Gibson, R. Observations on the Feeding Mechanism, Observations on the feeding mechanism, structure of the gut, and digestive physiology of the European lobster Homarus gammarus (L.) (Decapoda: Nephropidae). J. Exp. Mar. Bio. Ecol. 26, 297–324 (1977).
Reynolds, E. S. The use of lead citrate at high pH as an electron-opaque stain in electron microscopy. J. Cell Biol. 17, 208–212 (1963).
Holt, C. et al. Halioticida noduliformans infection in eggs of lobster (Homarus gammarus) reveals its generalist parasitic strategy in marine invertebrates. J. Invertebr. Pathol. 154 (2018).
Simon, A. FastQC: A quality control tool for high throughput sequence data, http://WwwBioinformaticsBabrahamAcUk/Projects/Fast (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics. 30, 2114–2120 (2014).
Bushnell, B. BBMap short read aligner, and other bioinformatic tools (2016).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol. 13 (2017).
Coil, D., Jospin, G. & Darling, A. E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31, 587–589 (2015).
Darling, A. E., Mau, B. & Perna, N. T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 5 (2010).
Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics. 30, 2068–2069 (2014).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2014).
Huson, D. H. et al. MEGAN community edition - Interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 12, 1–12 (2016).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Robinson, J. T. et al. Integrative Genome Viewer. Nat. Biotechnol. 29, 24–6 (2011).
Okonechnikov, K., Conesa, A. & García-Alcalde, F. Qualimap 2: Advanced multi-sample quality control for high-throughput sequencing data. Bioinformatics 32, 292–294 (2015).
Hunt, M. et al. REAPR: A universal tool for genome assembly evaluation. Genome Biol. 14 (2013).
Besemer, J., Lomsadze, A. & Borodovsky, M. GeneMarkS: a self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 29, 1–12 (2001).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Bailey, T. L. et al. MEME S UITE: tools for motif discovery and searching. Nucleic Acids Res. 37, 202–208 (2009).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Yu, Y., Ouyang, Y. & Yao, W. ShinyCircos: An R/Shiny application for interactive creation of Circos plot. Bioinformatics 34, 1229–1231 (2018).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Miller, M. A., Pfeiffer, W. & Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. 2010 Gatew. Comput. Environ. Work. GCE 2010 (2010).
Bojko, J. et al. ‘Candidatus Aquirickettsiella gammari’ (Gammaproteobacteria: Legionellales: Coxiellaceae): A bacterial pathogen of the freshwater crustacean Gammarus fossarum (Malacostraca: Amphipoda). J. Invertebr Pathol. 156, 41–53 (2018).
Acknowledgements
This work was conducted within the Centre for Sustainable Aquaculture Futures, a joint initiative between the University of Exeter and the Centre for Environment, Fisheries and Aquaculture Science (Cefas) and funded by a Cefas-Exeter University Alliance PhD Studentship to CH, in collaboration with the National Lobster Hatchery (Padstow, UK). Work was also supported through the Agri-Tech Catalyst, Industrial Stage Awards, Lobster Grower 2 project funded by Innovate UK (102531) and BBSRC (BB/N013891/1). We would like to thank Dr. Charlie Ellis, Sally Cuthbertson, Emma Theobald, Jake Scolding, Matthew Green, Patrick Hooper and Ander Urrutia for aiding in sample collection. Furthermore, we would like to thank the staff at the Exeter Sequencing Service and Computational core facilities at the University of Exeter, supported by the Medical Research Council Clinical Infrastructure award (MR/M008924/1), Wellcome Trust Institutional Strategic Support Fund (WT097835MF), Wellcome Trust Multi User Equipment Award (WT101650MA) and BBSRC LOLA award (BB/K003240/1).
Author information
Authors and Affiliations
Contributions
C.C.H.: Sample collection, molecular analysis, in-situ hybridisation, bioinformatic analysis, data visualisation, writing of the manuscript. M.S.: Sample collection, histological processing and analysis, prevalence data. D.B.: Supervision to C.C.H., contributed to manuscript. K.S.B.: Histological/TEM imaging, contributed to manuscript. R.v.A.; Bioinformatic analysis. C.L.D.: Sample collection, supervision to C.C.H., contributed to manuscript. M.v.d.G.: Supervision to C.C.H., contributed to manuscript. S.R.: Sample collection, TEM processing. C.H.: Molecular analysis. G.D.S.: Supervision to C.C.H., histological analysis, contributed to manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Holt, C.C., Stone, M., Bass, D. et al. The first clawed lobster virus Homarus gammarus nudivirus (HgNV n. sp.) expands the diversity of the Nudiviridae. Sci Rep 9, 10086 (2019). https://doi.org/10.1038/s41598-019-46008-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-46008-y
This article is cited by
-
Spatial and temporal axes impact ecology of the gut microbiome in juvenile European lobster (Homarus gammarus)
The ISME Journal (2020)
-
A novel nudivirus infecting the invasive demon shrimp Dikerogammarus haemobaphes (Amphipoda)
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
