
Abstract
Larval connectivity among and within coral reefs is important for sustaining coral metapopulations, enhancing ecosystem resilience through species and genetic diversity, and maintaining reef ecosystems’ structure and functions. This study characterized genetic structure and assessed horizontal and vertical connectivity among populations of the ubiquitous gonochoric broadcast spawning coral Montastraea cavernosa in Belize. Using nine polymorphic microsatellite loci, we genotyped M. cavernosa colonies from four depth zones at four study sites within Belizean marine management zones. Study sites were selected within South Water Caye Marine Reserve (3 sites) and Glover’s Reef Marine Reserve (1 site). Strong contemporary genetic differentiation was observed between relatively shallow M. cavernosa populations (10 m, 16 m) and relatively deep (25 m, 35 m) populations, coinciding with a transition from reef crest to reef slope. These results were consistent across both marine reserves. Vertical and horizontal migration models suggest that all populations were historically panmictic, with little unidirectional migration. The relative local isolation of shallow and mesophotic M. cavernosa populations in Belize, coupled with the importance of Belize’s upper mesophotic populations as potential larval sources for other areas in the Tropical Western Atlantic, reinforces the need for management strategies that conserve coral populations across all depth zones.
Similar content being viewed by others

Introduction
Given the multitude of anthropogenic threats to coral reefs and steady declines of shallow coral cover observed over the past several decades1,2,3,4,5, deeper reefs have been proposed as potential refuges for at-risk, shallow reefs1,6,7. Glynn1 first introduced a hypothesis positing that deep reefs may function as refuge habitats for shallow reefs, which has been further developed and termed the Deep Reef Refugia Hypothesis6,8,9,10. This hypothesis relies heavily on the assumption that deeper coral reefs are able to provide viable offspring to recolonize degraded shallow coral reefs8. In particular, depth-generalist coral species found along depth gradients encompassing both shallow and mesophotic coral ecosystems (MCEs; ~30–150 m) are potential candidates for contributing larvae to shallow coral reef areas. In the Tropical Western Atlantic (TWA), it is estimated that at least 25–40% of coral species found on shallow coral reefs also occur on MCEs8,9. While quantification of larval dispersal, recruitment, and survivorship in situ is a challenging feat, assessing similarities in genomic DNA among shallow and mesophotic coral populations can provide evidence of contemporary and historical patterns of connectivity, and evaluate the potential for mesophotic coral populations to replenish declining communities of degraded shallow coral reefs.
Polymorphic molecular markers, including microsatellites, can be used to infer genetic differences among coral populations and to estimate genetic structure within populations. Studies in the TWA have largely examined horizontal connectivity through the estimation of gene flow among sites across broad spatial scales11,12,13,14. Multiple studies have also implemented genetic methodologies to quantify vertical connectivity of depth-generalist scleractinian coral species in order to address the refuge potential of MCEs9,15,16,17,18. Studies focusing on the scleractinian coral species Montastraea cavernosa (Linnaeus, 1767) have demonstrated varying degrees of connectivity between MCEs and shallow coral reefs. M. cavernosa is found throughout the TWA, including most reefs off the coast of Belize. An extreme depth-generalist, M. cavernosa is found across a variety of habitats ranging from 1–113 m in depth8,19. In a broad-scale study of M. cavernosa populations in the TWA, populations in the U.S. Virgin Islands and Bermuda showed no differentiation across depth zones, while populations in South Florida demonstrated evidence of genetic structure across depth zones16. Similarly, while M. cavernosa populations in both Little Cayman Island and San Salvador, Bahamas were differentiated by depth, populations in Lee Stocking Island, Bahamas were relatively well-connected15. M. cavernosa populations in the northwestern Gulf of Mexico (NW GOM) are likely a single panmictic population, with no genetic structure between shallow and mesophotic depth zones, while in Belize and southwest Florida, populations were genetically differentiated by depth18. While these studies have focused on large geographic scales (100 s of km), there has been little work focused on small scale, local population structure and connectivity of scleractinian species in Belize.
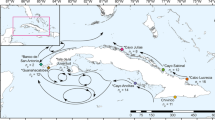
The barrier reef off the coastline of Belize (i.e. the Belize Barrier Reef) connects the reefs of Guatemala and Honduras to the south and east with the reefs of Mexico to the north, collectively forming the greater Mesoamerican Reef. The shoreward side of the reef is separated from the coast by a lagoon which ranges from 20–40 km in width and 1–65 m in depth, containing hundreds of patch reefs20,21. Reef margins on the barrier reef surrounding Carrie Bow Cay and the seaward side of Glover’s Reef Atoll (Fig. 1) typically exhibit spur and groove structures to a depth of 20–33 m, with a near vertical step from 30–37 m, and a slope to a reef wall extending to depths beyond 100 m20 (Fig. 2). A network of seventeen multi-use Marine Protected Areas (MPAs) has been established in Belize since 1982. These MPAs span over 3,600 km2 (~10%) of the country’s territorial seas, ~530 km2 of which are designated no-take zones22. The barrier reef surrounding Carrie Bow Cay and all of Glover’s Reef Atoll lie within South Water Caye and Glover’s Reef Marine Reserves, respectively. Both reserves have multiple levels of management ranging from general use to highly restrictive preservation or wilderness zones. Sites within these marine reserves benefit from restrictions to access, fishing, and anchoring23,24. Despite the large body of literature available on the Belize Barrier Reef, there have been relatively few studies of scleractinian corals within the depth range of MCEs18,25, and even fewer focused on population genetics within this depth range. The majority of information available on scleractinian distribution below 30 m was published in the 1970s from submersible dives by James and Ginsberg20 and in the 1980s from open circuit SCUBA dives near Carrie Bow Cay26,27. Furthermore, only one study18 of the several recent studies examining scleractinian population genetics and connectivity13,14 included samples from Belizean MCEs.

Map of Belize Barrier Reef with sampling sites within South Water Caye and Glover’s Reef Marine Reserves overlaid. Coral reef habitat and MPA shapefiles adapted from Meerman and Clabaugh72.

Diagram of a typical sampling site showing generalized reef geomorphology along sampling depths. Inset photographs are representative of typical habitat at sampling depths.
This study investigated the patterns of genetic connectivity among populations of M. cavernosa across a small spatial scale, specifically targeting reefs within the nearby South Water Caye and Glover’s Reef Atoll Marine Reserves. A multi-regional analysis of M. cavernosa connectivity reported little connectivity between mesophotic (~35 m) and relatively shallow (~15 m) sites in Belize, despite short lateral separation between populations (~10 m of horizontal distance)18. Through the sampling of an additional site and depth zones we sought to describe small scale vertical and horizontal connectivity across a depth gradient within existing MPAs to evaluate if populations coincided with depth-based habitat characterization (i.e. “shallow” and “mesophotic”) as potentially indicated in previous work from the region18.
Results
Genetic differentiation and population structure along a depth gradient
Analysis of molecular variance showed low but significant differentiation across sampled M. cavernosa populations (AMOVA; 1.60%, df 15,239, SS = 78.81, p = 0.0001), with the highest genotypic variation being among individuals within populations (12.26%, df 223,239, SS = 817.80). Principal coordinates analysis (PCoA) revealed similarities and clustering of the 10 m and 16 m depth zones and 25 m and 35 m depth zones, respectively (Fig. 3). Pairwise population FST comparison supported the PCoA patterns, revealing that 10 m and 16 m populations were similar to one another but significantly differentiated from 25 m and 35 m populations (Supplementary Fig. S1). When samples were combined into “shallow” (10 m and 16 m) and “deep” (25 m and 35 m) populations, every deep-shallow pairwise comparison demonstrated significant differentiation (Figs. 4 and S1). Additionally, FST comparisons between populations at Raph’s Wall and all other populations were among the most highly differentiated pairwise comparisons (Fig. 4). A Mantel test suggested no significant correlation between geographic and genetic distances in the sampling region (R2 = 0.0069, p = 0.21); geographic distances between sampling sites ranged from 0.796–32.322 km.

Principal coordinates analysis (PCoA) based on Nei genetic distance (DA) matrix of pairwise population comparisons. Shape denotes depth zone and color denotes the dominant genetic cluster (K = 3) within each population, as determined by structure analysis. Populations are labeled next to points, formatted as SITE.DEPTH (site abbreviations listed in Table 2).

Heat map showing pairwise population differentiation through estimation of fixation index (FST) between combined “shallow” (10 m and 16 m) and “deep” (25 m and 35 m) populations. Values within cells are estimated FST; intensity of red coloration corresponds with increasing FST; bolded values denote significant differentiation between populations (after FDR correction; α = 0.05). Site abbreviations listed in Table 2.
Population structure analysis using the Evanno method28,29 estimated the most likely number of genetic clusters (K) for M. cavernosa populations on the Belize Barrier Reef to be K = 3. K selection through log model likelihood (L(K)) was in agreement with the Evanno method (Supplementary Fig. S2). Based on these results, a structure bar plot was generated for K = 3 (highest (L(K)) and ∆K value). In the K = 3 bar plot, the Blue or Green genetic cluster was the primary genetic cluster in all but one population (Raph’s Wall–35 m). Shallow populations (10 m and 16 m) primarily grouped to the Green genetic cluster (73.71–95.14% individual membership probabilities) while deep populations (25 m and 35 m) primarily grouped to the Blue genetic cluster (42.88–74.71%), with the exception of Raph’s Wall–35 m, which predominantly grouped to the Yellow genetic cluster (50.5%). While 25 m sample populations were genetically distinct from shallow populations, they had a stronger signal of admixture with shallow populations (Green cluster) than did 35 m populations (Fig. 5). Deep populations also had higher levels of admixture with the Yellow genetic cluster compared to shallow populations, and levels of admixture increased in prevalence from 25 m to 35 m sample populations.

Genetic structure plots for Montastraea cavernosa populations from 4 depth zones across 4 reef sites in Belize. Each vertical bar is an individual M. cavernosa sample showing percent membership to each of 3 proposed genetic clusters (K) represented as Blue, Green, and Yellow (structure; 10 replicate simulations, K = 1–19, 103 burn-in, 106 MCMC replicates, LOCPRIOR).
Vertical and horizontal migration models
At all sampling sites, vertical migration tests resulted in Panmixia models as the most likely scenario, yielding the highest model probabilities (Table 1). All other models (Symmetric, Upward, Downward) had probabilities near zero. Estimated mutation scaled population sizes (Θ) for all vertical migration tests at all four sites were an order of magnitude larger at Glover’s Reef and Raph’s Wall (15.30 and 21.43, respectively) compared to South Reef and Tobacco Reef (1.10 and 3.96, respectively). In horizontal migration tests a similar pattern was found, where Panmixia models resulted in model probabilities of one for both East/West (all sites) and North/South (South Reef, Raph’s Wall, Tobacco Reef) models with all other models (Symmetric, Southward, Northward, Eastward, Westward) having model probabilities near zero.
Discussion
Populations of M. cavernosa in this study demonstrated a distinct shallow/deep genetic break, with strong genetic differentiation between a shallower population (10 m and 16 m) and adjacent deeper population (25 m and 35 m). This apparent genetic breaking point between 16 m and 25 m depth zones was consistent across all sites within South Water Caye Marine Reserve and also at Glover’s Reef Marine Reserve (~35 km east of South Water Caye Marine Reserve). All sampling sites had similar geomorphology, with “shallow” populations atop the reef (i.e. back reef and reef crest) and “deep” populations on the reef slope (fore reef and wall; Fig. 2). Additionally, the 35 m samples had higher probability of membership to a Yellow genetic cluster as compared to all other samples from other depth zones, potentially indicating a “depth-specialist” cluster with additional genetic structure beyond our sampling depths in the upper mesophotic depth zone. M. cavernosa have historically been identified deeper than 35 m in the region20,26, but additional samples beyond 35 m are required to assess any potential structure or specificity further into the mesophotic zone of Belize, as has been discovered in similar studies of other regions15,30.
While vertical connectivity was relatively low in the region, there was strong evidence for horizontal genetic connectivity within depth zones across the region. Results of all analyses indicated that M. cavernosa populations along the Belize Barrier Reef exhibit little horizontal structuring among sites. Pairwise FST comparisons revealed that Raph’s Wall populations were the most highly differentiated from populations at all other sampling sites (Fig. 4). This was unexpected given the similarity in geomorphology to other sites and the proximity of Raph’s Wall to South Reef and Tobacco Reef (0.796 km and 6.268 km, respectively) in comparison to the proximity of Glover’s Reef to all barrier reef sites (31.194–32.322 km; Fig. 1). Overall, it is evident that even though M. cavernosa populations in Belize are relatively well-mixed in terms of horizontal connectivity, deeper populations do not appear to be major larval contributors to shallow populations within the region, or vice versa. The similarity in population structure across these sampled sites may be driven by a number of regional abiotic and/or biotic factors, but more research is needed to elucidate the drivers of population differentiation across depth zones on the Belize Barrier Reef.
The coincidence of the genetic break with the shift in reef geomorphology may be indicative that the physical transition from reef crest to reef wall has an effect on the genetic structuring of M. cavernosa populations. Though it is unlikely that the geomorphology of the reef in this region is a direct physical barrier to gene flow, differences in geomorphology between depth zones likely influence abiotic and biotic factors which in turn contribute to the genetic structuring observed here. One such abiotic factor that may impede gene flow between M. cavernosa populations are small-scale oceanographic (i.e. <1 km) currents. There is previous evidence for spatial and temporal divergence in surface current regimes based on location on the Belize Barrier Reef. Hydrodynamic models suggest that eddy circulation direction affects cyclonic circulation around the Belize Barrier Reef subsequently influencing flow direction31,32. Models based on drifter releases have suggested that shoreward of the Belize Barrier Reef, currents typically demonstrate a westward motion, while seaward of the reef they show variability in motion (northeast to west) depending on the patterns of the predominant Yucatan current33. Additionally, surface current models from the region have demonstrated that small particles near Glover’s Reef Atoll tend to move northward while particles shoreward of South Water Caye tend to be retained on the Belize shelf 34. The discrepancy between shoreward and seaward currents is further accentuated during late summer as offshore current velocity increases33, which has potential implications for dispersal of larvae during spawning in late July through September. Given the available data on current dynamics and population structure of M. cavernosa, it is likely that deep gametes and larvae are transported northward, away from shallow gametes and larvae which are likely retained on the barrier reef18.
Temperature and light are among other abiotic factors that may influence M. cavernosa population structure on the Belize Barrier Reef. Temperature is an important abiotic factor for many marine species, especially scleractinian corals which are susceptible to bleaching events when water temperatures are elevated for prolonged periods1,35,36. Corals in MCEs have been found to have lower bleaching thresholds as compared to nearby shallow reefs37, though it is unclear whether this is a genotypic or phenotypic response. Light is also an important abiotic driver of ecological structure and function, especially in MCEs6,10. It has been suggested that rather than strict depth definitions of MCEs, light may be a better metric for defining MCEs, either directly or through the absence of light-dependent coral species10,38. Reef geomorphology can affect the optical properties between shallow and mesophotic habitat, with higher-relief reef sites having a significantly shallower 1% optical depth (i.e. the photosynthetic compensation point) than low-relief reef sites10. On the Belize Barrier Reef where reef slope may be near vertical or even inverted in some areas, the implications of geomorphology on optical characteristics result in shallower 1% and 10% (i.e. the midpoint of the euphotic zone) optical depths (58 and 28 m, respectively) as compared to other high-relief reef sites10. While some temperature and light data are available for Belize39,40,41, additional in situ data are needed from all depth zones, particularly into the upper mesophotic zone. This data coupled with ex situ experimentation would prove useful in evaluating these factors as potential drivers of genetic differentiation.
Asynchronicity in spawning between shallow and mesophotic M. cavernosa colonies may act as a prezygotic barrier, driving population differentiation and structure. As gonochoric broadcast spawners, male and female M. cavernosa synchronously release gametes that mix and fertilize in the water column or near the surface approximately one week after the full moon between the months of July and September42,43. Colonies at mesophotic depths have previously been observed to spawn simultaneously with shallow conspecifics, potentially allowing for inter-breeding between depth zones44. Only one study has examined spawning synchronicity of M. cavernosa across shallow and mesophotic depth gradients to date. At Flower Garden Banks National Marine Sanctuary in the NW GOM, shallow and mesophotic M. cavernosa colonies have been documented spawning in synchrony44. In Belize there is evidence for asynchronous spawning of broadcast spawning macrobenthic organisms between shallow and mesophotic depths, including the sponge Xestospongia muta45 and the scleractinian coral Orbicella franksi25. Mesophotic X. muta were observed spawning while no shallow conspecifics spawned45 and mesophotic O. franksi were found to spawn an average of 40 minutes prior to shallow conspecifics25. Asynchronicity in spawning can lead to reproductive isolation in coral populations, but temporal differences between conspecifics at different depths but may also allow deeper gametes time to reach the surface and mix with shallow gametes46. There is presently little information on spawning of M. cavernosa in MCEs in Belize, including any details of synchronicity with shallow conspecifics. There is also the possibility of interactive effects of spawning time and currents on vertical connectivity in this region. Gametes from mesophotic M. cavernosa may be susceptible to opposing currents on the seaward reef margin, transporting them away from local reefs while shallow gametes are being retained locally onto the barrier reef 33,34, leading to little chance of cross fertilization between shallow and deep conspecifics. This would result in a lack of gene flow between populations, even in the absence of other biological barriers between MCE and shallow conspecifics.
Genetic structure may also be affected through contemporary postzygotic barriers. It is possible that larvae of parent colonies from one depth zone may settle in depth zones different from their origin (e.g. deep larvae may settle on shallow reefs or vice versa). These larvae may be maladapted for environmental or biological factors at this alternate depth which may lead to decreased survivorship47,48, creating a lack of gene flow such as that which we have seen in M. cavernosa populations in Belize. In contrast, postzygotic genetic barriers could also occur pre-settlement. Coral larvae are known to use multiple cues to strategically settle on suitable reef habitat49,50,51. Coral larvae may be exhibiting microhabitat selection between sites on the reef crest or the reef slope to minimize phenotype–environment mismatches, thus increasing survivorship47,52,53. However, in a study on O. franksi Noren25 found that larvae from upper mesophotic colonies instead demonstrated preferential settlement on aragonite tiles conditioned at shallow depths. Although Noren25 found no evidence of postzygotic barriers to settlement in O. franksi, these data were recorded after only 4 weeks, which may be too soon to detect any effects of maladaptation of deep larvae to shallow habitats on the reefs in this region.
Although we were able to detect significant contemporary genetic differentiation between relatively deep and shallow M. cavernosa populations in Belize, migration analyses found little evidence for directional historical migration in any vertical or horizontal models, instead selecting panmixia as the most likely scenario in all instances. The generation time for M. cavernosa is unknown, but based on the size threshold for sexual maturity and the long lifespan of colonies43,54 predicted patterns of migration are likely estimated over the order of thousands of years55. These results suggest that barriers to migration across these depth zones may have developed more recently than migration analyses are able to predict. Again, this may be due to differences in oceanographic current regimes between depth zones as well as larval responses to environmentally or biologically driven selection. These analyses also highlight the fact that despite significant genetic differentiation across depth zones, as indicated by FST values, overall these levels of genetic differentiation were relatively low. Studivan and Voss found similar results when modeling M. cavernosa vertical migration between shallow and mesophotic habitats in the NW GOM55, Belize, and southwest Florida18. However, Serrano et al.16 found in their study of M. cavernosa populations across “shallow” (≤10 m), “intermediate” (15–20 m) and “deep” (≥25 m) habitat that models with shallow to deep migration were most probable, even in sites with apparently panmictic population structure.
This study provides evidence that it may be important to sample more frequently between depth zones in future studies, as in some regions population structure and genetic differentiation may not coincide precisely with the habitat labels researchers assign them, such as “mesophotic” or “shallow”. Here we found that even though populations were separated by ~10 m in both depth and horizontal distance, we could see strong evidence of genetic structuring. Although the M. cavernosa colonies we sampled at 25 m belong to “shallow reef” habitat based upon their depth zonation, from a genetic perspective they are distinct from other shallower (10 m and 16 m) colonies, which grouped together as we might expect given their depth zonation. At all sampling sites the 25 m populations had similar genetic structure to that of populations found in the upper mesophotic zone (35 m). This is potential evidence that habitat designations assigned to reef zones may be less dependent upon depth zone and should perhaps place more emphasis on differences in habitat characteristics and environmental factors which may vary across regions.
Our results indicate there is strong genetic differentiation between relatively shallow and deep M. cavernosa populations in Belize, therefore we suggest these populations should be managed to conserve and maintain coral populations across all depths. We observed little evidence of contemporary gene flow between deep and shallow populations on the Belize Barrier Reef, therefore it seems that in Belize neither local deep nor shallow populations are likely to serve as a viable refuge for the other. It is important to continue monitoring shallow communities while also incorporating deeper communities into assessments of ecosystem services. The need for monitoring of deeper populations may be especially important, since deep and shallow populations may not respond similarly to stress and disturbance. Although this study demonstrates impeded gene flow between relatively shallow and deep M. cavernosa populations in Belize, there is evidence of strong connectivity between mesophotic M. cavernosa populations in Belize and relatively shallow M. cavernosa populations nearly 1000 km away in the Dry Tortugas off the coast of Florida18. Therefore, Belize’s deeper reefs may be serving as refuges, but for distant shallow reefs rather than local shallow reefs. Considering these findings, international management efforts may be required to help maintain coral metapopulations across broad geographic scales. Overall, both “shallow” and “deep” M. cavernosa populations will likely continue to be important to long-term health and resilience of coral reefs through their contributions to genetic diversity in Belize and beyond.
Methods
Study site selection
This study focused on the Belize Barrier Reef surrounding Carrie Bow Cay, off the Belizean coast. The reefs in this area typically contain a steep slope which minimizes horizontal distance among sampling depth zones (Fig. 2). Similar to many locations in the TWA, M. cavernosa is ubiquitous along the Belize Barrier Reef, found from <1–95 m20,27]. M. cavernosa colonies were sampled from four sites for this study: three sites in South Water Caye Marine Reserve, near Carrie Bow Cay (South Reef, Raph’s Wall, and Tobacco Reef), and one site in Glover’s Reef Marine Reserve (Glover’s Reef), ~30 km southeast of Carrie Bow Cay (Fig. 2; Table 2). Sites were only selected for sampling if there was suitable reef habitat spanning all targeted depth zones and a sufficient abundance of M. cavernosa colonies to allow statistically robust sampling efforts while minimizing the likelihood of sampling clones.
Coral sample collection
Approximately fifteen M. cavernosa colonies were sampled at each of four depth zones (back reef ~10 m; reef crest ~16 m; fore reef ~25 m; upper mesophotic ~35 m; Fig. 2) per reef site (N = 242). Sampled colonies were >1 m apart, which has been found to sufficiently reduce the likelihood of sampling clones from asexual fragmentation16. A small skeletal fragment containing coral tissue (~6 cm2) was removed from each colony by SCUBA divers with hammer and masonry chisel and subsequently placed into an individual, uniquely numbered zip-top bag. Colonies were photographed prior to sampling with scaled lasers (15 cm) for size reference. Upon returning to the surface, sample bags were placed into a cooler at ambient seawater temperature and transported back to the Smithsonian Carrie Bow Cay Field Station for processing within 3 hours. At Carrie Bow Cay each M. cavernosa fragment was photographed and subsamples were generated via chisel fragmentation. Excess coral skeleton, sponge tissue, crustose coralline algae, and macroalgae were removed to reduce the introduction of polymerase chain reaction (PCR) inhibitors and contaminating DNA into samples. M. cavernosa samples were preserved in TRIzol reagent and stored at −20 °C until transportation back to FAU Harbor Branch on ice. Once in the laboratory, samples were stored at −80 °C until DNA extraction. Samples were collected over two field expeditions; samples from 35 m depth zones within South Water Caye Marine Reserve at (i.e. Tobacco Reef, Raph’s Wall, and South Reef) were collected in March 2016 by Studivan and Voss18, and samples from the remaining depth zones in South Water Caye Marine Reserve were collected in March 2017 (including resampling of 16 m depth zones). Samples from all depth zones in Glover’s Reef Marine Reserve were collected in March 2017.
Genomic DNA extraction
Genomic DNA was extracted from the 2016 samples using a modified TRIzol extraction56 as described in Studivan and Voss18. For all 2017 samples a cetyl trimethylammonium bromide (CTAB) extraction was implemented following Mieog et al.57 with the following modifications: coral fragments were removed from thawed TRIzol and tissue (~0.1 g) was scraped from several polyps with a sterile scalpel. Tissue was placed into a 2.0 mL microcentrifuge tube with 800 µL of 2% CTAB extraction buffer, ~0.075 g of 0.5 mm glass beads, and 20 µg mL−1 proteinase K. The sample was macerated in a FastPrep®−24 homogenizer (MP Biomedicals) at 6 m s−1 for three 45 s intervals with a 2 min cool down period between intervals. The sample was incubated in a thermomixer at 60 °C while mixing (1000 rpm for 5 s, 1.5 min intervals) for 90 min. Following incubation, DNA extraction was performed according to the procedures outlined in Mieog et al.57, and samples were eluted in 100 µL 55 °C 1X TE (pH 8.0) and incubated for 10 min at 55 °C. Extracted DNA was cleaned using the Zymo Research DNA Clean & Concentrator™−5 kit to remove polysaccharides and other PCR inhibitors. Concentration and quality of cleaned DNA were measured on a NanoDrop™ 2000 (Thermo Fisher Scientific) spectrophotometer and samples were subsequently diluted for PCR amplification.
Montastraea cavernosa microsatellite genotyping
Nine previously developed microsatellite loci16 were amplified to genotype M. cavernosa samples. Triplexed PCRs were used with self-labeled fluorescent primers (6FAM, VIC, and NED), which anneal to universal tails included in forward primers55,58,59, (Supplementary Table S1) using the QIAGEN Type-It Microsatellite PCR kit. PCRs were run following the methods outlined in Studivan and Voss55, except with 30 cycles per reaction. Amplified samples were visualized on a 2% agarose gel and diluted to 1:30–1:50 in deionized water based on band intensity from agarose gel images, prior to sequencing on an Applied Biosystems ABI 3130xl genetic analyzer with 500 ROX size standard. Alleles were scored from returned electropherograms using GeneMapper v3.7 (Applied Biosystems). Any electropherograms with ambiguous or non-existent allele peaks, or low quality size standard scores were re-amplified and re-screened (including re-extraction of DNA as necessary) to ensure consistent allele scoring, resulting in the most complete dataset possible. Samples were run an average of 1.1 times on the genetic analyzer.
Assumption testing and population structure analyses
After alleles were scored, GenAlEx v6.50360 was used to identify 3 clonal multi-locus genotypes (MLGs) within sites, which were removed from all subsequent analyses (unique MLGs per population listed as ng in Table 2). MLGs were determined to be the result of asexual reproduction rather than resampling of the same colony using reference photographs of sampled colonies or random chromosome recombination (all p-values < 0.0001) using MLGsim v2.061,62 (1000 simulations). GenAlEx was used to calculate allele frequencies, evaluate populations for Hardy-Weinberg equilibrium for all loci, and calculate fixation indices (FST). Linkage disequilibrium was calculated for all pairwise comparisons of loci among populations using Arlequin v3.563. Hardy-Weinberg equilibrium and linkage disequilibrium p-values were corrected for false discovery rate (FDR) with the R package fdrtool64. No populations or loci were found to have significant patterns in violations of Hardy-Weinberg equilibrium (Supplementary Table S2) or linkage disequilibrium among loci within any populations. FreeNA65 was used to test the effects of null alleles on deviations from Hardy-Weinberg equilibrium. Null allele corrected and raw FST values by locus and population were tightly correlated (R2 = 0.996 and R2 = 0.985, respectively) therefore uncorrected FST values were used in subsequent analyses. Population differentiation was assessed with an analysis of molecular variance (AMOVA) in GenAlEx using fixation indices (FST; 9999 model and population permutations). Pairwise FST estimates and p-values were first calculated for all sites and depths. Based on patterns from initial analysis of all sites and depths as discrete populations, additional analyses were conducted combining the relatively shallow sampling depths (10 m and 16 m) and the relatively deep sampling depths (25 m and 35 m) into “shallow” and “deep” populations, respectively, increasing population sample sizes and simplifying pairwise comparisons. Population differentiation was visualized with principal coordinates analysis (PCoA) using Nei genetic distance (DA) in GenAlEx. Isolation by geographic distance was assessed between all sample populations with a Mantel test (9999 permutations) in GenAlEx, using general site GPS coordinates to calculate linear distance between sites.
Population structure (i.e. genetic clusters; K) was estimated using Bayesian model-based clustering with the program structure v2.3.466. The R package ParallelStructure67 was used to run structure in parallel on FAU’s high performance computing cluster, testing 10 replicate simulations for all values of K between 1–19 (maximum K = sampling sites + 3) to identify any possible cryptic genetic clusters within populations. All simulations used 103 burn-in iterations and 106 Markov Chain-Monte Carlo replicates, including the LOCPRIOR option which aids simulation testing based on sampling location. The web-based version of structure harvester29 was used to determine the most likely value of K through implementation of the Evanno method of K selection28, including the ad hoc test statistic ∆K. A mean model simulation was generated for the most likely value of K as determined by structure harvester using clumpp v1.1.268 based on replicate simulation runs, and population structure bar plots showing individual percent membership to genetic clusters were generated using distruct v1.169.
Vertical and horizontal migration modeling
Historical (~4 Ne generations) migration rates across vertical and horizontal spatial scales were estimated using migrate v3.6, which uses a Bayesian approach to estimate the mutation rate and determine the likelihood of population gene flow models through coalescence theory70,71. A priori migration models were developed to test (1) vertical migration within individual sites, (2) North/South horizontal migration along the barrier reef sites (South Reef, Raph’s Wall, Tobacco Reef), and (3) East/West horizontal migration between Glover’s Reef Atoll and the Belize Barrier Reef. When testing vertical migration, populations were combined into “shallow” (10 m and 16 m) and “deep” (25 m and 35 m) populations based on PCoA and FST results. The following set of migration models were used to determine (1) vertical migration at each sampling site: (A) Symmetric; full model with symmetric migration between depths, (B) Upward; asymmetric migration from deep to shallow depths, (C) Downward; asymmetric migration from shallow to deep depths, (D) Panmixia. Tests of (2) North/South horizontal migration used the following set of models: (A) Symmetric; full model with symmetric migration between sites, (B) Southward; asymmetric migration from northern to southern sites, (C) Northward; asymmetric migration from southern to northern sites, (D) Panmixia. Tests of (3) East/West horizontal migration used the following set of models: (A) Symmetric; full model with symmetric migration between sites, (B) Westward; asymmetric migration from eastern (Glover’s Reef) to western sites (South Reef, Raph’s Wall, Tobacco Reef), (C) Eastward; asymmetric migration from western to eastern sites, (D) Panmixia.
All migration simulations were run with the following parameters: long-inc 100, long-sample 15000, 20 replicates, burn-in 20000, and four heated chains of 1, 1.5, 3, 105. Prior distributions for Θ (mutation-scaled population size) and M (mutation-scaled immigration rate) were set from 0–100 and 0–1000, respectively. For each set of migration models, the best model simulation was chosen based upon comparison and ranking of Bezier log marginal likelihoods using the thermodynamic integration method detailed by Beerli and Palczewski70.
Data Availability
Microsatellite genotype data and structure output files are available as a Supplementary Dataset file. Additional analysis scripts and documentation are available on GitHub (https://github.com/mstudiva/Mcav-microsats.git).
References
Glynn, P. W. Coral reef bleaching: facts, hypotheses and implications. Glob. Chang. Biol. 2, 495–509, https://doi.org/10.1111/j.1365-2486.1996.tb00063.x (1996).
De’ath, G., Fabricius, K. E., Sweatman, H. & Puotinen, M. The 27-year decline of coral cover on the Great Barrier Reef and its causes. Proc. Natl. Acad. Sci. USA 109, 17995–17999, https://doi.org/10.1073/pnas.1208909109 (2012).
Mumby, P. J. et al. Fishing, trophic cascades, and the process of grazing on coral reefs. Science 311, 98–101, https://doi.org/10.1126/science.1121129 (2006).
Hughes, T. P. Catastrophes, phase shifts, and large-scale degradation of a Caribbean coral reef. Science 265, 1547–51, https://doi.org/10.1126/science.265.5178.1547 (1994).
Gardner, T. A., Côté, I. M., Gill, J. A., Grant, A. & Watkinson, A. R. Long-term region-wide declines in caribbean corals. Science 301, 958–960, https://doi.org/10.1126/science.1086050 (2003).
Lesser, M. P., Slattery, M. & Leichter, J. J. Ecology of mesophotic coral reefs. J. Exp. Mar. Bio. Ecol. 375, 1–8, https://doi.org/10.1016/j.jembe.2009.05.009 (2009).
Riegl, B. & Piller, W. E. Possible refugia for reefs in times of environmental stress. Int. J. Earth Sci. 92, 520–531, https://doi.org/10.1007/s00531-003-0328-9 (2003).
Bongaerts, P., Ridgway, T., Sampayo, E. M. & Hoegh-Guldberg, O. Assessing the ‘deep reef refugia’ hypothesis: focus on Caribbean reefs. Coral Reefs 29, 309–327, https://doi.org/10.1007/s00338-009-0581-x (2010).
Bongaerts, P. et al. Deep reefs are not universal refuges: reseeding potential varies among coral species. Sci. Adv. 3, e1602373, https://doi.org/10.1126/sciadv.1602373 (2017).
Lesser, M. P., Slattery, M. & Mobley, C. D. Biodiversity and functional ecology of mesophotic coral reefs. Annu. Rev. Ecol. Evol. Syst. 49, 49–71, https://doi.org/10.1146/annurev-ecolsys-110617-062423 (2018).
Goodbody-Gringley, G., Woollacott, R. M. & Giribet, G. Population structure and connectivity in the Atlantic scleractinian coral Montastraea cavernosa (Linnaeus, 1767). Mar. Ecol. 33, 32–48, https://doi.org/10.1111/j.1439-0485.2011.00452.x (2012).
Nunes, F., Norris, R. D. & Knowlton, N. Implications of isolation and low genetic diversity in peripheral populations of an amphi-Atlantic coral. Mol. Ecol. 18, 4283–4297, https://doi.org/10.1111/j.1365-294X.2009.04347.x (2009).
Nunes, F. L. D., Norris, R. D. & Knowlton, N. Long distance dispersal and connectivity in amphi-Atlantic corals at regional and basin scales. PLoS One 6, e22298, https://doi.org/10.1371/journal.pone.0022298 (2011).
Rippe, J. P. et al. Population structure and connectivity of the mountainous star coral, Orbicella faveolata, throughout the wider Caribbean region. Ecol. Evol. 7, 9234–9246, https://doi.org/10.1002/ece3.3448 (2017).
Brazeau, D. A., Lesser, M. P. & Slattery, M. Genetic structure in the coral, Montastraea cavernosa: assessing genetic differentiation among and within mesophotic reefs. PLoS One 8, e65845, https://doi.org/10.1371/journal.pone.0065845 (2013).
Serrano, X. et al. Geographic differences in vertical connectivity in the Caribbean coral Montastraea cavernosa despite high levels of horizontal connectivity at shallow depths. Mol. Ecol. 23, 4226–4240, https://doi.org/10.1111/mec.12861 (2014).
Serrano, X. M. et al. Long distance dispersal and vertical gene flow in the Caribbean brooding coral Porites astreoides. Sci. Rep. 6, 21619, https://doi.org/10.1038/srep21619 (2016).
Studivan, M. S. & Voss, J. D. Population connectivity among shallow and mesophotic Montastraea cavernosa corals in the Gulf of Mexico identifies potential for refugia. Coral Reefs 37, 1183–1196, https://doi.org/10.1007/s00338-018-1733-7 (2018).
Reed, J. K. Deepest distribution of Atlantic hermatypic corals discovered in the Bahamas. Proc 5th Int Coral Reef Symp 6, 249–254 (1985).
James, N. P. & Ginsburg, R. N. The seaward margin of Belize barrier and atoll reefs (Blackwell Scientific, 1979).
Miller, J. A. & Macintyre, I. G. Field guidebook to the reefs of Belize (Atlantic Reef Committee, University of Miami, 1977).
Kramer, P. et al. 2015 Report Card for the Mesoamerican Reef. Healthy Reefs Initiative (www.healthyreefs.org) (2015).
Belize Fisheries Department. Management plan Glover’s Reef Marine Reserve World Heritage Site (2007).
Belize Fisheries Department. Management plan South Water Caye Marine Reserve World Heritage Site (2009).
Noren, H. K. G. Can twilight reefs usher in a new dawn for depauperate shallow coral reefs? (Nova Southeastern University, 2016).
Rützler, K. & Macintyre, I. G. The habitat distribution and community structure of the barrier reef complex at Carrie Bow Cay, Belize. In The Atlantic Barrier Reef Ecosystem at Carrie Bow Cay, Belize, I: Structure and Communities (eds Rützler, K. & Macintyre, I. G.) 9–45 (Smithsonian Institution Press, 1982).
Cairns, S. D. Stony corals (Cnidaria: Hydrozoa, Scleractinia) of Carrie Bow Cay, Belize. In The Atlantic Barrier Reef Ecosystem at Carrie Bow Cay, Belize, I Structure and Communities (eds Rützler, K. & Macintyre, I. G.) 271–302 (Smithsonian Institution Press, 1982).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620, https://doi.org/10.1111/j.1365-294X.2005.02553.x (2005).
Earl, D. A. & VonHoldt, B. M. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361, https://doi.org/10.1007/s12686-011-9548-7 (2012).
Bongaerts, P. et al. Deep down on a Caribbean reef: lower mesophotic depths harbor a specialized coral-endosymbiont community. Sci. Rep. 5, 7652, https://doi.org/10.1038/srep07652 (2015).
Chérubin, L. M., Kuchinke, C. P. & Paris, C. B. Ocean circulation and terrestrial runoff dynamics in the Mesoamerican region from spectral optimization of SeaWiFS data and a high resolution simulation. Coral Reefs 27, 503–519, https://doi.org/10.1007/s00338-007-0348-1 (2008).
Ezer, T., Thattai, D. V., Kjerfve, B. & Heyman, W. D. On the variability of the flow along the Meso-American Barrier Reef system: a numerical model study of the influence of the Caribbean current and eddies. Ocean Dyn. 55, 458–475, https://doi.org/10.1007/s10236-005-0033-2 (2005).
Lindo-Atichati, D., Curcic, M., Paris, C. B. & Buston, P. M. Description of surface transport in the region of the Belizean Barrier Reef based on observations and alternative high-resolution models. Ocean Model. 106, 74–89, https://doi.org/10.1016/j.ocemod.2016.09.010 (2016).
Tang, L., Sheng, J., Hatcher, B. G. & Sale, P. F. Numerical study of circulation, dispersion, and hydrodynamic connectivity of surface waters on the Belize shelf. J. Geophys. Res. 111, C01003, https://doi.org/10.1029/2005JC002930 (2006).
Coles, S. L., Jokiel, P. L. & Lewis, C. R. Thermal tolerance in tropical versus subtropical Pacific reef corals. Pacific Sci. 30, 159–166 (1976).
Goreau, T. J. & Hayes, R. L. Coral Bleaching and Ocean “Hot Spots”. Ambio-Journal Hum. Environ. 23, 176–180, https://doi.org/10.2307/4314195 (1994).
Smith, T. B. et al. Caribbean mesophotic coral ecosystems are unlikely climate change refugia. Glob. Chang. Biol. 22, 2756–2765, https://doi.org/10.1111/gcb.13175 (2016).
Laverick, J. H., Andradi-Brown, D. A. & Rogers, A. D. Using light-dependent scleractinia to define the upper boundary of mesophotic coral ecosystems on the reefs of Utila, Honduras. PLoS One 12, 1–18, https://doi.org/10.1371/journal.pone.0183075 (2017).
Leichter, J. J., Helmuth, B. & Fischer, A. M. Variation beneath the surface: quantifying complex thermal environments on coral reefs in the Caribbean, Bahamas and Florida. J. Mar. Res. 64, 563–588, https://doi.org/10.1357/002224006778715711 (2006).
Shick, J. M., Lesser, M. P. & Jokiel, P. L. Effects of ultraviolet radiation on corals and other coral reef organisms. Glob. Chang. Biol. 2, 527–545, https://doi.org/10.1111/j.1365-2486.1996.tb00065.x (1996).
Banaszak, A. T., Michael, P., Kuffner, I. B. & Ondrusek, M. Relationship between ultraviolet (UV) radiation and mycosporine-like amino acids (MAAs) in marine organisms. 63, 617–628 (1998).
Vize, P. D., Embesi, J. A., Nickell, M., Brown, D. P. & Hagman, D. K. Tight temporal consistency of coral mass spawning at the Flower Garden Banks, Gulf of Mexico, from 1997-2003. Gulf Mex. Sci. 23, 107–114, https://doi.org/10.18785/goms.2301.08 (2005).
Szmant, A. Sexual reproduction by the Caribbean reef corals Montastrea annularis and M. cavernosa. Mar. Ecol. Prog. Ser. 74, 13–25, https://doi.org/10.3354/meps074013 (1991).
Vize, P. D. Deepwater broadcast spawning by Montastraea cavernosa, Montastraea franksi, and Diploria strigosa at the Flower Garden Banks, Gulf of Mexico. Coral Reefs 25, 169–171, https://doi.org/10.1007/s00338-005-0082-5 (2006).
Ritson-Williams, R., Becerro, M. A. & Paul, V. J. Spawning of the giant barrel sponge Xestospongia muta in Belize. Coral Reefs 24, 160–160, https://doi.org/10.1007/s00338-004-0460-4 (2005).
Levitan, D. R. et al. Mechanisms of reproductive isolation among sympatric broadcast-spawning corals of the Montastraea annularis species complex. Evolution 58, 308–323, https://doi.org/10.1111/j.0014-3820.2004.tb01647.x (2004).
Marshall, D. J., Monro, K., Bode, M., Keough, M. J. & Swearer, S. Phenotype-environment mismatches reduce connectivity in the sea. Ecol. Lett. 13, 128–140, https://doi.org/10.1111/j.1461-0248.2009.01408.x (2010).
Andrade, S. C. S. & Solferini, V. N. Fine-scale genetic structure overrides macro-scale structure in a marine snail: nonrandom recruitment, demographic events or selection? Biol. J. Linn. Soc. 91, 23–36, https://doi.org/10.1111/j.1095-8312.2007.00782.x (2007).
Harrington, L., Fabricius, K., De’ath, G. & Negri, A. Recognition and selection of settlement substrata determine post-settlement survival in corals. Ecology 85, 3428–3437 (2004).
Price, N. Habitat selection, facilitation, and biotic settlement cues affect distribution and performance of coral recruits in French Polynesia. Oecologia 163, 747–758, https://doi.org/10.1007/s00442-010-1578-4 (2010).
Raimondi, P. T. & Morse, A. N. C. The consequences of complex larval behavior in a coral. Ecology 81, 3193–3211 (2000).
Kisdi, É. Dispersal: risk spreading versus local adaptation. Am. Nat. 159, 579–596, https://doi.org/10.1086/339989 (2002).
Vigliola, L. et al. Genetic identity determines risk of post-settlement mortality of a marine fish. Ecology 88, 1263–1277, https://doi.org/10.1890/06-0066 (2007).
Soong, K. Colony size as a species character in massive reef corals. Coral Reefs 12, 77–83, https://doi.org/10.1007/BF00302106 (1993).
Studivan, M. S. & Voss, J. D. Assessment of mesophotic coral ecosystem connectivity for proposed expansion of a marine sanctuary in the Northwest Gulf of Mexico: population genetics. Front. Mar. Sci. 5, 1–11, https://doi.org/10.3389/fmars.2018.00152 (2018).
Chomczynski, P. & Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction: twenty-something years on. Nat. Protoc. 1, 581–585, https://doi.org/10.1038/nprot.2006.83 (2006).
Mieog, J. C., van Oppen, M. J. H., Berkelmans, R., Stam, W. T. & Olsen, J. L. Quantification of algal endosymbionts (Symbiodinium) in coral tissue using real-time PCR. Mol. Ecol. Resour. 9, 74–82, https://doi.org/10.1111/j.1755-0998.2008.02222.x (2009).
Schuelke, M. An economic method for the fluorescent labeling of PCR fragments. Nat. Biotechnol. 18, 233–234, https://doi.org/10.1038/72708 (2000).
Blacket, M. J., Robin, C., Good, R. T., Lee, S. F. & Miller, A. D. Universal primers for fluorescent labelling of PCR fragments-an efficient and cost-effective approach to genotyping by fluorescence. Mol. Ecol. Resour. 12, 456–463, https://doi.org/10.1111/j.1755-0998.2011.03104.x (2012).
Peakall, R. & Smouse, P. E. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research–an update. Bioinformatics 28, 2537–2539, https://doi.org/10.1093/bioinformatics/bts460 (2012).
Stenberg, P., Lundmark, M. & Saura, A. MLGsim: a program for detecting clones using a simulation approach. Mol. Ecol. Notes 3, 329–331, https://doi.org/10.1046/j.1471-8286.2003.00408.x (2003).
Theoretical research in evolutionarylife sciences, software and downloads. Available at, https://www.rug.nl/research/gelifes/tres/software. (Accessed: 12th October 2018).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567, https://doi.org/10.1111/j.1755-0998.2010.02847.x (2010).
Strimmer, K. fdrtool: a versatile R package for estimating local and tail area-based false discovery rates. Bioinformatics 24, 1461–1462, https://doi.org/10.1093/bioinformatics/btn209 (2008).
Chapuis, M. P. & Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 24, 621–631, https://doi.org/10.1093/molbev/msl191 (2007).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–59 (2000).
Besnier, F. & Glover, K. A. ParallelStructure: a R package to distribute parallel runs of the population genetics program STRUCTURE on multi-core computers. PLoS One 8, e70651, https://doi.org/10.1371/journal.pone.0070651 (2013).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806, https://doi.org/10.1093/bioinformatics/btm233 (2007).
Rosenberg, N. A. DISTRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138, https://doi.org/10.1046/j.1471-8286.2003.00566.x (2004).
Beerli, P. & Palczewski, M. Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics 185, 313–26, https://doi.org/10.1534/genetics.109.112532 (2010).
Beerli, P. Comparison of Bayesian and maximum-likelihood inference of population genetic parameters. Bioinformatics 22, 341–345, https://doi.org/10.1093/bioinformatics/bti803 (2006).
Meerman, J. & Clabaugh, J. Biodiversity and environmental resource data system of Belize. Available at, http://www.biodiversity.bz (2017).
Acknowledgements
We thank Z. Foltz and S. Jones for facilitating access to Smithsonian Carrie Bow Cay Field Station; D. Dodge, A. Shatters, and A. Alker for assistance in field collections; A. Reaume and A. Sturm for assistance in DNA extraction; and G. O’Corry-Crowe and T. Ferrer for sequencing runs. Computation capacity was provided by Research Computing Services at Florida Atlantic University. Funding for this research was awarded to J. Voss through the NOAA Office of Ocean Exploration and Research under awards NA09OAR4320073 and NA14OAR4320260 to the Cooperative Institute for Ocean Exploration, Research and Technology (CIOERT) at Florida Atlantic University’s Harbor Branch Oceanographic Institute. Additional funding was provided to R. Eckert through grant 33070 from the PADI Foundation. All corals were collected under CITES permits 7123 and 000023-17 from the Belize Fisheries Department. This is contribution 2202 from Harbor Branch Oceanographic Institute.
Author information
Authors and Affiliations
Contributions
J.D.V. designed the study and sampling scheme. R.J.E. and M.S.S. performed laboratory and microsatellite data analyses. R.J.E. created figures. All authors collected samples and contributed to the final edited manuscript prepared by R.J.E.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Eckert, R.J., Studivan, M.S. & Voss, J.D. Populations of the coral species Montastraea cavernosa on the Belize Barrier Reef lack vertical connectivity. Sci Rep 9, 7200 (2019). https://doi.org/10.1038/s41598-019-43479-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43479-x
This article is cited by
-
Breviolum and Cladocopium Are Dominant Among Symbiodiniaceae of the Coral Holobiont Madracis decactis
Microbial Ecology (2022)
-
Distinct lineages and population genomic structure of the coral Pachyseris speciosa in the small equatorial reef system of Singapore
Coral Reefs (2022)
-
Population genetic structure of the broadcast spawning coral, Montastraea cavernosa, demonstrates refugia potential of upper mesophotic populations in the Florida Keys
Coral Reefs (2022)
-
Coral distribution and bleaching vulnerability areas in Southwestern Atlantic under ocean warming
Scientific Reports (2021)
-
Depth-dependent parental effects create invisible barriers to coral dispersal
Communications Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
