
Abstract
Due to the relation between lipids and Hepatitis C virus (HCV) life-cycle, we aimed to explore the existence of single nucleotide polymorphisms (SNPs) associated with low susceptibility to HCV-infection within lipid metabolism genes. This was a case-control study in three phases: (I) allelic frequencies of 9 SNPs within 6 genes were compared in 404 HCV-infected patients and 801 population controls; (II) results were validated in 602 HCV-infected individuals and 1352 controls; (III) results were confirmed in 30 HCV-exposed uninfected (EU) individuals. In phase I, only the LDLRAP1-rs4075184-A allele was differentially distributed in patients and controls (358 of 808 alleles [44.3%] and 807 of 1602 alleles [50.3%], respectively) (p = 0.004). In phase II, the A allele frequency was 547 of 1204 alleles (45.4%) in patients and 1326 of 2704 alleles (49.0%) in controls (p = 0.037). This frequency in EU was 36 of 60 alleles (60%), which was higher than that observed in patients from phase I (p = 0.018) and phase II (p = 0.027). The LDLRAP1-mRNA expression was lower in AA carriers than in non-AA carriers (median [Q1-Q3]: 0.85 [0.17–1.75] relative-units [ru] versus 1.71 [1.00–2.73] ru; p = 0.041). Our results suggest that LDLRAP1-rs4075184-A allele is associated with lower susceptibility to HCV-infection and with reduced expression of LDLRAP1-mRNA.
Similar content being viewed by others


Introduction
Few individuals exposed to the hepatitis C virus (HCV) remain uninfected without seroconversion1.
The low susceptibility or total resistance to HCV-infection could be determined genetically. As a biological proof of principle, an in vitro study reported that three naturally occurring single nucleotide polymorphisms (SNPs) in the peptidyl-prolyl isomerase A (PPIA) gene abrogated HCV replication2. In spite of this, no subject homozygous for the minor alleles of these mutations was found in a cohort of HCV-exposed uninfected (EU) individuals3.
Because of the intimate relation between the lipids and viral life cycle4, variations in genes encoding for proteins related to lipid metabolism could be involved in the susceptibility to HCV infection. In fact, it has been recently observed that the lipidomics profiling of EU individuals is distinct from HCV-infected individuals, even those who clear viremia, either spontaneously or after antiviral therapy5. However, there are scarce genetic association studies focused in genes involved in the lipid metabolism in this setting6. Specifically, rs59257 and rs6888 genetic markers within LDLR gene as well as rs10420349 polymorphism linked to APOB gene have been associated with HCV-infection susceptibility. But none of them has been validated so far. The identification of genetic markers related to the HCV-infection susceptibility in genes involved in the lipid metabolism could help to understand some aspects of the virus life cycle as well as, to design new preventive or therapeutic strategies against HCV-infection.
Because of this, the aim of this study was to explore the existence of genetic factors associated with the low susceptibility to HCV infection within genes related to the lipid metabolism.
Results
Study populations
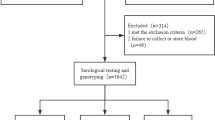
A total of 3251 individuals were included in this work to perform genetic association analyses in the three phases.
In phase I, the HCV-infected group I was originally composed of 442 HCV-chronically infected individuals. Among them, 38 (8.6%) were discarded because they showed genetic relationships with other individuals included in this group. Therefore, this dataset was finally constituted by 404 individuals. The control group I was constituted by 801 individuals.
In phase II, only 609 (48.4%) subjects from a total of 1258 HCV-chronically infected individuals attended in our Unit since 1999 had an available sample. Among them, 7 (1.1%) individuals were discarded because their ethnicity could not be probed. Therefore, 602 individuals were finally included in the HCV-infected group II. The control group II was constituted by 1352 individuals randomly selected from 6000 individuals collected in the genome research program at ACE foundation. The 1352 controls were those necessary to have 80% power to detect differences in allelic frequencies taking into account (i) 15% of genotype losses, (ii) the number of individuals included in the HCV-infected group II and, (iii) the difference in allelic frequencies described in phase I for the rs4075184 in controls and HCV-infected individuals.
In phase III, a total of 30 individuals were recruited as EU. All of these EU individuals were HIV-infected and users of illegal drugs during a median (Q1-Q3) of 11.5 (9.0–13.5) years. During that period, they reported to share injection devices during 6 (4.0–8.25) months. Moreover, 62 (52.5%) individuals from 118 subjects who were unequivocally identified as cases of spontaneous clearance of HCV infection were also included in this study.
Main characteristics of all those individuals are depicted in Table 1.
Phase I. Discovery
A total of 81 SNPs linked to genes involved in the lipid metabolism were selected from those previously genotyped in HCV-infected group I. Among them, only 16 (19.7%) SNPs linked to LDLRAP1, PCSK9, HMGCR, LPL, VLDLR and LDLR genes had genotype data in the control group I (Supplementary Table 3). However, the genotypic distribution of those 7 SNPs linked to LDLR gene, which were in high LD (d′ > 0.9) (Supplementary Fig. 2), were not in accordance with the HWE law in the control group I (Supplementary Table 4) and, consequently, they were discarded. Finally 9 (11.1%) SNPs were available for genetic association studies. The genotypic distributions and allelic frequencies of these SNPs in the control group I and in the HCV-infected group I are depicted in Table 2.
Only the SNP rs4075184 showed a significant different allelic distribution between the two groups after multiple testing correction. Specifically, there was a lower frequency of the A allele in the HCV-infected population (358 out of 808 alleles) than in the control population (807 out of 1602 alleles) (p = 0.004, OR = 0.78, 95%CI = 0.66–0.93) (Table 2).
To test if these differences were affected by age or gender, we performed separated stratified studies by gender or by the median age of the entire population included in phase I (48 years). Differences in the allelic frequencies on each strata did not reach the statistical significance. Moreover, the ORs and the allelic frequencies did not vary more than 8% from that observed in the study performed in the entire population (Fig. 1). After data adjustment by age the OR (95%CI) was 0.78 (0.64–0.94) (adjusted p = 0.012). The corresponding value after data adjustment by gender was 0.79 (0.67–0.95) (adjusted p = 0.013).

Comparison of rs4075184 A allele frequencies in controls and in HCV-infected individuals from phase I stratified by age 48 (A) or by gender (B).
Phase II. Validation
To validate the result obtained in phase I, the rs4075184 genetic marker was genotyped in a different sample of population controls (control group II) and HCV-infected patients (HCV-infected group II). The genotypic distributions in controls and HCV-infected individuals were 340 (25.1%) AA, 646 (47.8%) AG and 366 (27.1%) GG; and, 132 (22.0%) AA, 283 (47.0%) AG, and 187 (31%) GG, respectively (HWE p value > 0.10 in both groups). Accordingly, the frequency of rs4075184 A allele was 49.3% (1326 out of 2704 alleles) in the control group II and 45.4% (547 out of 1204 alleles) in the HCV-infected group II (p = 0.037, OR = 0.86, 95%CI = 0.76–0.99).
Phase III. Confirmation
To confirm the association of rs4075184 with the low susceptibility to HCV infection we analysed the genotypic distribution of this marker in 30 EU individuals. Nine (30%) individuals were AA, 18 (60.0%) were AG, and 3 (10%) were GG (HWE p value > 0.100). The frequency of rs4075184 A allele in this sample was 60.0% (36 out of 60 alleles). This frequency was significantly higher than that observed in the HCV-infected group I or that reached in the HCV-infected group II (Fig. 2). The frequencies of the rs4075184 A allele in EU were also numerically higher than those observed in both control groups, but the differences were not statistically significant (Fig. 2).

Comparison of rs4075184 A allele frequencies in controls groups, HCV-infected groups, HCV spontaneous resolvers and exposed uninfected individuals.
To explore if the rs4075184 A allele could be related to spontaneous viral clearance, this marker was also genotyped in 62 spontaneous resolvers of HCV infection. Among them, 14 individuals (22.6%) were AA, 30 (48.4%) were AG, and 18 (29.0%) were GG (HWE p value > 0.800). The A allele frequency was similar to those found in both HCV-infected groups. Although this frequency was not statistically different to that observed in EU individuals there was a trend to a significant statistical association (Fig. 2).
LDLRAP1 exons sequencing analyses
The coding region of LDLRAP1 gene was bi-directionally sequenced in 17 EU individuals: 9 homozygous for the rs4075184 A allele, 7 heterozygous and 1 individual with the GG genotype. There were two common SNPs; rs6687605 and rs28969504 located in exon 6 and 7 respectively, that showed complete LD with the rs4075184 genetic marker (d′ = 1). It was not detected any variation related to dysfunction of the LDLRAP1 protein.
LDLRAP1 mRNA expression analysis
The expression of LDLRAP1 mRNA gene in PBMCs was analysed in 32 anonymous voluntaries. Among them, the genotypic distribution of rs4075184, was 8 (25%) AA, 16 (50%) AG and 8 (25%) GG (HWE p value = 1.000). All these individuals were non-related healthy Spanish subjects. The expression of LDLRAP1 mRNA was lower in individuals carrying the AA genotype than in those carrying the AG or GG genotype (median [Q1-Q3]: 0.85 [0.17–1.75] relative units [ru] versus 1.71 [1.00–2.73] ru; respectively) (p = 0.041) (Fig. 3).

LDLRAP1 mRNA expression in peripheral blood mononuclear cells from healthy people according to the genotype of rs4075184.
Discussion
In this study, we found a genetic association of LDLRAP1 rs4075184 A allele with the low susceptibility to HCV infection and with reduced expression of LDLRAP1 mRNA.
LDLRAP1 is an adaptor that interacts with the cytoplasmic tail of low density lipoprotein receptor (LDLR), phospholipids, and component of the clatrin endocytic machinery10. LDLRAP1 is recruited to the plasma membrane after LDL binding facilitating endocytosis of the LDL-LDLR complex in hepatocytes11. Consequently, mutations in this gene can cause hypercholesterolemia (OMIM*605747).
LDLR acts as HCV-LDL complex receptor. Although, the role of LDLR in post-binding HCV entry is controversial12,13, its involvement in HCV infection is well established13,14. In addition, LDLR expression is stimulated by the HCV to promote the lipid uptake and facilitate viral proliferation. Consequently, it has been suggested that patients with familial hypercholesterolemia due to mutations in the LDLR gene may be protected against HCV infection15. Indeed, polymorphisms linked to LDLR have been associated with Hepatitis C viral load16 and with the capacity for responding to interferon-based antiviral therapy17,18,19,20. Thus, it could be hypothesized that a dysfunction of LDLRAP1 would affect to the capacity of LDLR to favour the entry of HCV or, alternatively, it could interfere with the metabolic requirements of the HCV life cycle.
We found a lower expression of LDLRAP mRNA in those individuals carrying the rs4075184 AA genotype. This finding was in accordance with that reported in the Genotype-Tissue Expression (GTEX) project (dbGAP Accesion phs000424.v7.p2) (www.gtexportal.org/home/snp/rs4075184). This fact could be the functional basis of the observed genetic association in this study. Interestingly, this SNP seems not related to spontaneous HCV clearance. It does not necessarily deny the role of LDLRAP1 expression in the low susceptibility to HCV infection, but it suggests the possible existence of other unknown mechanisms involved in this setting.
A possible explanation for the association observed is that rs4075184 A allele could be in LD with other causative SNP that would not be present in HCV-infected individuals but present in EU individuals. To explore this hypothesis we sequenced the coding region of the LDLRAP1 gene, mainly in those rs4075184 AA EU individuals. Nevertheless, we did not find any genetic alteration that could be related to LDLRAP1 dysfunction. In spite of this, we cannot discard the existence of mutations linked to rs4075184 A allele in other DNA regions that could confer protection against the HCV-infection. More functional experiments and DNA sequencing analyses are necessary to explain the basis of the association observed.
Polymorphisms within genes related to the lipid metabolism have been previously associated with low susceptibility to HCV infection such as the rs5925 and rs688 within LDLR gene7,8 and the rs1042034 within APOB gene9. Unfortunately, none of them were included in our study and, consequently, we were not able to validate those findings. Since these SNPs, as well as rs4075184, belong to genes involved in LDL and HCV uptake, it would be very interesting to explore whether there is a genetic interaction between them for better explaining the low susceptibility to HCV infection.
Recently, SNPs linked to RELA21 and CD20922 genes have been related to the low HCV-infection susceptibility. Although these works, as the ours, need replication in other series, taken together they suggest that resistance to HCV may be a multi-genetic trait, as was also suggested for resistance to HIV-infection23.
This study has some limitations. First, there were differences in the distribution of age and gender among groups of cases and controls included in phase I and II. Case/control allelic association analysis is more powerful than other ones, but it does not allow adjusting by covariables. To overcome this limitation we performed stratified analyses by age and gender in phase I. In these studies, the odds ratios obtained were almost identical to those obtained in the entire population. Indeed, in spite of the differences in age and gender between all groups, the rs4075184 A allele frequency was very similar among the two groups of controls, as well as among in the two groups of HCV-infected individuals included in both I and II phases, suggesting that neither age nor gender are confounding variables. Second, due to the scarce number of EU individuals, the sample size of these subjects in our study, like in others22, was relatively small to carry out genetic association analyses. However, they constituted a high quality population as HCV EU taking into account the high prevalence of coinfection among parenteral drug users infected by HIV in our population24. This is a strength of our work. Besides, in phase I and II we employed an indirect strategy to identify genetic markers associated with low susceptibility to HCV infection. Thus, we assumed that if a genetic factor has a role in the protection against viral infection, it should be differentially represented in the infected population with respect to its frequency in the general population. This strategy has been previously used by others in the setting of HIV or HCV infection susceptibility7,9,23. The inclusion of a sample of EU individuals as a confirmation phase enforces our findings. This is another strength of our study. Third, the inclusion of HCV-infected individuals in phase II was conditioned to the existence of stored blood or DNA samples. Similarly, individuals included as controls in phase II were aged individuals to be used as controls in genetic analyses focussed in Alzheimer’s disease. Therefore, selection bias cannot be excluded. In spite of this, (i) the homogeneity of the allelic frequencies obtained in this phase with respect to the same groups included in phase I and, (ii) the fact that LDLRAP1 has not been previously associated with Alzheimer´s disease (according to the GWAS catalogue data from NHGRI-EBI available at www.ebi.ac.uk/gwas), suggest that the allelic frequencies obtained in phase II are not deviated. Finally, the analysis of the LDLRAP1 expression was carried out in peripheral blood mononuclear cells (PBMCs). Because the target cells of HCV are hepatocytes, these analyses should have been performed in liver tissue. In spite of this, they were performed in a relative large number of people obtaining robust results.
In conclusion, our results suggest a role of LDLRAP1 gene in the low susceptibility to HCV-infection. Because the protective allele is associated with a reduced mRNA expression of LDLRAP1 gene, and taking into account that we did not found any DNA mutation in the coding sequence of this gene in EU individuals, this could be the mechanism underlying this protection against HCV infection. However, further studies are needed to explore the exact role of LDLRAP1 in this setting. If our results are confirmed, LDLRAP1 could be considered a target for preventing the HCV infection in individuals with high-risk habits.
Material and Methods
Design
This was a retrospective case-control study developed in three phases: discovery (phase I), validation (phase II) and confirmation (phase III). All individuals included in this study were non-related Spanish Caucasians.
Phase I
Patients and controls
Two genetic data sets from two groups of individuals were used in phase I: control group I and the HCV-infected group I. The control group I was constituted by a sample of the Spanish population which was recruited by a random sampling approach from a cross-sectional population-based epidemiological survey performed in eight different cities of Spain as previously described25. More details about the recruitment of these subjects are described elsewhere26. The HCV-infection status of these individuals was not known. All of them had been genotyped using the Affymetrix Nsp I 250 K chip (Affymetrix, Santa Clara, USA)25. In this group a high number of genotypes were also imputed as described elsewhere27. These individuals have been used as controls in several genome-wide association studies (GWASs)27,28,29. The HCV-infected group I was constituted by HCV chronically infected individuals who consecutively initiated a pegylated interferon plus ribavirin treatment from May 2000 to December 2010 in the Infectious Diseases Units of 5 tertiary care hospitals in Spain and who were genotyped for 116 SNPs16,17,30.
Single nucleotide polymorphisms selection
From the two available datasets, all those SNPs linked to genes related to the lipid metabolism that were previously genotyped in the HCV-infected group I and that were also genotyped or imputed in the control group I were selected (see supplementary Table 3). Genotypic data from these SNPs were extracted from those datasets using Plink software31. Those SNPs whose genotypic distribution did not fulfil the Hardy-Weinberg equilibrium law (p < 0.05) in any of these groups were discarded.
Statistical analyses
The program Graphical Representation of Relationships (GRR)32 was used to check sample relatedness and to correct potential sample mislabelling, duplications, or contaminations in phase I. Thus, due to the differences in the amount of SNPs genotyped in the two datasets, those individuals who showed an identity by state (IBS) > 0.1875 with other individuals in the control group I, or an IBS > 1.750 with other individuals in the HCV-infected group I, were removed.
Plink software31 was used to carry out standard case/control allelic association (1 degree of freedom) as well as to perform the Hardy-Weinberg equilibrium (HWE) test in phase I. In this phase, multiple testing correction was applied taking into account the number SNPs selected. Thus, the p-value threshold was established by the following formula: p = 0.05/number of SNPs. Finally, 9 SNPs were selected, establishing the p-value cut-off for statistical significance at 0.005. For the rest of comparisons the p-value threshold was set at 0.05.
Haploview software (version 4.1) (https://www.broad.harvard.edu/haploview/haploview) was employed to define the linkage disequilibrium (LD) blocks using the ‘solid spine of LD’ algorithm. Pair-wise LD was measured with Lewontin’s standardized deviation coefficient (d′).
The stratified analyses in phase I to calculate adjusted p and odds ratios (OR) values were performed by mean of Mantel-Haenszel test using the statistical calculator Statcalc from Epi Info™ 7 (available at https://www.cdc.gov/epiinfo/index.html).
Phase II
Patients and controls
Two groups of individuals were included in phase II: Control group II and HCV-infected group II. Control group II was constituted by individuals randomly selected from those that are being collected since 2012 under the genome research program of the ACE foundation (http://www.fundacioace.com/grace/) to be used as controls in genome wide association studies (GWASs) focussed in Alzheimer’s disease. The HCV-infection of these individuals was not known. All of them were cognitively healthy according to the neuropsychological test battery (NBACE) used in Fundació ACE33. More details about the recruitment of these subjects are described elsewhere34.
The HCV-infected group II was constituted by those individuals not included in the HCV-infected group I who were attended since 1999 due to a chronic HCV infection in the Infectious Disease Units of our Hospital and who received or not therapy against Hepatitis C. They were included if they had available a frozen blood or a DNA sample.
DNA isolation and genotyping
DNA was isolated from frozen whole blood samples using the Quick Pure Blood DNA extraction Kit (Macherey-Nagel, Düren, Germany), Magna Pure system (Roche Diagnostics, Mannheim, Germany) or Qiacube system (Qiagen, Hilden, Germany) in accordance with the manufacturers’ instructions.
The rs4075184 genetic marker was genotyped using the LightCycler 480 instrument (Roche Diagnostics, Mannheim, Germany). Amplification primers and sequence-specific probes are depicted in Supplementary Table 1. Real-time PCRs conditions and melting curve analyses were performed as previously described by Martinez-Mir et al.35 except for annealing temperature that was at 61 °C.
Statistical analyses
The sample size of the control group II was calculated with the ENE 3.0 software (GlaxoSmithKline, Madrid, Spain).
For the comparison of rs4075184 allelic frequencies between groups and to carry out the HWE test, we used the online resource at the Institute for Human Genetics, Munich, Germany (http://ihg.gsf.de).
Phase III
Exposed uninfected individuals and spontaneous resolvers of HCV infection
Those HIV-infected individuals who reported to have been using parenteral drugs with shared injection devices for longer than three months were included as EU subjects if they were seronegative for HCV. EU individuals are being collected from four tertiary hospital in Spain since 2016. Besides, individuals identified as spontaneous resolvers of HCV infection since 1999 in our hospital and who had available a frozen blood or a DNA sample were also included as an additional control sample. Spontaneous resolvers were considered to those individuals with positive anti-HCV antibodies who did not show HCV-ARN in plasma and who were never treated against HCV.
DNA isolation, genotyping and DNA sequencing
DNA was isolated from frozen whole blood samples using the Qiacube system (Qiagen, Hilden, Germany) in accordance with the manufacturers’ instructions.
The rs4075184 genetic marker was genotyped as described above.
Purified PCRs products covering the 9 exons of LDLRAP1 and part of the 5′ and 3′ untranslated regions (Supplementary Fig. 1) were bi-directionally sequenced using standard capillary electrophoresis techniques. Primers used for DNA amplification, which were the same used for sequencing, are depicted in Supplementary Table 2. All PCRs were performed in 20 μL of reaction volume containing 20 ng of DNA, 0.5 μM of each primer, 1x My Taq reaction buffer (Bioline, London, UK) and 0.5 Units of My Taq DNA Polymerase (Bioline). PCR conditions were as follows: denaturation at 95 °C for 3 min, followed by 35 cycles at 95 °C for 60 s, 61 °C for 60 s, and 72 °C for 60 s, and a final extension at 72 °C for 3 min. Purification of PCR products were performed using the Illustra ExoProstar (GE Healthcare, Chicago, Illinois, USA) in accordance with manufacturer’s instructions.
Statistical analyses
Data were analysed as described in phase II.
LDLRAP1 mRNA quantification
A total of 32 anonymous individuals from the staff of our hospital, who voluntarily donate a blood sample for participating in this study, were used to perform mRNA expression analyses. All of them were genotyped for the rs4075184 genetic marker as described above.
Total RNA was purified from 5 × 106 of PBMCs using Tripure reagent (Roche). RNA was quantified by spectrophotometry and dissolved at 1 μg/μl. Reverse transcription of total RNA was performed using the Superscript III (Invitrogen), following manufacturer’s instructions. All the RNA samples were also subjected to control reactions without reverse transcriptase to check for potential contamination. LDLRAP1 mRNA expression was normalized by the human β-actin mRNA expression as an endogenous control. Both quantitative PCRs were carried out in the same tube using the LDLRAP1 Taqman gene expression Assay ID Hs00296701_m1 (Applied Biosystems, Waltham, Massachusetts, USA) and the Human ACTB (Beta Actin) endogenous control (Applied Biosystems), following the manufacturer’s instructions. Each cDNA sample was amplified in triplicate wells. Parallel negative control experiments were done with reverse transcriptase negative cDNAs. The relative expression of LDLRAP1 mRNAs compared to endogenous β-actin controls were calculated using the MX-pro V4.0 software (Agilent Technologies, Santa Clara, California, USA) according to manufacturer’s instructions.
Ethics
This study was in compliance with the national legislation and it was performed according to the ethical guidelines of the Declaration of Helsinki. The study was approved by the Ethics Committee of the Hospital Universitario de Valme (internal reference number: 0422-N-16). Informed consent was obtained from all individuals before sampling.
Data Availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
References
Hagan, H., Pouget, E. R., Des Jarlais, D. C. & Lelutiu-Weinberger, C. Meta-regression of hepatitis C virus infection in relation to time since onset of illicit drug injection: the influence of time and place. Am J Epidemiol 168, 1099–1109, https://doi.org/10.1093/aje/kwn237 (2008).
von Hahn, T. et al. Hepatocytes that express variants of cyclophilin A are resistant to HCV infection and replication. Gastroenterology 143, 439–447.e1, https://doi.org/10.1053/j.gastro.2012.04.053 (2012).
Sugden, P. B., Cameron, B., Luciani, F. & Lloyd, A. R. Exploration of genetically determined resistance against hepatitis C infection in high-risk injecting drug users. J Viral Hepat e65–73, https://doi.org/10.1111/jvh.12232 (2014).
Bassendine, M. F. et al. Semin Immunopathol 35, 87–100, https://doi.org/10.1007/s00281-012-0356-2 (2013).
Shawa, I., Sheridan, D., Felmlee, D. & Cramp, M. Lipid Interactions Influence Hepatitis C Virus Susceptibility and Resistance to Infection. Clin Liv Dis 10, 17–20, https://doi.org/10.1002/cld.643 (2017).
Shawa, I. T., Felmlee, D. J., Hegazy, D., Sheridan, D. A. & Cramp, M. E. Exploration of potential mechanisms of hepatitis C virus resistance in exposed uninfected intravenous drug users. J Viral Hepat 24, 1082–1088, https://doi.org/10.1111/jvh.12720 (2017).
Li, H., Liu, Z., Han, Q., Li, Y. & Chen, J. Association of genetic polymorphism of low-density lipoprotein receptor with chronic viral hepatitis C infection in Han Chinese. J Med Virol 78, 1289–1295, https://doi.org/10.1002/jmv.20693 (2006).
Steba, G. S. et al. SNP rs688 within the low-density lipoprotein receptor (LDL-R) gene associates with HCV susceptibility. Liver Int 39, 463–469, https://doi.org/10.1111/liv.13978 (2019).
Harada, R. et al. APOB codon 4311 polymorphism is associated with hepatitis C virus infection through altered lipid metabolism. BMC Gastroenterol 18, 24, https://doi.org/10.1186/s12876-018-0747-5 (2018).
Garuti, R. et al. The modular adaptor protein autosomal recessive hypercholesterolemia (ARH) promotes low density lipoprotein receptor clustering into clathrin-coated pits. J Biol Chem 280, 40996–41004, https://doi.org/10.1074/jbc.M509394200 (2005).
Sirinian, M. I. et al. Adaptor protein ARH is recruited to the plasma membrane by low density lipoprotein (LDL) binding and modulates endocytosis of the LDL/LDL receptor complex in hepatocytes. J Biol Chem 280, 38416–38423, https://doi.org/10.1074/jbc.M504343200 (2005).
Albecka, A. et al. Role of low-density lipoprotein receptor in the hepatitis C virus life cycle. Hepatology 55, 998–1007, https://doi.org/10.1002/hep.25501 (2012).
Yamamoto, S. et al. Lipoprotein Receptors Redundantly Participate in Entry of Hepatitis C Virus. PLoS Pathog 12, e1005610, https://doi.org/10.1371/journal.ppat.1005610 (2016).
Molina, S. et al. The low-density lipoprotein receptor plays a role in the infection of primary human hepatocytes by hepatitis C virus. J Hepatol 46, 411–419, https://doi.org/10.1016/j.jhep.2006.09.024 (2007).
Ye, J. Hepatitis C virus: a new class of virus associated with particles derived from very low-density lipoproteins. Arterioscler Thromb Vasc Biol 32, 1099–1103, https://doi.org/10.1161/ATVBAHA.111.241448 (2012).
Caruz, A. et al. Association of low-density lipoprotein receptor genotypes with hepatitis C viral load. Genes Immun 15, 16–24, https://doi.org/10.1038/gene.2013.56 (2014).
Neukam, K. et al. Variations at multiple genes improve interleukin 28B genotype predictive capacity for response to therapy against hepatitis C infection. AIDS 27, 2715–2724, https://doi.org/10.1097/01.aids.0000432459.36970.a9 (2013).
Di Lello, F. A. et al. Effects of the genetic pattern defined by low-density lipoprotein receptor and IL28B genotypes on the outcome of hepatitis C virus infection. Eur J Clin Microbiol Infect Dis 32, 1427–1435, https://doi.org/10.1007/s10096-013-1894-9 (2013).
Pineda, J. A. et al. Low-density lipoprotein receptor genotyping enhances the predictive value of IL28B genotype in HIV/hepatitis C virus-coinfected patients. AIDS 25, 1415–1420, https://doi.org/10.1097/QAD.0b013e328348a7ac (2011).
Rivero-Juarez, A. et al. LDLr genotype modifies the impact of IL28B on HCV viral kinetics after the first weeks of treatment with PEG-IFN/RBV in HIV/HCV patients. AIDS 26, 1009–1015, https://doi.org/10.1097/QAD.0b013e3283528b1c (2012).
Tian, T. et al. Genetic variations in NF-kappaB were associated with the susceptibility to hepatitis C virus infection among Chinese high-risk population. Sci Rep 8, 104, https://doi.org/10.1038/s41598-017-18463-y (2017).
Steba, G. S. et al. DC-SIGN Polymorphisms Associate with Risk of Hepatitis C Virus Infection Among Men who Have Sex with Men but not Among Injecting Drug Users. J Infect Dis 217, 353–357, https://doi.org/10.1093/infdis/jix587 (2018).
McLaren, P. J. et al. Association study of common genetic variants and HIV-1 acquisition in 6,300 infected cases and 7,200 controls. PLoS Pathog 9, e1003515, https://doi.org/10.1371/journal.ppat.1003515 (2013).
Serrano-Villar, S. et al. Decreasing prevalence of HCV coinfection in all risk groups for HIV infection between 2004 and 2011 in Spain. J Viral Hepat 22, 496–503, https://doi.org/10.1111/jvh.12353 (2015).
Gayan, J. et al. Genetic structure of the Spanish population. BMC Genomics 11, 326, https://doi.org/10.1186/1471-2164-11-326 (2010).
Lorenzo, C. et al. Was the historic contribution of Spain to the Mexican gene pool partially responsible for the higher prevalence of type 2 diabetes in mexican-origin populations? The Spanish Insulin Resistance Study Group, the San Antonio Heart Study, and the Mexico City Diabetes Study. Diabetes Care 24, 2059–2064 (2001).
Antunez, C. et al. The membrane-spanning 4-domains, subfamily A (MS4A) gene cluster contains a common variant associated with Alzheimer’s disease. Genome Med 3, 33, https://doi.org/10.1186/gm249 (2011).
Matesanz, F. et al. Genome-wide association study of multiple sclerosis confirms a novel locus at 5p13.1. PLoS One 7, e36140, https://doi.org/10.1371/journal.pone.0036140 (2012).
Real, L. M. et al. A colorectal cancer susceptibility new variant at 4q26 in the Spanish population identified by genome-wide association analysis. PLoS One 9, e101178, https://doi.org/10.1371/journal.pone.0101178 (2014).
Real, L. M. et al. A polymorphism linked to RRAS, SCAF1, IRF3 and BCL2L12 genes is associated with cirrhosis in hepatitis C virus carriers. Liver Int 34, 558–566, https://doi.org/10.1111/liv.12330 (2014).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575, https://doi.org/10.1086/519795 (2007).
Abecasis, G. R., Cherny, S. S., Cookson, W. O. & Cardon, L. R. GRR: graphical representation of relationship errors. Bioinformatics 17, 742–743, https://doi.org/10.1093/bioinformatics/17.8.742 (2001).
Alegret, M. et al. Cut-off scores of a brief neuropsychological battery (NBACE) for Spanish individual adults older than 44 years old. PLoS One 8, e76436, https://doi.org/10.1371/journal.pone.0076436 (2013).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet 51, 414–430, https://doi.org/10.1038/s41588-019-0358-2 (2019).
Martinez-Mir, A. et al. Genetic study of neurexin and neuroligin genes in Alzheimer’s disease. J Alzheimers Dis 35, 403–412, https://doi.org/10.3233/JAD-122257 (2013).
Acknowledgements
We would like to thank Elisabet Pérez, Paola Micaela Ozzano and Maria Jesús Alvarez-Ossorio for technical assistance. Professor Manuel Serrano Rios, is acknowledged for recruiting individuals representative of the Spanish population that constituted the control group I. This work was supported by grants from the Consejería de Salud de la Junta de Andalucía (PI-0001/2017), the Grupo de Estudio de Hepatitis Víricas from the Sociedad Española de Enfermedades Infecciosas y Microbiología Clínica (GEHEP-SEIMC) (GEHEP-012), the Plan Nacional de I + D + I cofinanced by ISCIII-Subdirección General de Evaluación and the Fondo Europeo de Desarrollo Regional (FEDER) (www.red.es/redes/inicio) (RD16/0025/0040, RD12/0017/0012), the Acción Estratégica en Salud, integrated in the Spanish National R + D + I Plan and financed by ISCIII-Subdirección General de Evaluación and the Fondo Europeo de Desarrollo Regional (FEDER- “Una manera de Hacer Europa”) (PI13/02434 and PI16/01861), the Ministerio de Economía Industria y Competitividad (SAF2016–8015-R), Fundación Bancaria “La Caixa” and Grifols SA (GR@ACE project). LMR and JM are the recipients of grants from the Servicio Andaluz de Salud de la Junta de Andalucía (C-0009-2015 and B-0037, respectively). JAP has received a research extension grant from the Programa de Intensificación de la Actividad de Investigación del Servicio Nacional de Salud Carlos III (I3SNS). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Author notes
A comprehensive list of consortium members appears at the end of the paper
Consortia
Contributions
Study concept and design: L.M.R., J.M., N.M., A.C., J.A.P. Acquisition of data: L.M.R., J.M., A.R.J., F.T., D.M., M.F., A.C.G., N.M., A.R., J.A.P. Analysis and interpretation of data: L.M.R., J.M., S.M.G., A.O., J.G.S., M.E.S., A.C., J.A.P. Drafting of the manuscript: L.M.R., J.M., N.M., J.A.P. Critical revision of the manuscript: All authors. Final approval: All authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Real, L.M., Macías, J., Rivero-Juárez, A. et al. Genetic markers of lipid metabolism genes associated with low susceptibility to HCV infection. Sci Rep 9, 9054 (2019). https://doi.org/10.1038/s41598-019-45389-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-45389-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
