
Abstract
The definition of the gene expression profile of genes encoding Ion Channels and Transporters (ICT-GEP) represents a novel and attracting aspect in cancer. We determined the ICT-GEP of Follicular Lymphoma (FL), and compared it with that of the more aggressive Diffuse Large B Cell Lymphoma (DLBCL). cDNA microarray data were collected both from patients enrolled for this study, and from public datasets. In FL the ICT-GEP indicated the overexpression of both the K+ channel encoding gene KCNN4, and SLC2A1, which encodes the Glut1 glucose transporter. SLC2A1 turned out to represent the hub of a functional network, connecting channels and transporters in FL. Relapsed FL patients were characterised by 38 differentially expressed ICT genes, among which ATP9A, SLC2A1 and KCNN4 were under-expressed, indicating a down-regulation of both excitability and glycolysis. A completely different profile of K+ channel encoding genes emerged in DLBCL accompanied by the over-expression of the fatty acid transporter-encoding gene SLC27A1 as well as of the metabolism regulator NCoR1. This indicates a change in excitability and a shift towards an oxidative metabolism in DLBCL. Overall, the ICT-GEP may contribute to identifying novel lymphoma biomarkers related to excitability and metabolic pathways, with particular relevance for drug resistant, relapsed FL.
Similar content being viewed by others


Introduction
The two most common Non-Hodgkin lymphomas (NHLs) in developed countries are diffuse large B-cell lymphoma (DLBCL) and follicular lymphoma (FL), collectively accounting for nearly half of all NHLs1,2. FL is a low-grade germinal centre-derived tumour, characterised by a prolonged clinical course, with frequent relapses after treatment, 10% of which will not respond to chemo-immunotherapy. Furthermore, over the course of their illness, roughly half FL patients develop a more aggressive disease, which may eventually transform into a DLBCL3,4.
The identification of the gene expression profile (GEP) has been proven to be a powerful clinical tool in several types of cancers5,6,7,8 including lymphomas9,10,11. In these, the GEP has strongly contributed not only to the understanding of neoplastic B-cell biology but also for the diagnostic and prognostic characterisation of the disease. The GEP of DLBCL is now well characterised and validated for clinical applications7,8. In particular, GEP studies accelerated our understanding of the molecular complexity of DLBCL, and led to the identification of two molecularly distinct forms of DLBCL with gene expression patterns indicative of different stages of B-cell differentiation, resembling either germinal center B-cells (GCB) or activated B-cells (ABC)7. The GEP of FL has also been intensely studied in the last ten years10,11,12,13,14, and several driver genes and signalling pathways involved in the pathogenesis and progression of the tumour have been identified9,10,11,12. It remains still unsolved whether the aggressiveness of transformed FL occurs from the prevalent growth of an existing subclone, or results from novel genetic/epigenetic hits in the neoplastic B cells, or even to changes in the immune cells present within the tumour microenvironment13,14.
Besides their undiscussed role in the regulation of many physiological functions in both excitable and not excitable cells, ion channels and transporters (ICTs) are emerging as novel cancer biomarkers15,16. Recently, an ICT-based signature has been produced for gliomas, breast and lung cancers16,17,18 and has been proposed as a novel tool to predict survival and clinical outcome. The expression and role of ion channels in lymphocytes, mostly T-cells, has been investigated since almost thirty years, leading to the uncontested role of different K+ and Ca2+ currents in the regulation of lymphocyte excitability, which in turn drives their activation19,20. Few studies have addressed the expression and role of ICTs in lymphoma, and all of them have led to the identification and functional characterization of single molecular entities7,21,22,23. Overall, no lymphoma ICT-GEP has been provided so far.
Based on these premises, the purpose of the present study was to determine the ICT-GEP of FL, to be compared with that of the more aggressive relapsed FL and DLBCL, with the aim to identifying different profiles related to disease progression and therefore their potential translational relevance. We studied both samples from a cohort of patients specifically enrolled for this study as well as public datasets.
Results
Analysis of differentially expressed (DE) genes in FL samples
We collected 54 consecutive diseased lymph node samples, 13 of which were diagnosed as FL and six as DLBCL. After RNA extraction, cDNA microarray analysis was applied only to those samples (11 FL and 2 DLBCL) whose RNA integrity number (RIN) was >6 (Supplementary Table S1). The clinico-pathological characteristics of the patients enrolled are shown in Table 1. FL samples displayed different histological grades, and only the subtype A of grade III was present. Besides the expected Bcl2 alterations24, all the samples showed Bcl6 over expression (indicated by the pathologists’ diagnosis and confirmed by microarray data, see Table 1 and Supplementary Table S2) and 6 out of 11 experienced disease relapse towards a similar FL disease. The two DLBCL patients belonged to IIIA and IVA stages, showed different response to therapy, and neither relapsed.
The DNA microarray analysis was first focused on FL samples: the GEP was determined, and compared to that of normal lymph nodes from pooled healthy donors. In particular, a gene was assumed to be DE when the corrected p-value was lower than 0.01 and the fold change (log2 fold change) was ≥2. Applying such thresholds, 3988 DE genes, mostly under expressed, were detected and classified in 3 different clusters: over expressed genes (n = 925), moderately under expressed genes (n = 1642) and highly under expressed genes (n = 1421) (Fig. 1; raw data are deposited into the Gene Expression Omnibus (GEO) database (series entry GSE126247) (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126247).

Heatmap of all the DE genes. Heatmap of 3,988 DE genes. DE genes were first filtered by removing all the genes that had an average expression level two times smaller or two times greater than control samples. After filtering, we obtained 3,988 DE genes that were used for cluster analysis by a Ward hierarchical clustering algorithm separately for samples and genes. To cluster samples, we used the matrix of the Pearson’s correlation coefficient; for genes, we used the matrix of the Euclidean distance. The cluster analysis and heat-map analysis were both performed using the R statistical environment. Clustering analysis revealed groups of genes and samples (reported on the bottom and expressing the tumour grade in brackets when available) with similar average expression levels, according to the colour key. Depending on the expression level, genes were segregated into 3 different clusters: over expressed genes (green, n = 925), moderately under expressed (dark red, n = 1642) and highly under expressed (brilliant red, n = 1421) compared with the normal lymph node.
No relevant difference in the stratification of the DE genes within the 11 FL samples emerged (see the upper dendrogram in Fig. 1), indicating a substantial homogeneity of the molecular characteristics of the samples, in agreement with the roughly homogenous clinical and pathological characteristic of our cohort. Microarray results were validated on those samples (nine) with enough residual RNA after the microarray analysis, performing RQ-PCR on some selected genes. The Pearson correlation coefficient values indicated a good correlation between the RQ-PCR and microarray expression data (Supplementary Table S3).
We next performed a functional annotation analysis (FAA) to identify the most altered biological processes. Particularly, looking at the Gene Ontology Biological Processes annotations (GOBP) we found that the DE genes are significantly associated to 474 groups (called “terms”) of potential functional distinction (Supplementary Dataset 1).
The functional annotation analysis (FAA) on the DE genes associated to the three clusters of the heatmap in Fig. 1, indicated that the biological processes associated to the over expressed DE genes were related to immune response, cell death, transport, chemotaxis and some peculiar signalling pathways such as TNF and NF-κB. Moderately under expressed genes were associated to biological processes related to cellular development, cell differentiation, cell motility and cytoskeleton organization, while the highly under expressed genes were associated with reproductive processes and cell cycle terms (Supplementary Datasets 2–4).
We then compared the gene expression profile of our FL cohort (henceforth addressed as the Florence Cohort) with other FL datasets, deposited into the GEO database (http://www.ncbi.nlm.nih.gov/geo/). Three different datasets (GSE32018, GSE9327 and GSE65135) that mostly matched our cohort’s characteristics were selected, and the microarray expression raw data were used to identify common differentially expressed genes. When comparing the profile of each dataset with that of the Florence Cohort, it emerged that the GSE65135 dataset had 641 DE genes, out of a total of 4151 DE genes, in common with the GEP of our cohort, hence displaying the higher similarity with the Florence Cohort (see the Venn diagram in Supplementary Fig. S1). The GSE65135 dataset was hence used for further comparisons.
Analysis of DE genes associated with the transporter classification database (TCDB)
We then performed a more focused analysis selecting, among the DE genes, the probes associated with ICT-encoding genes, according to the Transporter Classification Database (TCDB) (http://www.tcdb.org/hgnc_explore.php) as in25. Selecting those genes that presented an average expression level higher than 1 compared to the control, we identified 46 DE genes (Supplementary Table S4 and Supplementary Dataset 5) included in two main groups: those coding for transporters (n = 39; 85%) and those coding for ion channels (n = 7; 15%). Most of the DE genes encoded solute carriers (SLC, n = 25). Other categories included ATP-binding cassettes (ABC, n = 6), ATPases (n = 7), potassium channels (n = 5), calcium channels (n = 2), and one annexin (ANXA11). Seventy-six percent of these 46 DE ICT genes were under expressed, in line with the percentage of under expressed genes in the whole set of DE genes. Only 11 out of 46 genes were over expressed (Supplementary Table S4). An additional heatmap, restricted on the representation of ICT, was generated (Fig. 2). The profile confirms homogeneity among samples, as occurred for the whole GEP.

Heatmap of DE genes associated with transporters and ion channels. Heatmap of 48 DE genes associated with the TCDB, presented as an average of the expression level higher than 1 compared with the control. Samples were ordered in columns and genes in rows, gene names were reported on the right and samples on the bottom expressing the tumour grade in brackets when available. The samples were organized based on the FL stage. Expression levels are reported according to the colour key. Three different clusters were obtained: over expressed genes (green, n = 11), moderately under expressed genes (dark red, n = 22) and highly under expressed genes (brilliant red, n = 15). DE ICT genes shared with the GSE65135 dataset are highlighted in red boxes and named FL-8-ICT signature. Among them, SLC2A1 and SLC9A9, marked with * are over expressed in both datasets.
We then analysed whether and how the 46 DE ICT genes were related to each other. Using the PQ software, we found co-occurring ICT DE genes in either the same article abstracts or the image captions26. The final network, constituted by 50 nodes and 136 edges, was visualized through the Cytoscape platform (Fig. 3). Over- and under-expressed genes (nodes) were evidenced by a colour code (light-green and light-red coloured, respectively). A main network emerged, in which all the genes were found to be linked, except two (CACNA2D2 and CACNA2D3, both under expressed) that were connected to each other but disconnected from the other ICT genes. SLC2A1, encoding the glucose transporter 1 (Glut1), represents the core of this connection network and is the most cited ICT in the literature, as indicated by the node size. Most of the solute carriers and potassium channels surrounded the edge of the network, and all of them were strictly connected. ATPases, on the other hand, were peripheral in the network and had fewer links with the other ICT functions and roles.

Ion channel and transporter gene network based on the literature data. Literature-based network of our DE ion channel and transporter genes performed using the PQ algorithm and generated using the Cytoscape tool. Gene connections are based on and filtered by expression/localization methods including: microscopy techniques, electrophoresis, western, Southern and northern blots, fluorometry, brain mapping, GE profiling, gene silencing, ligase chain reaction, PCR techniques, luminescent measurements, protein array analysis, in situ hybridization and histochemistry. Genes were represented by nodes (over expressed genes are shown in light green, and under expressed genes are shown in light red), connections (edges) represented the co-occurrence of at least two different genes in GEP papers. The node size is proportional to the number of occurrences related to the gene; SLC2A1 (GLUT-1) emerged as the hottest ion channel and transporter gene in the literature.
The 46 DE ICT genes identified in the Florence Cohort (FC-46-ICT signature) were then compared with the ICT DE genes of the GSE65135 dataset. 103 ICT genes were differentially expressed in the GSE65135 dataset (Supplementary Table S5). The overlap between the two datasets showed 8 commonly DE ICT genes (highlighted in red in Fig. 2 , and named FL-8-ICT signature), which hence potentially represent the ICT gene signature of Follicular Lymphoma. Among them, SLC2A1 and SLC9A9 (encoding the Na+/H+ antiporter NHE9) were present and over expressed in both datasets.
Correlation of the ICT gene signature with clinical parameters
We then evaluated whether any relationship existed between the FL-DE-ICT genes and clinical data. Due to the small size of our cohort, it was impossible to perform survival analyses, using state-of-the-art methods for microarray experiments (rbsurv Package, 1, from Bioconductor in R). On the contrary, an association analysis with clinical parameters based on a generalized linear model was applicable, although limited to the Florence Cohort. In fact the clinical parameters relative to the datasets GSE32018, GSE9327 and GSE65135 were not available. No statistically significant association emerged between clinical parameters and DE-ICT genes. The extension to the whole set of DE genes led to identify 27 DE genes (involved in different processes such as cell differentiation, regulation of transcription and cytoskeleton organization) associated with the disease stage (Supplementary Table S6). Although these genes showed a significant nominal p-value, they did not pass the canonical threshold (p < 0.05) after the multiple test correction, required to correct the p-value when testing the statistically significance of a large group of genes.
We then analyzed the DE genes in relapsed vs non-relapsed FL patients. Once applied to the whole set of DE gene, this analysis evidenced 1580 differentially expressed genes between the two cohorts, with a p value of 0.05. When restricted to the ICT genes, it emerged that 38 DE ICT genes (16 over expressed and 22 under expressed) characterized relapsed patients (Table 2). All the ICT categories (ABC transporters, ATPases, K+ channels and solute carriers) were roughly equally distributed between over and under expressed genes, whereas only a sodium channel encoding gene (SCN2A2) was over expressed. Notably, KCNN4 encoding for KCa3.1 potassium channels was under expressed, at difference from what observed in the whole set of FL samples (over expressed in Fig. 2). Seven out of the 38 DE ICT genes overlapped with the FC-46-ICT signature and two (SLC2A1 and SLC6A8, encoding for a creatine transporter) with the FL-8-ICT signature. At difference from what occurs in the whole cohort of FL patients, SLC2A1 was highly under expressed in relapsed FL patients (see the highlighted genes in Table 2).
Comparison of ICT gene signature in FL and DLBCL samples
Since FL can progress either to a similar, although more aggressive, FL or to a DLBCL3,4, we also determined the ICT-GEP of DLBCL. To this purpose, we first analysed the two primary DLBCL samples present in our cohort (Table 1), whose RIN values allowed the accomplishment of whole genome analysis (Supplementary Table S1). Their ICT signature was determined, using the same analytical procedure used in FLs’ GE analysis. 26 DE ICT genes emerged, 19 of which overlapped with the FC-46-ICT signature (Table 3) and four (SLC22A16, SLC30A1, SLC6A8 and CACNA2D3) with the FL-8-ICT signature. All the overlapping genes (underlined in Table 3) showed the same expression trend in both cohorts. The 19 overlapping genes encoded solute carriers (SLC, n = 10), ATP-binding cassettes (ABC, n = 3), ATPases (n = 3) and ion channels (n = 3). Eighty-nine percent (17/19) of these 19 genes were under expressed. The seven ICT DE genes not overlapping between the DLBCL and the FC-46-ICT signature (reported in bold in Table 3) were the followings: ANXA8, ATP9A, CACNA1E, CACNA1I, SLC26A1, SLC27A1, SLC7A4. This is apparently a DLBCL-specific ICT signature. Because of the limited number of DLBCL samples present in our cohort the DLBCL-ICT profile was validated with the ICT GEP obtained from a publicly available dataset deposited into the GEO database. We chose (on the basis of the characteristic of our cohort) the dataset GSE12195 that contains microarray data from 71 DLBCL, and compared the GEP with that of centrocytes purified from the tonsils of 5 healthy subjects from the same dataset27.
The expression analysis was performed as for the other datasets and 42 DE ICT genes emerged, 20 being under expressed, and 34 over expressed (Table 4). In the GSE12195 dataset the DE ICT genes are compared to centrocytes, i.e. “activated” B lymphocytes, while the data obtained from our patients’ cohort compares microarray data from patients to those of normal, resting B cells. Nevertheless, 4 out of the 26 DE ICT genes of our two primary DLBCL samples overlapped with the ICT profile in the GSE12195 dataset: ATP2A3, ATP9A, CACNA1I and SLC27A1 (highlighted in bold in Tables 3 and 4). Among them, ATP9A, CACNA1I and SLC27A1 matched the 7 DLBCL-DE ICT specific genes identified in our cohort (reported in bold and squared in Table 3), and may hence represent the true DLBCL-specific ICT signature.
Discussion
In the present study we determined the expression profile of ICT genes (ICT-GEP) of Follicular Lymphoma (FL) and of Diffuse Large B Cell Lymphoma (DLBCL), with the aim to identifying different profiles related to disease progression and therefore with their potential translational relevance. cDNA microarray data were collected both from samples of a patients’ cohort enrolled for this study (the Florence cohort, FC), and from public datasets that matched the clinical and technical characteristics of our cohort. For the first time, it is here provided the ICT signature of FL, its variations during disease relapse and the different ICT profile of DLBCL.
The GEP of 11 FL consecutive samples from the FC was determined, from which 46 DE ICT genes emerged in the Florence Cohort and 8 of them were in common between the FC and the GSE65135 dataset (Fig. 2). The ICT profile of FLs showed an upregulation of K+ channels, witnessed by the over expression of KCNN4 and KCNAB2, which encode for the alpha and beta subunits of Ca2+-dependent K+ channels, respectively. Notably, the Ca2+-dependent K+ channel KCa3.1 encoded by KCNN4, is one of the two K+ currents (the other being Kv1.3) involved in lymphocyte activation and proliferation, and its expression marks the differentiation step characterizing activated naïve B cells and IgD+ CD27+ memory B cells19. Thus KCNN4 over expression supports the notion that B lymphocytes present in FLs are neoplastic germinal centre cells in an activated state. Other potassium channel encoding gene, such as KCNA2 and KCNH2 (encoding KV1.2 and Kv11.1, respectively) were under expressed, at difference from what occurs in other human cancers including leukemias28. Calcium channels encoding genes, in particular CACNA2D2 and CACNA2D3, were also under expressed, suggesting that Ca2+ signalling is sustained by other Ca2+ transport mechanisms, possibly including ATPases, as witnessed by the over expression of ATP2A3. The over expression of two solute carriers, SLC2A1 (encoding the glucose transporter member 1 (Glut-1)) and SLC9A9 (encoding the Na+/H+ antiporter NHE9), merits attention. While NHE proteins contribute to determine the inverted pH characteristics of cancer cells extruding the protons deriving from (anaerobic) metabolism29, Glut-1 is the hallmark of the main metabolic pathway of cancers, i.e. aerobic glycolysis (the so-called Warburg effect). In lymphomas, Glut-1 expression is apparently involved in FL transformation30 and it is related to the NF-κB pathway, which turned out to be upregulated in FL from our FAA. Also the TNF pathway results upregulated in FL, and both of them are associated with the control of the metabolism31,32.
Notably, SLC2A1 was also found at the main edge of the network involving all the DE ICT genes (Fig. 3), meaning that the other solute carriers and potassium channels surrounding the edge of the network are strictly connected with it. Overall, the ICT profile emerging from our data indicates that the main metabolic pathway of FL is represented by glycolysis, and that neoplastic B cells are “excitable”, although at a lesser extent compared to activated normal B lymphocytes. Indeed, in neoplastic B cells, the only relevant K+ channel is KCa3.1, which could contribute to sustain cell proliferation15,20.
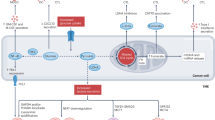
Conversely, both SLC2A1 and KCNN4 turned out to be under expressed in relapsed FL cases (Table 2). This suggests that, upon relapse, neoplastic B cells become even less activated and undergo a metabolic shift, leading to down regulation of glycolysis (Fig. 4). FL often acquire chemoresistance after relapse, and fatty acid metabolism has been linked with resistance to chemotherapeutics in multiple cancer types33. The glycolytic to oxidative shift is also confirmed by the overexpression (log2 fold change value 5.52) of ATPAF2, one of the factors involved in mitochondrial functioning34, and by the under expression of ATP9A (Table 2). A depletion of ATP9A has been found to cause retaining of Glut-1 in endosomes, probably by inhibiting its recycling, and hence reduces its expression on the cell surface35. ATP9A was also DE in DLBC patients, although with different directions in the Florence Cohort and in the GSE12195 dataset, respectively (Tables 3 and 4). Such discrepancy could be traced back to the fact that DE genes are compared to different controls (normal lymphocytes and centrocytes, respectively) in the two datasets. Nevertheless, the common dysregulation of this gene in both DLBCL and in the relapsed FL subgroup suggests its possible involvement in the progression of FL to the more aggressive DLBCL disease.

Overview of the ICT profile along lymphoma progression. Summary figure indicating the changes in the expression of the different ICT and the associated metabolic pathways along lymphoma progression. The represented ICT are described in detail in the text.
The changes in excitability and the shift from a glycolytic to an oxidative metabolic profile observed in relapsed FL is even more evident when analysing the DE-ICT profile of DLBCL (Tables 3, 4, Fig. 4). In DLBCL, in fact, both CACNA1I (and the correlated CACNA1E) which encodes the alpha subunit of the low voltage-activated, T-type calcium channel, and the SLC27A1 solute carrier, fatty acid transporter, implicated in the uptake of fatty acids for further beta oxidation36, turned out to be DE. The dysregulation of Ca2+ channels encoding genes is also accompanied by a drastic shift of the profile of K+ channels. In fact, the two K+ channels which mark normal B lymphocytes (KV1.3 and KCa3.1) are substituted by the over expression of genes encoding inward rectifier K+ channels (Kir) and Big KCa channels (BK) (KCNJ8, KCNJ10, KCNJ5, KCNMA1 (Table 4 and Fig. 4).
SLC27A1 deregulation (i.e. over expression in both the Florence cohort and the GSE12195 dataset) merits attention. SLC27A1 has been associated with tumorigenesis37, since its increased levels would properly supply fatty acids from the surrounding adipocytes for β-oxidation, in turn providing an alternative pathway for acetyl CoA to be metabolized in the TCA cycle when cancer cells switch from a glycolytic to an oxidative metabolism37. Thus, the downregulation of Glut-1 and the upregulation of the fatty acid transporter SLC27A1 in DLBCL indicate a shift from a glycolytic to an oxidative metabolism during lymphoma progression (Fig. 4). In agreement, oxidative enzymes (ACAD10, ACAD8, ECHS1, HADHB) turned out to be over expressed (log2FC > 0) in DLBCL (Supplementary Table S7). Consistently, the nuclear corepressor-encoding gene NCoR1, whose down-regulation has been reported to drive the switch towards oxidative metabolism38, results under expressed in DLBCL (Supplementary Table S8). In agreement with our data, an OxPhos subset of DLBCL, identified by a lower expression of NCoR1, has been recently described in39,40. Patients belonging to this subset might benefit from treatments perturbing the fatty acid oxidation program39. Although further functional studies must be conducted to confirm the therapeutic potential of the fatty acid oxidative pathway in aggressive lymphomas, SLC27A1, and the OxPhos pathway, might constitute a promising therapeutic target.
On the whole, starting from a wide transcriptomic analysis, our data indicate the occurrence of a progressive decrease in excitability and a metabolic shift during neoplastic progression of FL to more aggressive diseases. In particular, for the first time, we have characterized the ICT genes deregulated in relapsed FL patients, showing a decrease in excitability and glycolysis, which precedes the already described change towards an oxidative metabolism in DLBCL.
This finding might have a translational relevance through the identification of novel ICT related therapeutic targets that might overcome the chemoresistance that characterizes relapsed FL.
Methods
Patients
Lymph node surgical specimens were collected from patients at Careggi University Hospital (AOUC), Firenze (n = 50) and from San Jacopo Hospital, Pistoia (n = 4) during 3 years after informed written consent. The study was approved by the Ethical Committee of the AUOC and all research was performed in accordance with relevant guidelines/regulations. FL diagnosis was made according to 2008 World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues41. FL grading was assessed according to the REAL and WHO 3rd edition classifications according to the number of centroblasts (0–5; 6–15, >15 per high power field, respectively). Staging was assessed with the Ann Arbor classification system42.
Primary samples
Part of each surgical specimen was cut into two corresponding pieces, one was processed for histological analysis and diagnosis, the other for total RNA extraction (Trizol, Invitrogen). 11 FL and 2 DLBCL samples displaying a RIN (RNA integrity number) >6 (Supplementary Table S1) at the Agilent 2100 Bioanalyzer (Agilent Technologies) were further processed for gene expression cDNA microarray analyses, and the GEP was compared with that of two available human lymph nodes (male and female donors, Maximum Value and Purity, MVP, Total RNA, Human Lymph Node, Stratagene) pooled and used as control reference.
Real-time quantitative PCR (rqPCR)
Total RNA extraction, reverse transcription (RT) and RQ-PCR were performed as in43. Primers and detailed experimental procedure are reported online in Supplementary Table S3.
Microarray data analyses
Purified RNA’s samples were prepared and compared as previously described in25. Each spot was first corrected for background intensities using the “normexp” approach of the background correct function. The between-array normalization and the quantile approach were performed according to the limma package’s functions. To choose the DE genes, we considered a threshold of 0.01 on the corrected p-value, plus an average cut-off of twofold changes. To find groups of co-regulated genes, we applied a hierarchical clustering algorithm (Ward method) to genes using the Euclidean distance as a measure of similarity.
All the genes analysed were annotated for their role in biological processes using the GO package of Bioconductor, (Gene Ontology consortium). Due to the small number of genes in some categories, two-sided Fisher’s exact test was applied.
DLBCL data analysis, due to the small samples size, was performed using the single slide method developed previously44 and implemented in the sma package (https://cran.r-project.org/package=sma) in R. Statistically significant differential expression was defined as an LOD score larger than 1.
Enrichment and functional annotation analysis (FAA) of the FL samples cohort
FAA of DE genes was based on Gene Ontology (GO) and (Kyoto Encyclopedia of Genes and Genomes (KEGG) terms enrichment, using the GOStats R package. For each GO term, a p-value is calculated representing the probability that the observed numbers of genes could have resulted from randomly distributing this GO term between the tested and reference groups.
We first found the set of all unique GO terms within the ontology that are associated with one or more of the genes of interest. Next, we determined how many of the selected DE genes are annotated at each term and how many of the genes represented on the microarray are annotated at the term. The test evaluated if there are more interesting genes at the term than one might expect by chance. For each GO term, a p-value is calculated representing the probability that the observed numbers of genes could have resulted from randomly distributing this GO term between the tested group and the reference group.
Analysis of microarray datasets
DE-ICT genes were identified as previously described.
GSE32018 contains data from 27 FLs compared to 7 lymph nodes. GSE9327 contains data from 33 FL, compared with 8 reactive lymph nodes. The GSE65135 contains data from 14 FLs, compared to the RNA from B cells from the tonsils of 5 healthy subjects.
The GSE12195 dataset, chosen on the basis of the characteristic of our cohort, contains data from 71 DLBCL and their DE genes are compared to centrocytes purified from the tonsils of 5 healthy subjects.
TCGA meta-analysis
Was performed using the Cbioportal website45,46.
Literature-based molecular network generation
Was performed using ProteinQuest (PQ, BioDigitalValley s.r.l.; www.proteinquest.com/pq/), a web-based platform for biomedical literature retrieval and analysis that retrieves PubMed abstracts and the text of the image captions from free full-text articles26.
References
Novelli, S., Briones, J. & Sierra, J. Epidemiology of lymphoid malignancies, last decade update. Springerplus 2, 70 (2013).
Morton, L. M. et al. Lymphoma incidence patterns by WHO subtype in the United States, 1992–2001. Blood 107, 265–276 (2006).
Swenson, W. T. et al. Improved survival of follicular lymphoma patients in the United States. J. Clin. Oncol. 23, 5019–5026 (2005).
Irish, J. M. et al. B-cell signaling networks reveal a negative prognostic human lymphoma cell subset that emerges during tumor progression. Proc. Natl. Acad. Sci. USA 107, 12747–12754 (2010).
Acker, B. et al. Histologic conversion in the non-Hodgkin’s lymphomas. J. Clin. Oncol. 1, 11–6 (1983).
Horning, S. J. & Rosenberg, S. A. The natural history of initially untreated low-grade non-Hodgkin’s lymphomas. N. Engl. J. Med. 311, 1471–5 (1984).
Alizadeh, A. A. et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511 (2000).
Rosenwald, A. et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 3, 185–197 (2003).
Glas, A. M. et al. Gene expression profiling in follicular lymphoma to assess clinical aggressiveness and to guide the choice of treatment. Blood 105, 301–307 (2005).
Glas, A. M. et al. Gene-expression and immunohistochemical study of specific T-cell subsets and accessory cell types in the transformation and prognosis of follicular lymphoma. J. Clin. Oncol. 25, 390–398 (2007).
Schwaenen, C. et al. Microarray-based genomic profiling reveals novel genomic aberrations in follicular lymphoma which associate with patient survival and gene expression status. Genes Chromosomes Canc. 48, 39–54 (2009).
Husson, H. et al. Gene expression profiling of follicular lymphoma and normal germinal center B cells using cDNA arrays. Blood 99, 282–289 (2002).
Martinez, A., Carreras, J. & Campo, E. The follicular lymphoma microenvironment, From tumor cell to host immunity. Curr. Hematol. Malig. Rep. 3, 179–186 (2008).
Kridel, R., Sehn, L. H. & Gascoyne, R. D. Pathogenesis of follicular lymphoma. J. Clin. Invest. 122, 3424–3431 (2012).
Lastraioli, E., Iorio, J. & Arcangeli, A. Ion channel expression as promising cancer biomarker. Biochim. Biophys. Acta. 1848(10 Pt B), 2685–702 (2015).
Ko, J. H. et al. Expression profiling of ion channel genes predicts clinical outcome in breast cancer. Mol. Cancer. 12, 106 (2013).
Ko, J. H. et al. Ion channel gene expression in lung adenocarcinoma, potential role in prognosis and diagnosis. PLoS One 9(1), e86569 (2014).
Wang, R. et al. Ion channel gene expression predicts survival in glioma patients. Sci. Rep. 5, 11593 (2015).
Feske, S., Wulff, H. & Skolnik, E. Y. Ion Channels in Innate and Adaptive Immunity. Annu. Rev. Immunol. 33, 291–353 (2015).
Arcangeli, A., Pillozzi, S. & Becchetti, A. Targeting ion channels in leukemias, a new challenge for treatment. Curr. Med. Chem. 19, 683–696 (2012).
Suguro, M. et al. Expression profiling analysis of the CD5+ diffuse large B-cell lymphoma subgroup, development of a CD5 signature. Cancer Sci. 97, 868–874 (2006).
Overes, I. M. et al. Expression of P2X5 in lymphoid malignancies results in LRH-1-specific cytotoxic T-cell-mediated lysis. Br. J. Haematol. 141, 799–807 (2008).
Birerdinc, A. et al. Pro-apoptotic and antiproliferative activity of human KCNRG, a putative tumor suppressor in 13q14 region. Tumour Biol. 31, 33–45 (2010).
Tsujimoto, Y., Finger, L. R., Yunis, J., Nowell, P. C. & Croce, C. M. Cloning of the chromosome breakpoint of neoplastic B cells with the t(1418) chromosome translocation. Science. 226, 1097–9 (1984).
Masselli, M. et al. Deregulation of ion channel and transporter encoding genes in pediatric gliomas. Front. Oncol. 2, 53 (2012).
Gatti, S. et al. Gene expression profiling of HGF/Met activation in neonatal mouse heart. Transgenic Res. 22, 579–593 (2013).
Compagno, M. et al. Mutations of multiple genes cause deregulation of NF-kB in diffuse large B-cell lymphoma. Nature 459, 717–21 (2009).
Pillozzi, S. et al. HERG potassium channels are constitutively expressed in primary human acute myeloid leukemias and regulate cell proliferation of normal and leukemic hemopoietic progenitors. Leukemia 16, 1791–1798 (2002).
Grinstein, S. & Cohen, S. Role of Membrane Potential and Volume-activated Na+/H+ Exchange. J Gen Physiol. 89, 185–213 (1987).
Sommermann, T. G., O’Neill, K., Plas, D. R. & Cahir-McFarland, E. IKKbeta and NF-kappaB transcription govern lymphoma cell survival through AKT-induced plasma membrane trafficking of GLUT1. Cancer Res. 71, 7291–7300 (2011).
Mauro, C. et al. NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nature Cell Biology. 13, 1272–1279 (2011).
Samavati, L., Lee, I., Mathes, I., Lottspeich, F. & Hüttemann, M. Tumor Necrosis Factor α Inhibits Oxidative Phosphorylation through Tyrosine Phosphorylation at Subunit I of Cytochrome c Oxidase. JBC. 283, 21134–21144 (2008).
Zhao, Y., Butler, E. B. & Tan, M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 4, e532 (2013).
Wang, Z. G., White, S. P. & Ackerman, S. H. Atp11p and Atp12p Are Assembly Factors for the F1-ATPase in Human Mitochondria. The Journal of Biological Chemistry 276, 30773–30778 (2001).
Tanaka, Y. et al. The phospholipid flippase ATP9A is required for the recycling pathway from the endosomes to the plasma membrane. Mol Biol Cell. 27, 3883–3893 (2016).
Nath, A. & Chan, C. Genetic alterations in fatty acid transport and metabolism genes are associated with metastatic progression and poor prognosis of human cancers. Scientific Reports. 6, 18669 (2016).
Navarro, P. et al. Targeting Tumor Mitochondrial Metabolism Overcomes Resistance to Antiangiogenics. Cell Reports 15, 2705–18 (2016).
Lima, T. I. et al. Role of NCoR1 in mitochondrial function and energy metabolism. Cell Biology International. 42, 734–741 (2018).
Caro, P. et al. Metabolic Signatures Uncover Distinct Targets in Molecular Subsets of Diffuse Large B-Cell Lymphoma. Cancer Cell. 22, 547–560 (2012).
Maneix, L. & Catic, A. Touch and go, nuclear proteolysis in the regulation of metabolic genes and cancer. FEBS Lett. 590, 908–923 (2016).
Swerdlow, S. H. et al. (eds). WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, (4th edn., volume 2). (IARC Publications, 2008).
Shankland, K. R., Armitage, J. O. & Hancock, B. W. Non-Hodgkin lymphoma. Lancet. 380, 848–857 (2012).
Pillozzi, S. et al. EGFR-1 (FLT-1), β1 integrin, and hERG K+ channel for a macromolecular signaling complex in acute myeloid leukemia, role in cell migration and clinical outcome. Blood. 110, 1238–1250 (2007).
Newton, M. A., Kendziorski, C. M., Richmond, C. S., Blattner, F. R. & Tsui, K. W. On differential variability of expression ratios, improving statistical inference about gene expression changes from microarray data. J Comput Biol. 8, 37–52 (2001).
Cerami, E. et al. The cBio cancer genomics portal, an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–4 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2, 11 (2013).
Acknowledgements
This work was supported by grants from the Associazione Genitori contro le Leucemie e Tumori Infantili “Noi per Voi”, the Associazione Italiana per la Ricerca sul Cancro (AIRC). The PhD fellowship of Dr. M. Masselli (at the Experimental Oncology Doctorate School of the University of Florence) was jointly funded by Associazione Italiana contro le Leucemie, Linfomi e Mieloma (AIL) sections of Pistoia, Firenze and Prato. The PhD fellowship of Dr. A. Guerriero (at the Information Engineering Doctorate School of the University of Florence) was supported by Ente Cassa di Risparmio di Firenze (ECRF). We thank the Fondazione CR Pistoia for financing the Agilent workstation used for microarray analysis. We thank Dr. Breschi C. for the Pistoia Hospital sample, Dr. Lorenzoni A. for the technical support and Dr. L. Tofani and Dr. S. Di Giorgio (ITT) for the support in the statistical analysis. The data discussed in this publication have been deposited in NCBI’s Gene xpression Omnibus (Magi, A. et al. 2019) and are accessible through GEO Series accession number GSE 126247 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126247).
Author information
Authors and Affiliations
Contributions
A.M. performed the computational data analysis, interpreted it and revised the manuscript. M.M. performed microarray experiments, performed network analyses and contributed to write the manuscript. C.S. contributed in performing microarray experiments, performed computational analysis on public datasets, contributed to write the manuscript. A.G., P.L. collaborated to the computational analysis. B.P., L.R. and C.B. provided patients’ samples. O.C. supervised the experiments. S.P. collaborated in performing microarray experiments. A.A. conceived and coordinated the work, wrote the manuscript and approved the final version to be submitted.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Magi, A., Masselli, M., Sala, C. et al. The ion channels and transporters gene expression profile indicates a shift in excitability and metabolisms during malignant progression of Follicular Lymphoma. Sci Rep 9, 8586 (2019). https://doi.org/10.1038/s41598-019-44661-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-44661-x
This article is cited by
-
IKCa channels control breast cancer metabolism including AMPK-driven autophagy
Cell Death & Disease (2022)
-
Fatty acid metabolism in aggressive B-cell lymphoma is inhibited by tetraspanin CD37
Nature Communications (2022)
-
Impaired ion homeostasis as a possible associate factor in mucopolysaccharidosis pathogenesis: transcriptomic, cellular and animal studies
Metabolic Brain Disease (2022)
-
Transgenic mice overexpressing the LH receptor in the female reproductive system spontaneously develop endometrial tumour masses
Scientific Reports (2021)
-
CTCF loss mediates unique DNA hypermethylation landscapes in human cancers
Clinical Epigenetics (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
