
Abstract
In recent years, a 2D graphene-like sheet: monolayer C2N was synthesized via a simple wet-chemical reaction. Here, we studied the stability and electronic properties of bilayer C2N. According to a previous study, a bilayer may exist in one of three highly symmetric stacking configurations, namely as AA, AB and AB′-stacking. For the AA-stacking, the top layer is directly stacked on the bottom layer. Furthermore, AB- and AB′-stacking can be obtained by shifting the top layer of AA-stacking by a/3-b/3 along zigzag direction and by a/2 along armchair direction, respectively, where a and b are translation vectors of the unit cell. By using first-principles calculations, we calculated the stability of AA, AB and AB′-stacking C2N and their electronic band structure. We found that the AB-stacking is the most favorable structure and has the highest band gap, which appeared to agree with previous study. Nevertheless, we furthermore examine the energy landscape and translation sliding barriers between stacking layers. From energy profiles, we interestingly found that the most stable positions are shifted from the high symmetry AB-stacking. In electronic band structure details, band characteristic can be modified according to the shift. The interlayer shear mode close to local minimum point was determined to be roughly 2.02 × 1012 rad/s.
Similar content being viewed by others
Introduction
Since graphene was discovered1,2,3,4,5, two-dimensional (2D) materials have attracted tremendous attention due to their novel electric, thermal, mechanical and optoelectronic properties6,7,8. The intrinsic zero-band gap of graphene is, however, unsuitable for many nanoelectronic device applications1. A number of modifications have been demonstrated to open a gap in graphene, such as doping and truncating to nanoribbons9,10,11,12,13,14. Other 2D materials have also been identified which possess a band gap, such as hexagonal boron nitride (h-BN) monolayer15,16, graphitic carbon nitride (C3N4)17, transition-metal dichalcogenides (TMDs)18, group-IV (silicene and germanene)19, group-V (phosphorene)20, group III–V (such as monolayer GaAs, InN and AlN)21,22 and especially molybdenum disulfide (MoS2)23. Recently, a monolayer carbon with nitrogen named C2N was synthesized via a bottom-up wet-chemical reaction24. C2N possesses an optical band gap of 1.96 eV which can be used for photocatalytic and electronic devices. Consequently, it is worthwhile to study their physical and electronic properties in order to gain a deeper understanding of this promising material for nanoelectronic devices.
The physical and electronic properties of C2N have received considerable attention in terms of density functional theory (DFT) calculations since it was synthesized24. Zhang et al.25 performed DFT-GGA calculations to investigate the physical and electronic structures of monolayer and bilayer C2N. In monolayer, they found that the optimized lattice parameter of C2N is a = b = 8.330 Å, two different types of C-C bonds are 1.429 and 1.470 Å, and the C-N bond is 1.336 Å. In electronic structure, the calculated band gap is 1.66 eV at the Γ point in DFT-GGA calculation24,25,26. As commonly known, DFT-GGA severely underestimate band gap attributed to the self-interaction error (SIE). Zhang et al. also25 performed band structure calculation using the hybrid functional (HSE). This calculated HSE band gap is 2.47 eV25 which is much higher than 1.96 eV of the experimental value24. In their research25, they are the first group who reported the structure and the stability of bilayer C2N, which provides three high-symmetry stacking orders (AA, AB and AB′-stacking) with different energy gaps. However, to identify the most favorable configuration, high-symmetry configuration is insufficient. Asymmetric configurations must also be taken into account. In fact, this issue is very important for evaluating the stacking stability of the bilayers. For example in MoS2, Tao Peng et al.27 provided the conclusion that the high symmetry stacking order with Mo and S superposed from both layers is the most stable stacking order. Similarly, bilayer h-BN gives the minimum energy sliding path only in AA′ high symmetry stacking with an energy barrier of 3.4 meV per atom28. Obviously, in most typical bilayer systems, the most stable configurations involve high symmetry or nearly high symmetry stacking order. However, in bilayer silicene, Huixia Fu et al.29 found several meta-stable configurations which are slightly shifted from the high symmetry AA-stacking structure, namely slide-2AA.
Here, we performed first-principles calculations to investigate the stability and electronic structure of bilayer C2N based on density functional theory (DFT). For a benchmark, we first studied the electronic properties of the monolayer with DFT-GGA and found that monolayer C2N has a direct band gap of 1.665 eV at Γ points, which agrees with previous works24,25. The relaxed structure and covalent bonds are agreed with previous work25. For bilayer calculations, the stability of symmetric and non-symmetric configuration of bilayer C2N were explored. Interestingly, we found that the high symmetry AB-configuration of bilayer C2N reported in previous study25 is not the most favorable structure. The most stable stacking is slightly shifted from AB-configuration. In this work, the stability and energy barriers have been also identified.
Computational Method
Our calculations were based on the DFT as implemented in the Vienna ab-initio simulation package (VASP)30,31. The projector-augmented wave (PAW) method were used32 to treat the core electrons. For monolayer calculation, in order to obtain a more accurate description of the electronic states, the optimized lattice parameter and band structures were calculated using the hybrid functional (HSE). For bilayer calculation, The generalized gradient approximation of Perdew, Burke, and Ernzerhof (GGA-PBE) were used. The kinetic energy cut-off was set to 520 eV for the plane-wave expansions. The gamma centred 5 × 5 × 1 and 7 × 7 × 1 k-point sampling are adopted for monolayer and bilayer calculation parts, respectively. The total energy calculations were performed using the 18-atom C2N unit cell and 36-atom C2N unit cell for monolayer and bilayer C2N, respectively. The vacuum region of 15 Å was used to avoid the interactions between two adjacent periodic images. The electronic optimization and structural relaxations were performed until energy and forces converged to 10−4 eV (monolayer) and 10−6 eV (bilayer) and 0.02 eV/Å, respectively. In order to accurately describe the effect of van der Waals (vdW) interactions, we used the empirical correction method proposed by Grimme (GGA + D233 and GGA + D334) which is a good description of long-range vdW interactions35,36,37. To explore the nature of the long-range Hartree-Fock exchange interactions, the HSE + D3 approaches have been computed. Note that in order to compute bilayer, vdW interactions need to be considered. Finally, the rVV10 nonlocal correlation functional (SCAN + rVV10)38 has been computed for comparing our result.
Results and Discussion
At first, we explored the geometric properties and electronic structure of monolayer C2N. A unit cell of C2N consists of 12 carbon atoms and 6 nitrogen atoms with uniform holes in the layer. The atomistic ball-stick model of the monolayer C2N is illustrated in Fig. 1(a). In C2N, there are three types of covalent bonds: the in-plane C-N, C-C1 (C-C bonds in benzene rings) and C-C2 (C-C bonds in pyrazine rings) as shown in Fig. 1(a). As mentioned before, it is well-known that DFT underestimate band gap attributed to the self-interaction error (SIE), thus we used hybrid exchange-correlation functional to calculate the electronic band structure. Hybrid functionals, which mix a fraction of Hartree-Fock (HF) exchange with local or semilocal exchange, have become increasingly favorite in DFT calculation. In this work, we vary the percentage of HF exchange from the experimental value of α = 0.25 to 0.05 in order to mimic the experimental band gap. The mixing parameter 5% can perfectly match the experimental gap. In HSE, the lattice parameter of monolayer C2N is 8.317 Å which is little lower than that of GGA values of 8.330 Å25. In HSE calculation, the in-plane C-N, C-C1 and C-C2 bonds (see Fig. 1(a)) are 1.334, 1.468 and 1.427 Å, while the DFT-GGA in-plane C-N, C-C1 and C-C2 bonds are 1.336, 1.470 and 1.429 Å by Zhang et al.25 and of 1.337, 1.470 and 1.429 Å by Guan et al.26, respectively. Note that in all calculations, C-C1 bonds are about 3% longer than C-C2 bonds. The presence of N atoms results in the small distortion of the benzene rings. The lower of lattice parameter and covalent bond lengths comes from a strong localized wave-function in HSE.

(a) Unit cell of monolayer C2N crystal structure. C-C1 and C-C2 are C-C bonds presented in benzene and pyrazine ring, respectively. The C atoms are represented by brown balls and the N atoms are represented by grey balls. (b) Calculated band structures of monolayer C2N. M(0.5, 0.0, 0.0), Γ(0.0, 0.0, 0.0) and K(1/3, 1/3, 0.0) refer to special points in the first Brillouin zone.
The HSE calculated band structure of monolayer C2N is illustrated in Fig. 1(b). Note that in GGA calculation, the distribution of valence band maximum (VBM) and conduction band minimum (CBM) is well separated spatially with a direct energy gap of 1.962 eV; the former mainly contains the nitrogen px, py states and the latter is mainly from the nitrogen pz state. The pz states of VBM shift up over px, py when introduced HF exchange as in HSE calculation. Interestingly, when mixing parameter of 5% HF is included, the px, py and pz band states lie exactly the equal location at the VBM as seen in Fig. S1 in Supplementary information (SI). Since both of the valence and conduction bands are well dispersed and show no localized states to act as recombination centres for the photogenerated electron-hole pairs, C2N is good for the separation of photogenerated electron-hole pairs.
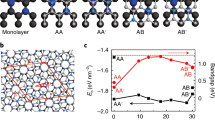
Considering the bilayer C2N, there are three high-symmetry stacking configurations, which were reported in the previous study by Zhang et al.25, namely AA, AB and AB′, as shown in Fig. 2(a–c). In AA-stacking, the top layer is stacked directly above the bottom layer. In AB stacking, the top layer is shifted along the zigzag direction by a/3-b/3, where a and b are translation vectors of the unit cell. For AB′ stacking, the top layer is shifted along the armchair direction by a/2. As presenting in Table 1, the total energy calculations indicate that AB-stacking is the most favorable stacking with the total energy of 13.292 and 2.58 meV/atom lower than that of AA- and AB′-stacking, respectively. Regarding of the previous work by Zhang et al.25, they proposed three high-symmetry structures including AA, AB and AB′ and suggested that the lowest energy configurations is AB, which agrees with our result. In Zhang et al.25, the energy difference of AA and AB′ with respect to the energy of AB stacking order are 16 and 3 meV/atom.

(a–d) AA-, AB-, AB′ and Min-stacking structures of 2 × 2 supercell bilayer C2N, respectively.
The calculated lattice constant (a = |a| = |b|), interlayer distance (h0), in-plane covalent bond lengths and energy gap of bilayer C2N with three different stacking orders are listed in Table 2. In symmetric AA, AB, and AB′ stacking orders, the in-plane covalent bond lengths C-N, C-C1 and C-C2 show no significant change with different stacking. Note that there is no meaningful difference of in-plane bond lengths between monolayer and bilayer due to the strong covalent bonding. The electronic band structures of bilayer AA, AB, and AB′-stacked C2N are shown in Fig. 3(a–c). It is clear that all of them are direct band gap, where VBM and CBM are located at the Γ point. Among these three different stacking orders, the AB-stacked bilayer possesses the widest band gap of 1.467 eV, while for the AA and AB′-stacked bilayer, the band gaps are 1.337 eV and 1.169 eV, respectively. Regarding the previous work by Zhang et al.25, the band gap of AB-stacking is 1.49 eV, while the band gap of AA and AB′-stacking are 1.34 eV and 1.21 eV. The three high symmetry bilayer configurations retained the direct band gap with different values according to stacking structure. Note that all energy gaps of the three bilayer stacking orders are smaller than the monolayer. The interlayer distance of AA, AB and AB′-stacking are 3.547, 3.203 and 3.242 Å, respectively. Consequently, the most energetically favourable, AB-stacking, gives the lowest interlayer distance.

(a–d) The electronic band structures of AA, AB, AB′ and Min-stacking order, respectively.
In this work, a more comprehensive energy landscape has been investigated in order to explore other possible equilibrium structures of bilayer C2N apart from the three proposed structures. We strive for more energetically stable configurations by performing two-dimensional energy scanning along both zigzag (ΔX) and armchair (ΔY) directions. The AA, AB, and AB′ stacking orders are equivalent to the shift of (ΔX = 0, ΔY = 0), (ΔX = 4.811 Å, ΔY = 0), and (ΔX = 3.609 Å, ΔY = 2.084 Å), respectively. For every given shift, the atomic position and the interlayer distance of the bilayer are allowed to relax. Figure 4 shows the 2D energy surface of bilayer C2N, referenced to AB stacking, as a function of relative shift (ΔX, ΔY). The discrete set of the sliding energy were interpolated to finer mesh by using Renka II procedure39,40,41. In Fig. 4, it is clearly shown that the high symmetry AB-stacking is not the most favourable stable configuration. Interestingly, there are six energetically favourable configurations surrounding the AB stacking. More specifically there are three symmetric regions where the energy is lower than the AB configuration. In each region, there are two actual minimum points (Min) marked as white dot in Fig. 4. The energy difference between Min and AB point in our calculation is approximately 1.81 meV/atom. The different choice of dispersion correction (GGA + D3) is also considered. Surprisingly, the actual minimum points calculated from GGA + D3 are all exactly the same position as GGA + D2 which shown in Fig. S2 in SI.

2D-energy profile surface of bilayer C2N depending on the inter-layer relative slide distance (ΔX, ΔY). The colour scale and contours represent the energy per atom relative to the energy per atom of AB. The solid contour line is 1 meV/atom increment. The minimum energy points are represented by white dots. Path I, Path II, Path III are the translation paths between two minimum point of intra-region, inter-region and cross over region, respectively.
The energy differences of the symmetric AA, AB, AB′ and Min point stacking with respect to the energy of AB-stacking are shown in Table 1. The calculated energy difference confirmed that the AB stacking configuration is not the most favourable structure for bilayer C2N. The local structure of one of the minimum energy points (Min) is shown in Fig. 2(d). Regarding the symmetric AA, AB, and AB′-stacking, all atoms on the top layer are located directly above the bottom layer. In Min point stacking of Fig. 2(d), all atoms on the top layer are not located directly above those of the bottom layer. The Min configuration is slightly shifted from the high symmetry AB-stacking structure. Unlike the symmetric AA, AB, and AB′-stacking, the in-plane covalent bond lengths C-N, C-C1 and C-C2 of the Min point are not equal, so we refer to the average bond length. The average bond length of C-C1, C-C2 and C-N are 1.469, 1.430 and 1.336 Å, respectively. The interlayer distance (h0) is 3.099 Å which is slightly lower than that of the AB-stacking structure. The average bond length and interlayer distance are listed in Table 2.
The electronic band structure corresponding to the configuration in Fig. 2(d) is presented in Fig. 3(d). Since the Min configurations are surrounding the AB stacking configuration, the calculated band structure of Min point looks similar to high-symmetry AB stacking order. The forbidden gap is still the direct-gap with a band gap of 1.444 eV. However, the band gap is lower than that of the high-symmetry AB stacking order by 23 meV.
The stability of the bilayer can be represented in terms of the formation of energy. We calculated the formation energy per atom (Eform) as a function of adjacent layer distance for each stacking order based on the GGA + D2 functional as shown in Fig. 5. The formation energy is defined as the total energy difference per atom between the bilayer and the component of two monolayers which are separated with adequate distance (Eform = Ebilayer − 2Emonolayer). The formation energy is the energy required to produce a bilayer from monolayers. The structures of bilayer C2N have similar trends of formation energy per atom. In Fig. 5, the Min stacking possesses the lowest formation energy of −23.85 meV/atom with the minimum interlayer distance of 3.099 Å. For the three high-symmetry stacking, AA, AB′ and AB-stacking, the formation energies are −9.8 −20 and −23 meV/atom which correspond the minimum interlayer distance of 3.547, 3.242 and 3.203 Å respectively. This demonstrates that the Min stacking configuration is the most favourable in term of stability. In order to speed up the exploration of exchange-correlation calculation (HSE + D3, SCAN + rVV10), we adopted the following Min-stacking configuration from GGA + D2/GGA + D3 to calculate the formation energy. The formation energy of AA, AB′, AB and Min-stacking of HSE + D3 and SCAN + rVV10 are shown in Fig. S3 in SI. It is clear that the formation energy of the Min position are lower than that of AB position in both HSE + D3 and SCAN + rVV10. These mean there are truly more favorable structure than AB.

The formation energy (Eform) of each stacking order as a function of inter-layer distance.
In Fig. 4 with deliberating at the Min points, the sliding paths between the Min points can be considered. There are three possible sliding paths. The first one is connected between two local minimum points in the same region called intra-region Path I. The second path is connected between two local minimum points in different regions called inter-region Path II. The third path is connected between two local minimum points between cross-over regions called Path III. The translated energy barrier demonstrates how difficult it is from one stacking to another by sliding one layer along a specific path. The translated energy barrier corresponding to path I, II and III as a function of distance r of the two local minimum points are shown in Fig. 6. Obviously, it is difficult to slide along path III. However, it might be possible that there is a transformation from two minimum point in path I and path II because they require small amount of energies of 0.076 and 0.319 meV/atom to overcome these energy barriers.

Translated energy barrier as a function of distance r between two minimum points along the Path I, II and III. The unit of energy barrier and distance r are meV/atom and Å, respectively.
Next, imaging the existing forces holding two layers together act like springs, linking one C2N layer to the next layer, it is possible to calculate the spring constant (k) at the minimum structure of bilayer C2N. For estimation, the spring constant in zigzag and armchair direction are equal at the Min point. Along armchair direction, the energy-strain relation equation can be fitted with the equation E(x) = kx2/2, where x is the displacement from Min Point. In this case, the fitted spring constant value is 0.0859 N/m per atom. This solution leads us to find the force and the angular frequency of the most stable C2N bilayer structure. In this present work, the calculated frequency of the interlayer shear mode around local minimum point is approximately 2.02 × 1012 rad/s or roughly 0.322 THz.
Summary
In summary, we performed first-principles calculations to explore the stacking stability and electronic properties of C2N bilayer. The stacking order and electronic band structure of three high-symmetry stacking configurations namely as AA, AB and AB′-stacking were calculated. Among these three high-symmetry stacking configurations, AB-stacking is the most favourable with smallest interlayer distance but highest band gap. Our results agree well with the previous study25. More comprehensive energy landscape has been studied to explore other possible configurations. We however found that there are three regions around AB which provide truly lower energy per atom than AB-stacking structures. In these regions, all atoms on the top layer are shifted from the bottom layer. Fascinatingly, the electronic band structures of the stable configuration express the same fashion as the high-symmetry AB stacking but they differ from AB-stacking in term of the energy gap and band line splitting. Importantly, the energy gap can be modified according to the stacking order. In terms of stability, the formation energy (Eform) per atom confirms the fact that these regions are more stable than AB stacking. Lastly, the frequency of the interlayer shear mode was determined to be 2.02 × 1012 rad/s or 0.322 THz.
Change history
03 March 2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
References
Geim, A. K. & Novoselov, K. S. The rise of graphene. Nat. materials 6, 183 (2007).
Novoselov, K. et al. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. United States Am. 102, 10451–10453 (2005).
Novoselov, K. S. et al. Two-dimensional gas of massless dirac fermions in graphene. nature 438, 197 (2005).
Xu, M., Liang, T., Shi, M. & Chen, H. Graphene-like two-dimensional materials. Chem. reviews 113, 3766–3798 (2013).
Butler, S. Z. et al. Progress, challenges, and opportunities in two-dimensional materials beyond graphene. ACS nano 7, 2898–2926 (2013).
Novoselov, K. S. et al. Electric field effect in atomically thin carbon films. science 306, 666–669 (2004).
Novoselov, K. S. et al. Room-temperature quantum hall effect in graphene. Sci. 315, 1379–1379 (2007).
Neto, A. H. C., Guinea, F., Peres, N. M. R., Novoselov, K. S. & Geim, A. K. The electronic properties of graphene. Rev. modern physics 81, 109 (2009).
Ni, Z. H. et al. Uniaxial strain on graphene: Raman spectroscopy study and band-gap opening. ACS nano 2, 2301–2305 (2008).
Fan, X., Shen, Z., Liu, A. & Kuo, J.-L. Band gap opening of graphene by doping small boron nitride domains. Nanoscale 4, 2157–2165 (2012).
Calvo, H. L., Pastawski, H. M., Roche, S. & Torres, L. E. F. Tuning laser-induced band gaps in graphene. Appl. Phys. Lett. 98, 232103 (2011).
Chung, H. C., Chang, C. P., Lin, C. Y. & Lin, M. F. Electronic and optical properties of graphene nanoribbons in external fields. Phys. Chem. Chem. Phys. 18, 7573–7616 (2016).
Chung, H. C., Lee, M. H., Chang, C. P. & Lin, M. F. Exploration of edge-dependent optical selection rules for grapheme nanoribbons. Opt. express 19, 23350–23363 (2011).
Dutta, S. & Pati, S. K. Novel properties of graphene nanoribbons: a review. J. Mater. Chem. 20, 8207–8223 (2010).
Kim, K. K. et al. Synthesis of monolayer hexagonal boron nitride on cu foil using chemical vapor deposition. Nano letters 12, 161–166 (2011).
Peng, Q., Ji, W. & De, S. Mechanical properties of the hexagonal boron nitride monolayer: Ab initio study. Comput. Mater. Sci. 56, 11–17 (2012).
Wang, X. et al. A metal-free polymeric photocatalyst for hydrogen production from water under visible light. Nat. materials 8, 76 (2009).
Chhowalla, M. et al. The chemistry of two-dimensional layered transition metal dichalcogenide nanosheets. Nat. chemistry 5, 263 (2013).
Matthes, L., Gori, P., Pulci, O. & Bechstedt, F. Universal infrared absorbance of two-dimensional honeycomb group-iv crystals. Phys. Rev. B 87, 035438 (2013).
Liu, H. et al. Phosphorene: an unexplored 2d semiconductor with a high hole mobility. ACS nano 8, 4033–4041 (2014).
Chung, H.-C., Chiu, C.-W. & Lin, M.-F. Spin-polarized magneto-electronic properties in buckled monolayer gaas. Sci. reports 9, 2332 (2019).
Zhuang, H. L., Singh, A. K. & Hennig, R. G. Computational discovery of single-layer iii-v materials. Phys. Rev. B 87, 165415 (2013).
Mak, K. F., Lee, C., Hone, J., Shan, J. & Heinz, T. F. Atomically thin mos2: a new direct-gap semiconductor. Phys. review letters 105, 136805 (2010).
Mahmood, J. et al. Nitrogenated holey two-dimensional structures. Nat. communications 6, 6486 (2015).
Zhang, R., Li, B. & Yang, J. Effects of stacking order, layer number and external electric field on electronic structures of few-layer c2n-h2d. Nanoscale 7, 14062–14070 (2015).
Guan, S. et al. Effects of strain on electronic and optic properties of holey two-dimensional c2n crystals. Appl. Phys. Lett. 107, 231904 (2015).
Peng, T., Huai-Hong, G., Teng, Y. & Zhi-Dong, Z. Stacking stability of mos2 bilayer: An ab initio study. Chin. Phys. B 23, 106801 (2014).
Constantinescu, G., Kuc, A. & Heine, T. Stacking in bulk and bilayer hexagonal boron nitride. Phys. review letters 111, 036104 (2013).
Fu, H., Zhang, J., Ding, Z., Li, H. & Meng, S. Stacking-dependent electronic structure of bilayer silicene. Appl. Phys. Lett. 104, 131904 (2014).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. materials science 6, 15–50 (1996).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. review B 54, 11169 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. review letters 77, 3865 (1996).
Grimme, S. Semiempirical gga-type density functional constructed with a long-range dispersion correction. J. computational chemistry 27, 1787–1799 (2006).
Grimme, S., Antony, J., Ehrlich, S. & Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (dft-d) for the 94 elements h-pu. The J. chemical physics 132, 154104 (2010).
Hu, W., Wu, X., Li, Z. & Yang, J. Helium separation via porous silicene based ultimate membrane. Nanoscale 5, 9062–9066 (2013).
Hu, W., Wu, X., Li, Z. & Yang, J. Porous silicene as a hydrogen purification membrane. Phys. Chem. Chem. Phys. 15, 5753–5757 (2013).
Zhang, R., Li, B. & Yang, J. A first-principles study on electron donor and acceptor molecules adsorbed on phosphorene. The J. Phys. Chem. C 119, 2871–2878 (2015).
Peng, H., Yang, Z.-H., Perdew, J. P. & Sun, J. Versatile van der waals density functional based on a meta-generalized gradient approximation. Phys. Rev. X 6, 041005 (2016).
Renka, R. J. Multivariate interpolation of large sets of scattered data. ACM Transactions on Math. Softw. (TOMS) 14, 139–148 (1988).
Renka, R. J. Algorithm 660: Qshep2d: Quadratic shepard method for bivariate interpolation of scattered data. ACM Transactions on Math. Softw. (TOMS) 14, 149–150 (1988).
Renka, R. J. Algorithm 790: Cshep2d: Cubic shepard method for bivariate interpolation of scattered data. ACM Transactions on Math. Softw. (TOMS) 25, 70–73 (1999).
Acknowledgements
K.D. was supported by the Graduate School, Kasetsart University, Thailand. S.J. was supported by Energy Conservation Promotion Fund (Energy Policy and Planning Office, Ministry of Energy) and National Science and Technology Development Agency (NSTDA), Thailand. A.B. was supported by the Institute for Promotion of Science and Technology (DPST Research Grant No. 009/2558). We wish to thank High-performance computing facilities King Mongkut’s University of Technology Thonburi (KMUTT, Thailand) for their hospitality.
Author information
Authors and Affiliations
Contributions
K.D. performed DFT calculations and contributed to analysing the first-principles calculations. J.T. contributed to analysing electric band structure. S.J. contributed to analysing 2D energy profile surface and frequency of shear mode. A.B. proposed the project, wrote the initial version of the manuscript and contributed to analyze data. All authors commented and reviewed on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dabsamut, K., T-Thienprasert, J., Jungthawan, S. et al. Stacking stability of C2N bilayer nanosheet. Sci Rep 9, 6861 (2019). https://doi.org/10.1038/s41598-019-43363-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43363-8
This article is cited by
-
A simple and efficient process for the synthesis of 2D carbon nitrides and related materials
Scientific Reports (2023)
-
Chirality effects on an electron transport in single-walled carbon nanotube
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
