
Abstract
Quantifying the genetic diversity of riparian trees is essential to understand their chances to survive hydroclimatic alterations and to maintain their role as foundation species modulating fluvial ecosystem processes. However, the application of suitable models that account for the specific dendritic structure of hydrographic networks is still incipient in the literature. We investigate the roles of ecological and spatial factors in driving the genetic diversity of Salix salviifolia, an Iberian endemic riparian tree, across the species latitudinal range. We applied spatial stream-network models that aptly integrate dendritic features (topology, directionality) to quantify the impacts of multiple scale factors in determining genetic diversity. Based on the drift hypothesis, we expect that genetic diversity accumulates downstream in riparian ecosystems, but life history traits (e.g. dispersal patterns) and abiotic or anthropogenic factors (e.g. drought events or hydrological alteration) might alter expected patterns. Hydrological factors explained the downstream accumulation of genetic diversity at the intermediate scale that was likely mediated by hydrochory. The models also suggested upstream gene flow within basins that likely occurred through anemophilous and entomophilous pollen and seed dispersal. Higher thermicity and summer drought were related to higher population inbreeding and individual homozygosity, respectively, suggesting that increased aridity might disrupt the connectivity and mating patterns among and within riparian populations.
Similar content being viewed by others
Introduction
Riparian trees are foundation species that support biodiversity and modulate key ecosystem functions through their interactions with flooding and sediment regimes in river channels and their floodplains1,2. Rivers have been exposed to long-lasting human pressures worldwide, and they are threatened by climate change and a resurgence of damming plans in response to freshwater and energy demands3. On the one hand, river regulation alters peak flows and creates physical barriers to gene flow, which hinder the regeneration dynamics of riverine plant communities4. On the other hand, climate-driven changes, such as precipitation shifts, might decouple seed development and dispersal from the discharge regime to which they evolved5. Improving our capacity to understand and anticipate changes in these fragile ecosystems is of the utmost importance if we are to mitigate the expected pervasive environmental and societal consequences of hydroclimatic alterations3. However, the current approaches used to monitor functional responses of riparian forests to global change do not fully accommodate the dendritic structures of hydrographic networks, which hinders the accurate management of these threatened ecosystems. Here, we combined spatial stream-network (SSN) models with landscape genetics tools to quantify the role of ecological factors in determining the amount and distribution of genetic diversity harboured in riparian forests. By doing so, we tested hypotheses on the main drivers of gene flow and connectivity at the population, basin and regional scales, and we showed that this monitoring tool is suitable to design science-based conservation plans.
Riparian forests are confined along dendritic hydrographic networks with typically directional water flow that disperses seeds and vegetative propagules downstream. Any attempt to investigate riparian genetic patterns requires accommodating the dendritic structure of hydrographic networks and its specific topology, connectivity and directionality within the hierarchical organization of riparian landscapes6. Spatial models previously applied to investigate genetic patterns across dendritic structures, such as rivers, have provided interesting but limited information because (1) Euclidean geographic distances disregard the complexities of hydrographic networks within nested watersheds7; (2) proxies of the basin hierarchy (i.e., stream order) poorly capture the topological properties of dendritic networks8; and (3) models do not tackle the joint effects of environmental and spatial components together, providing an incomplete picture of the multiple factors that drive genetic patterns9. SSN models7 provide a timely opportunity to investigate the joint effects of environmental drivers and spatial properties on a hydrographic network in riparian forests, and these models provide a complete and validated methodological toolbox10,11. These models integrate a set of explanatory variables into a single geostatistical model that can accommodate different spatial autocorrelation structures and use spatial weights to capture the influence of branching, flow direction, and discharge7. SSN models have been successfully applied to detect spatial patterns of water chemistry12 and the distribution of vagile organisms7,10,13, but they have not yet been applied to investigate the distribution of the genetic diversity of riparian forest species across different hydrographic networks.
In this study, we combined landscape genetic tools and SSN models to quantify the impacts of key ecological drivers on the distribution of the genetic diversity and structure of the riparian tree Salix salviifolia Brot. at various spatial scales. S. salviifolia is a foundation species that preferentially grows in intermediate to large order streams where natural flow, erosion and fluvial sedimentation processes create in-channel deposit bars14. In this species, gene flow occurs through pollen grains (transported by wind and insects), seeds (mobilized by wind and water) and vegetative propagules transported by water flow. Therefore, the disruption of the natural water flow and sediment regimes by river regulation threatens the dispersal ability and the genetic connectivity of these populations15,16. Furthermore, this taxon is endemic to the Western Iberian Peninsula, listed in the EU Habitats Directive, and it creates habitats that host IUCN Endangered Mediterranean fish species such as Anaecypris hispanica. The distribution of S. salviifolia encompasses a pronounced climatic gradient that spans from the southern Mediterranean edge to the Temperate ecoregion17. Hence, S. salviifolia is an ideal model species for studying the environmental and spatial drivers of genetic patterns in riparian species.
Water flow is the main vector that mobilizes propagules for riparian species downstream18, which causes the accumulation of riparian plant propagules downstream unless there are other means of upstream dispersal (the so-called drift-paradox hypothesis (Fig. 1)19. As a result, genetic diversity is expected to increase downstream. This observation has received empirical support that shows a dominant downstream gene flow direction6,20, although some studies have documented bidirectional gene flow9,21. Dominant winds or foraging preferences by pollinators can move pollen grains and genes upstream22. Moreover, local and regional landscape features, such as elevation or climatic gradients, might affect the gene flow patterns within or among basins (Fig. 2), and these features could potentially erase the expected genetic patterns derived from dominant downstream dispersal8. Applying SSN models enables us to dissect the contribution of environmental and spatial factors in determining the levels of genetic diversity across basins, populations and individuals. Specifically, this multiscale approach allows us to (i) investigate the genetic diversity levels of riparian populations in basins along an environmental gradient; (ii) quantify the relative contributions of ecological and spatial factors; and (iii) assess the spatial extent at which dominant factors impact the genetic patterns across dendritic networks. Finally, we further discussed the potential applications of SSN models to monitor the responses of riparian ecosystems to global change drivers.

(A) Hypothesis tested at different spatial scales for the main drivers of genetic patterns in Salix salviifolia populations. At the across-region scale, we expected higher genetic diversity in optimal climatic conditions. At the within-basin scale, we expected asymmetrical dispersal (“drift hypothesis”) resulting in the downstream increase in genetic diversity. (B) Within-basin spatial relationships (flow-connected/flow-unconnected) of the spatial stream-network model functions (tail-up/tail-down) adapted from Peterson & Ver Hoef7. The moving-average functions (MAF) for the tail-up (a, c) and tail-down (b, d) relationships are shown with varying widths representing the strength of the influence for each potential neighbouring site. Spatial autocorrelation occurs between sites when the MAF overlaps (grey), otherwise no spatial autocorrelation is considered (black). The black dots represent sites within the dendritic network.

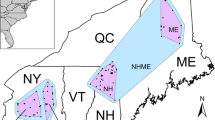
Study area within Europe (A), the three studied regions of Tua, Zêzere and Algarve (B) and sampled populations within hydrographic networks (C–E).
Results
Spatial genetic patterns in Salix salviifolia
Locally, ca. 15% of sites show signs of a deficit or excess of heterozygotes (Table 1), indicating that the allele frequencies at these sites depart from the Hardy-Weinberg Equilibrium (HWE). All estimators of genetic differentiation detected a significant genetic structure within regions (Supplementary Table S1), and the southern region showed the highest genetic structure levels and high proportions of private alleles (PA) (Supplementary Table S2).
Geostatistical modelling: covariates and covariance structures
The optimal sets of covariates were similar across the population-level response variables, and the hydrologic index (DA) was selected across the four estimators Ae, Fis, uHe and Ho (Ae, number of effective alleles; Fis, inbreeding; uHe, expected unbiased heterozygosity; Ho, observed heterozygosity) (Table 2) and had a positive impact on genetic diversity (in terms of Ae). Therefore, larger and wetter drainage basins tended to correlate with increased levels of genetic diversity. In addition, Ho was significantly and positively correlated with altitude (ALT). Climatic covariates were retained for Fis and HL (homozygosity level). The thermicity index (BIOC.TH) had a positive effect on Fis and, at the individual-level, the summer ombrothermic index (BIOC.SO) significantly affected the HL, with higher HL levels at decreasing BIOC.SO values. This result entails that locations undergoing intense summer droughts tend to host individuals and populations with increased levels of homozygosity.
The optimal covariance structure was a mixture of the possible structures, but with an overriding dendritic model (tail-up or tail-down) across most genetic estimators (Table 2, Fig. 3). The dominant covariance structures were the tail-up for Ae and HL, (47.9% and 75.4% explained variation, respectively); the tail-down for Fis and Ho (56.6% and 70.3% explained variation, respectively) and the Euclidean for uHe (64.5% of the variation explained).

Percentage of genetic variation explained by covariates (environmental factors) and by the different covariance structures within the final model mixture for the analysed genetic estimators at the population (Ae, number of effective alleles; Fis fixation index; uHe (unbiased expected heterozygosity; Ho, observed heterozygosity) and the individual (HL, homozygosity level) level. The nugget represents the unexplained variation.
Geostatistical modelling: Multiscale drivers of genetic diversity
Overall, the retained covariates together explained between 6 and 30% of the genetic variation at the population level for all genetic metrics with a strong hydrologic and climatic component (Table 2, Fig. 3). The percentage of residual variation corresponding to the covariance functions (tail-up, tail-down) accounts for most of the remaining variation. The nugget (variability that cannot be explained by the distance between observations, i.e. the unexplained variation) represented a low (≤10%) percentage of the variation. For all population-level estimators (Ae, Fis, uHe, Ho), autocovariance models captured intermediate-scale spatial patterns with spatial ranges of 0.2–20 km. For Ae and Fis, the covariance mixture also captured large-scale patterns of variation (ranges >100 km) (Table 2).
At the individual level, most of the residual variation corresponds to dendritic structures that fitted a tail-up model with a range close to 0 m in the best model for the HL (Table 2, Fig. 3). This result indicates that nearby individuals do not show increased HLs compared to individuals randomly drawn from distant locations in the population. In addition, the tail-down covariance structure captures 14.2% of the variation, and it detected a fine-scale structure (range = 11.9 m). The nugget represented 9.1% of the variability.
Discussion
The amount of genetic diversity and its spatial distribution within and among populations reflect complex interactions between intrinsic and extrinsic factors such as phenology, dispersal, topography and flood regime2. Lately, numerous landscape models have been pursued to disentangle the effects of different ecological and spatial factors in determining the distribution of the genetic diversity across complex landscapes23; however, these models fail to capture dendritic structures that characterize riparian habitats. SSN models proved to be suitable for quantifying the impact of environmental factors on shaping spatial genetic patterns, the spatial scale they operate, and their dominant direction (upstream, downstream or both).
The Iberian endemic Salix salviifolia displayed higher levels of genetic diversity (uHe) compared to other Salix species15,20, which is coherent with mating processes that favour outcrossing and suggests a relatively good genetic status in this species, at least under the current environmental conditions. Migration history might have modulated the current genetic patterns of these populations, as favourable microclimatic conditions within protected river valleys in the Iberian Peninsula offered refugia for tree species during glaciations24. The prevalence of private alleles may be interpreted as genetic evidence for persistence of S. salviifolia in the southern part of the peninsula during the last glacial period, as shown for other riparian species24.
The movements of propagules or individuals that inhabit riparian habitats are confined to the river flow, either upstream, downstream or both. Our results showed that a large proportion of the residual genetic variation is spatially structured within the basin (i.e., once the effects of ecological factors have been removed). Furthermore, by applying SSN models, we evidenced that the dendritic spatial structure accounted for majority (70–89%) of the variation observed within basins for most estimators of genetic diversity. Therefore, overlooking the dendritic structure of riparian habitats could lead to a misunderstanding of the main ecological drivers that underlie the biodiversity patterns within basins.
The distribution of the genetic diversity across populations is strongly determined by dispersal and gene flow patterns18. In Salix, the potential sources of gene flow are the movement of pollen grains, seeds and vegetative propagules. Seeds are dispersed by wind (upstream, downstream) and water (mainly downstream with some possible upstream events caused by massive flooding)4. The dendritic structure of a basin imposes that the volume of water that flows through the river channel (i.e., river discharge) increases downstream. Our models showed a positive correlation between river discharge and the genetic diversity level that also tended to increase downstream. In addition, the tail-up model explained significant proportions of Ae and uHe spatial variation. Both results suggest an important role of hydrochory in mobilizing propagules, as expected based on the drift hypothesis4. However, our models also noted a significant role of upstream processes that counteracted the dominant downstream movement. For example, the tail-down explained meaningful proportions of Ae, Fis and Ho, and the Euclidean model explained large proportion of uHe spatial variation. Upstream seed dispersal has been identified in other riparian species associated with zoochorous and human dispersal22. Salix spp. are wind and insect pollinated, and these vectors are reported to generate strong genetic patterns that follow dominant wind directions. In river valleys, topography and wind channelling constrain prevailing winds through the hydrographic network25, which is a phenomenon that can permit upstream gene movement, as reported in other Salicaceae species21. Despite the ability of Salicaceae to resprout from vegetative propagules26, the reduced presence of clones in our study discards the notion that they significantly contribute to natural S. salviifolia regeneration.
The population genetic diversity estimators (Ae, Fis, uHe, Ho) displayed patterns of spatial autocorrelation mostly at the intermediate scale (i.e., 1–100 km sensu Fausch27) through flow-connected (tail-up) and flow-unconnected (tail-down) spatial relationships (Fig. 1, Table 2). The ranges of these spatial autocorrelation structures suggest that genetic connectivity among S. salviifolia populations mainly occurs at the intermediate scale (<20 km), which is likely associated with the interaction of the Salix life history with the formation and distribution of hydrogeomorphological landforms (i.e., channel sediment deposits) where the species recruit and colonize2,28. Indeed, key biological and physical processes in riparian systems, such as metapopulation dynamics and disturbance regimes, are thought to operate at intermediate scales27.
Interestingly, Ae showed large-scale spatial autocorrelation (range = 154.6 km) that included flow-unconnected relationships among populations. This result suggests that within hydrographic networks, even remote populations would eventually become connected, possibly integrating a long-term effect of successive pollen and seed dispersal events or through rare long-distance pollen-mediated dispersal events4, as in other Salicaceae species21.
The HL revealed a limited influence of distance among individuals where only 14.2% of the residual variation in the HL exhibited fine-scale patterns of autocorrelation (range = 11.9 m). The fine-scale spatial aggregation may be primarily due to spatially structured variables, such as microenvironmental heterogeneity. Indeed, fine-scale soil-moisture gradients within riparian habitats are critical for the survival of Salix seedlings when the water level decreases after natural flooding events29. This finding is consistent with the significant correlation of summer drought with the HL, suggesting that increased aridity may constrain gene flow within basins as drought events become more extreme.
Spatial stream networks have been previously applied to investigate biodiversity patterns in riparian insect communities13. Here, we have extended the application of SSN models to identify the ecological drivers that underlie population genetic diversity patterns within basins and quantify how the impacts of these drivers change across an environmental gradient. The integration of these models with increasingly available datasets that survey different community compositions based on environmental DNA can be used to map biodiversity hotspots and depict connectivity networks. This application would assist in decision making to prioritize the conservation of biodiversity hotspots and can be applied to draw mitigation and restoration measures to enhance gene flow among disconnected populations1. In addition, quantifying connectivity changes after adding or removing multiple barriers is a top concern of catchment planning30; thus, simulating alternative scenarios is a promising application of the stream-network approach for riverine species management. Simulation techniques can also be used to optimize sampling strategies for different purposes in stream networks or provide recommendations about sample sizes needed to achieve study objectives. These applications can significantly aid in the design of efficient monitoring strategies at relatively low costs10.
Overall, refining our capacity to describe, predict and simulate the amount and distribution of genetic diversity harboured by riparian populations of foundation species can improve adaptive management through cost-effective monitoring designs for conservation31. Preserving their potential for future adaptation will enhance the resilience of riparian population networks with cascading effects on associated biological communities, ecosystem functions and services, contributing to ecologically successful river management.
Materials and Methods
Field sampling
In the summers of 2010–2012, we conducted field surveys in riparian forests in 24 river valleys located in the Western Iberian Peninsula, across eight independent catchment systems: Tua-Douro, Zêzere-Tagus, Aljezur, Seixe, Odiáxere, Arade, Quarteira, and Guadiana (Fig. 2). These basins are spatially distributed within three regions: the Tua and Zêzere regions, which exactly match the Tua and Zêzere river basins, respectively, and the Algarve region, which includes six basins. The study area spans from the southernmost distribution of S. salviifolia and largely covers its latitudinal range and approximately one-third of its longitudinal range. We sampled 30 sites (each site representing a population) totalling 605 trees that were georeferenced with a submetre precision handheld GPS (Ashtech MobileMapper100). We sampled 15 or more individuals along the river reaches, and we collected as many individuals as possible (up to six) in low-density reaches to survey a similar sample area per site. We collected six healthy leaves per tree, and we stored them in paper bags containing silica gel until further work in the lab.
Genotyping of biological samples
Genomic DNA was isolated from the dry leaf tissue per individual following standard methods (Supplementary Information S3). The samples were genotyped based on twelve polymorphic microsatellite markers; seven primers identified for S. salviifolia32 and five optimized from S. burjatica (cv. Germany)33 (Supplementary Information S3). We used samples from population 2 (N = 54) to test the performance of this set of polymorphic markers. Specifically, we examined the genetic corelation among loci (Supplementary Table S4A), tested for genotypic linkage disequilibrium (Supplementary Table S4B), and tested the ability of markers to discriminate among individuals (Supplementary Table S4C). Moreover, we estimated the expected number of individuals with the same genotype for an increasing number of loci (Supplementary Table S4D), estimated the probability of identical genotypes arising from sexual reproduction and random mating (Supplementary Table S4E), and estimated the incidence of null alleles and scoring error with a subset of N = 20 blind duplicated samples. Across loci, we detected low incidences of null alleles (2*10−4) and scoring errors (2*10−3) by applying Microchecker 2.2.334. We also applied pedant 1.035 to duplicated samples to estimate the per-allele maximum likelihood allelic dropout (ε1 = 0.2, CI = [0.00–0.5]) and false alleles (ε2, CI = 0.1 [0.00–0.3]). Our study species exhibits clonal reproduction; therefore, we used the R package Rclone36 to identify identical genotypes, estimate the probability that they have been generated by independent sexual reproduction events, and evaluate the discriminative power of our 12 polymorphic markers to identify unique multilocus genotypes (Supplementary Tables S4F, S4G). We identified 5 clones out of 605 individuals.
Environmental data
Based on previous studies14, we expected that spatial factors (Euclidean and dendritic spatial structures), bioclimatic variables (winter cold stress, summer drought stress), altitude and hydrology would determine the spatial distribution of the genetic variation of S. salviifolia at different spatial scales. We calculated environmental variables based on the GPS coordinates of the populations and individuals. We used two bioclimatological indices: the thermicity index (BIOC.TH) depicts the thermal envelope where plant species thrive, while the summer ombrothermic index (BIOC.SO) estimates the intensity of summer drought37. Geographic and hydrological variables were inferred from a digital elevation model (DEM) downloaded from the Shuttle Radar Topography Mission (SRTM) 90 m Digital Elevation Database v4.138. We projected the Iberian Peninsula territory to the Lambert azimuthal equal-area projection, which guarantees equal pixel areas (indispensable for the hydrologic calculations), and simultaneously interpolated it to 35 m resolution. The altitude (ALT) at each site was extracted from the produced DEM. Then, to estimate the potential discharge, we derived a hydrographic network and computed a hydrologic index (DA) consisting of the drainage area of each site that was weighted by the total annual precipitation (P) in its contributing area. This hydrologic index distinguishes catchments presenting similar dimensions but occurring in regions with different P, providing a surrogate for total discharge.
Spatial data
We generated the spatial data necessary for geostatistical modelling in ArcGIS 9.239 using the Functional Linkage of Water Basins and Streams (FLoWS) and the Spatial Tools for the Analysis of River Systems (STARS) geoprocessing toolboxes. We applied the FLoWS toolset40 to construct a landscape network, which is a spatial data structure that stores the topological relationships between nodes (stream confluences) and directed edges (stream segments). For the analyses at the individual level, we incorporated the position of each sampled tree into the landscape network. For the analyses at the population level, we used the position of the central tree to map each site. Based on the landscape network, we used the STARS toolset to generate41 (1) hydrologic distances, (2) weights for converging stream tributaries thought to have stronger influences downstream, and (3) the SSN objects that contain feature geometry, attribute data, and topological relationships of the dataset, which are intended for geostatistical modelling within the SSN R package42.
Estimates of genetic diversity and differentiation
To gain a comprehensive depiction of the genetic structure observed per study region, we calculated two types of estimators43: (i) fixation measures (Fst, Phist, Gst, G’st); and (ii) allelic differentiation measures (DJost). Given the controversy about the ability of Fst to quantify genetic structure44,45 when applying highly polymorphic genetic markers we opted for reporting four fixation measures (Supplementary Information S4) as implemented in GeneAlex 6.546. We tested for Hardy-Weinberg equilibrium (HWE) and linkage disequilibrium (LD) at each site by using GENEPOP47 and, for multiple tests, we applied the B-Y method48, which is a modified Bonferroni method recommended for conservation genetics studies49. We estimated the following population genetic diversity metrics by applying the R package gstudio50: observed and expected unbiased heterozygosity (Ho and uHe, respectively), mean number of alleles (AR), effective number of alleles (Ae), and mean number of private alleles (PA). We used INEST 2.051 to estimate the Fis per population while considering the frequency of null alleles. We calculated the homozygosity level (HL) for each individual as implemented in the adegenet R package v.3.2.2.52. HL works as a proxy of individual inbreeding and it provides insights on mating patterns at the population level that are expected to change across an environmental gradient, with increased HL levels expected in small populations and those poorly connected by water flow, partly due to genetic drift. Different components of the genetic diversity respond to the impact of ecological factors at a different pace; thus, allelic diversity typically changes fast after an ecological perturbation, whereas uHe and Ho show slow-paced changes53. Then, we evaluated the impacts of spatial and ecological factors on genetic diversity and structure by choosing a variety of estimators at the population level (Ae, Ho, uHe, Fis) and the individual level (HL).
Geostatistical modelling
We first performed an exploratory analysis to check for multicollinearity among environmental variables by inspecting the variance inflation factor (VIF). We retained all variables because they showed VIF values <2, suggesting no or little multicollinearity among study variables. We then modelled five genetic diversity estimators (Ae, Ho, uHe, Fis, HL) with spatially explicit stream-network models by applying the R package SSN42.
Each SSN model accommodates a mixture of covariances that capture multiple spatial relationships in the dendritic network, including clustered measurements10. This method allows the data to determine the variance components that have the strongest influence rather than making an implicit assumption about the spatial structure7. Stream-network models accommodate two classes of autocovariance models that use hydrologic rather than Euclidean distance and are referred to as tail-down and tail-up models7. These models are based on a moving-average construction; so, spatial autocorrelation between sites occurs when their moving-average functions overlap (Fig. 1). A flow-connected spatial relationship results from water flowing from the upstream to the downstream location. A flow-unconnected relationship exists when two locations share a common junction downstream but are not connected by flow. In the tail-down models, the moving-average function (MAF) points in the downstream direction and therefore, spatial correlation is permitted between both flow-connected and flow-unconnected locations. In contrast, the MAF for the tail-up model points upstream, and therefore, spatial correlation is restricted to flow-connected locations (Fig. 1). A given SSN model is fitted using a mixed-covariance structure that combines two or more autocovariance models that may include the tail-up and tail-down autocovariance models and a traditional model based on Euclidean distance7.
We used a two-step model selection procedure as in Frieden et al.13 to select the model containing the most suitable covariance structure along with a set of environmental variables (covariates) that better explained the observed genetic patterns. First, we fixed the covariance structure and focused on covariate selection through an exhaustive screening of the candidate models that resulted from every linear combination of covariates. In this stage, we applied maximum likelihood to estimate model parameters, and we used Akaike’s information criterion for covariate selection, which prevents over-fitting of the model54. Then, we fixed the selected covariates and compared every linear combination of tail-up, tail-down and Euclidean covariance structures, testing four different autocovariance functions for each model type: the spherical, exponential, Mariah and linear-with-sill functions for tail-down and tail-up models; and the spherical, exponential, Gaussian and Cauchy functions for the Euclidean model as recommended by Peterson & Ver Hoef7. Overall, we tested 125 models (see Supplementary information S5 for details). For each response variable, we used restricted maximum likelihood55 with the root-mean-square-prediction error for the observations and the leave-one-out cross-validation predictions to select the final model54. Once we identified the final model for each response variable, we examined the influence of each variance component (tail-up, tail-down, Euclidean and nugget effect)13.
Data Availability
Microsatellite genetic data are registered at GenBank (http://www.ncbi.nlm.nih.gov/genbank/), and the GenBank accession numbers are provided in Supplementary Information S3. Bioclimatic and hydrologic variables are available from http://home.isa.utl.pt/tmh/. The digital elevation model (SRTMv4.1) was downloaded from http://www.cgiar-csi.org/data/srtm-90m-digital-elevation-database-v4-1. STARS and FlOWS toolboxes for ArcGis, used to generate spatial data, were downloaded from “Tools for Spatial Statistical Modeling on Stream Networks” in the website https://www.fs.fed.us/rm/boise/AWAE/projects/.
References
Grady, K. C. et al. Genetic variation in productivity of foundation riparian species at the edge of their distribution: implications for restoration and assisted migration in a warming climate. Global Change Biology 17, 3724–3735 (2011).
Corenblit, D. et al. The biogeomorphological life cycle of poplars during the fluvial biogeomorphological succession: a special focus on Populus nigra L. Earth Surface Processes and Landforms 39, 546–563 (2014).
Vörösmarty, C. J. et al. Global threats to human water security and river biodiversity. Nature 467, 555–561 (2010).
Nilsson, C., Brown, R. L., Jansson, R. & Merrit, D. M. The role of hydrochory in structuring riparian and wetland vegetation. Biological Reviews of the Cambridge Philosophical Society 85, 837–858 (2010).
Stella, J. C., Battles, J. J., Orr, B. K. & McBride, J. R. Synchrony of seed dispersal, hydrology and local climate in a semi-arid river reach in California. Ecosystems 9, 1200–1214 (2006).
Paz-Viñas, I., Loot, G., Stevens, V. M. & Blanchet, S. Evolutionary processes driving spatial patterns of intraspecific genetic diversity in river ecosystems. Molecular Ecology 24, 4586–4604, https://doi.org/10.1111/mec.13345 (2015).
Peterson, E. E. & Ver Hoef, J. M. A mixed-model moving-average approach to geostatistical modeling in stream networks. Ecology 91, 644–651 (2010).
Cushman, S. A. et al. Landscape genetic conectivity in a riparian foundation tree is jointly driven by climatic gradients and river networks. Ecological Applications 24, 1000–1014 (2014).
Wei, X., Meng, H., Bao, D. & Jiang, M. Gene flow and genetic structure of a mountain riparian tree species, Euptelea pleiospermum (Eupteleaceae): how important is the stream dendritic network? Tree Genetics & Genomes 11, 1–11, https://doi.org/10.1007/s11295-015-0886-6 (2015).
Isaak, D. J. et al. Applications of spatial statistical network models to stream data. Wiley Interdisciplinary Reviews: Water 1, 277–294 (2014).
Rushworth, A. M., Peterson, E. E., Ver Hoef, J. M. & Bowman, A. W. Validation and comparison of geostatistical and spline models for spatial stream networks. Environmetrics 26, 327–338, https://doi.org/10.1002/env.2340 (2015).
McGuire, K. J. et al. Networks analysis reveals multiscale controls on streamwater chemistry. PNAS 111, 7030–7035 (2014).
Frieden, J. C., Peterson, E. E., Angus Webb, J. & Negus, P. M. Improving the predictive power of spatial statistical models of stream macroinvertebrates using weighted autocovariance functions. Environmental Modelling and Software 60, 320–330 (2014).
Amigo, J. Las saucedas riparias de Salicion salviifoliae en Galicia (Noroeste de España). Lazaroa 26, 67–81 (2005).
Sochor, M., Vašut, R. J., Bártová, E., Maeský, L. & Mráček, J. Can gene flow among populations counteract the habitat loss of extremely fragile biotopes? An example from the population genetic structure in Salix daphnoides. Tree Genetics & Genomes 9, 1193–1205 (2013).
Fink, S. & Scheidegger, C. Effects of barriers on functional connectivity of riparian plant habitats under climate change. Ecological Engineering 115, 75–90 (2018).
Olson, D. M. et al. Terrestrial ecoregions of the world: a new map of life on earth. Bioscience 51, 933–938 (2001).
Ennos, R. In Integrating Ecology and Evolution in a Spatial Context (eds Silvertown, J. & Antonovics, J.) 45–71 (Blackwell Science Oxford, 2001).
Pollux, B. J. A., Santamaria, L. & Ouborg, N. J. Differences in endozoochorous dispersal between aquatic plant species, with reference to plant population persistence in rivers. Freshwater Biology 50, 232–242, https://doi.org/10.1111/j.1365-2427.2004.01314.x (2005).
Kikuchi, S., Suzuki, W. & Sashimura, N. Gene flow in an endangered willow Salix hukaoana (Salicaceae) in natural and fragmented riparian landscapes. Conservation genetics 12, 79–89 (2011).
Imbert, E. & Lefèvre, F. Dispersal and gene flow of Populus nigra (Salicaceae) along a dynamic river system. Journal of Ecology 91, 447–456 (2003).
Honnay, O., Jacquemyn, H., Van Looy, K., Vandepitte, K. & Breyne, P. Temporal and spatial genetic variation in a metapopulation of the annual Erysimum cheiranthoides on stony river banks. Journal of Ecology 97, 131–141, https://doi.org/10.1111/j.1365-2745.2008.01452.x (2009).
Balkenhol, N., Cushman, S. A., Storfer, A. T. & Waits, L. P. Landscape Genetics: Concepts, Methods, Applications. (John Wiley & Sons Ltd., 2016).
Havrdová, A. et al. Higher genetic diversity in recolonized areas than in refugia of Alnus glutinosa triggered by continent-wide lineage admixture. Molecular Ecology 24, 4759–4777, https://doi.org/10.1111/mec.13348 (2015).
Carrera, M. L., Gyakum, J. R. & Lin, C. A. Observational Study of Wind Channeling within the St. Lawrence River Valley. Journal of Applied Meteorology and Climatology 48, 2341–2361, https://doi.org/10.1175/2009JAMC2061.1 (2009).
Karrenberg, S., Edwards, P. J. & Kollmann, J. The life history of Salicaceae living in the active zone of floodplains. Freshwater Biology 47, 733–748 (2002).
Fausch, K. D., Torgersen, C. E., Baxter, C. V. & Hiram, W. L. Landscapes to riverscapes: bridging the gap between research and conservation of stream fishes. Bioscience 52, 483–498 (2002).
Bendix, J. & Hupp, C. R. Hydrological and geomorphological impacts on riparian plant communities. Hydrological processes 14, 2977–2990 (2000).
Stella, J. C. & Battles, J. J. How do riparian woody seedlings survive seasonal drought? Oecologia 164, 579–590 (2010).
Branco, P., Segurado, P., Santos, J. M. & Ferreira, M. T. Prioritizing barrier removal to improve functional connectivity of rivers. Journal of Applied Ecology 51, 1197–1206, https://doi.org/10.1111/1365-2664.12317 (2014).
Rodríguez-González, P. M., Albuquerque, A., Martínez-Almarza, M. & Díaz-Delgado, R. Long-term monitoring for conservation management: Lessons from a case study integrating remote sensing and field approaches in floodplain forests. Journal of Environmental Management 202, 392–402, https://doi.org/10.1016/j.jenvman.2017.01.067 (2017).
Simões, F. et al. In Restauro fluvial e gestão ecológica: manual de boas práticas de gestão de rios e ribeiras (eds Camprodon, J., Ferreira, M. T. & Ordeix, M.) 338–345 (CTFC, 2012).
Barker, J. H. A., Pahlich, A., Trybush, S., Edwards, K. J. & Karp, A. Microssatellite markers for diverse Salix species. Molecular Ecology Notes 3, 4–6 (2003).
Oosterhouth, C. V., Hutchinson, W. F., Wills, D. P. M. & Shipley, P. MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes 4, 535–538 (2004).
Johnson, P. C. D. & Haydon, D. T. Software for quantifying and simulating microsatellite genotyping error. Bioinformatics and Biology Insights 1, 71–75 (2007).
Arnaud-Haond, S. & D., B. RClone: Partially clonal population analysis. R package version 1.0.2, https://CRAN.R-project.org/package=RClone (2016).
Monteiro-Henriques, T. et al. Bioclimatological mapping tackling uncertainty propagation: application to mainland Portugal. International Journal of Climatology 36(1), 400–411, https://doi.org/10.1002/joc.4357 (2016).
Jarvis, A., Reuter, H. I., Nelson, A. & Guevara, E. (ed. Hole-filled SRTM for the lobe Version 4 available form CGIAR-CSI SRTM 90m Database, http://www.cgiar-csi.org/data/srtm-90m-digital-elevation-database-v4-1 [accessed 9/April/2014]) (2008).
ESRI. ArcGis: Release 9.2 [software]. Environmental Systems Institute, Redlands, California (2006).
Theobald, D. M. et al. Functional linkage of water basins and streams (FLoWS) v1 User’s Guide. 43 (Fort Collins, 2006).
Peterson, E. E. & Ver Hoef, J. M. STARS: An ArcGIS toolset used to calculate the spatial information needed to fit spatial statistical models to stream network data. Journal of Statistical Software 56, 1–17 (2014).
Ver Hoef, J. M., Peterson, E. E., Clifford, D. & Shah, R. SSN: An R package for spatial statistical modeling on stream networks. Journal of Statistical Software 56, 1–45 (2014).
Jost, L. et al. Differentiation measures for conservation genetics. Evolutionary Applications, n/a–n/a, https://doi.org/10.1111/eva.12590 (2018).
Gerlach, G., Jueterbock, A., Kraemer, P., Deppermann, J. & Hardman, P. Calculations of population differentiation based on GST and D: forget GST but not all of statistics! Molecular Ecology 19, 3845–3852, https://doi.org/10.1111/j.1365-294X.2010.04784.x (2010).
Meirmans, P. G. & Hedrick, P. W. Assessing population structure: FST and related measures. Molecular Ecology Resources 11, 5–18, https://doi.org/10.1111/j.1755-0998.2010.02927.x (2011).
Peakall, R. & Smouse, P. E. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update. Bioinformatics 28, 2537–2539, https://doi.org/10.1093/bioinformatics/bts460 (2012).
Rousset, F. GENEPOP'007: a complete re-implementation of the GENEPOP software for Windows and Linux. Molecular Ecology Resources 8, 103–106 (2008).
Benjamini, Y. & Yekutieli, D. The Control of the False Discovery Rate in Multiple Testing under Dependency. The Annals of Statistics 29, 1165–1188 (2001).
Narum, S. R. Beyond Bonferroni: Less conservative analyses for conservation genetics. Conservation Genetics 7, 783–787, https://doi.org/10.1007/s10592-005-9056-y (2006).
gstudio: spatial utility funcitons from the Dyer laboratory (R package version 1.2, 2014).
Chybicki, I. J. & Burczyk, J. Simultaneous Estimation of Null Alleles and Inbreeding Coefficients. Journal of Heredity 100, 106–113, https://doi.org/10.1093/jhered/esn088 (2009).
Jombart, T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Futuyma, D. J. Evolutionary Biology. (Sinauer Associates, Massachussets, 1998).
Bennett, N. D. et al. Characterising performance of environmental models. Environmental Modelling & Software 40, 1–20, https://doi.org/10.1016/j.envsoft.2012.09.011 (2013).
Cressie, N. Statistics for Spatial data (revised edition). (Wiley, 1993).
Acknowledgements
We thank E. Peterson for kind support implementing the spatial stream-network models; J. Gonzalo for the Spanish climatic data; P. Jordano and J. Orestes for fruitful discussions; J. Amigo for distribution data on Salix salviifolia; A. Fabião for assistance in Algarve rivers sampling; and the landowners, who facilitated the terrestrial and boat access to study sites. This study was funded by Fundo EDP–Energias de Portugal (Tua, Zêzere) and INTERREG-IVB-SUDOE–Ricover project (Algarve). The Portuguese Foundation for Science and Technology (FCT) supported Centro de Estudos Florestais (UID/AGR/00239/2013), CITAB-UTAD (UID/AGR/04033/2019), P.M.R.G. (Post-doctoral SFRH/BPD/47140/2008 and FCT Investigator IF/00059/2015 grants), C.G. (FCT IF/01375/2012), and T.M.H. (Post-doctoral SFRH/BPD/115057/2016). PMRG was supported by COST Action (CA16208) – CONVERGES: Knowledge Conversion for Enhancing Management of European Riparian Ecosystems and Services.
Author information
Authors and Affiliations
Contributions
P.M.R.G., A.A., C.F., M.H.A., A.M. and M.T.F. conceived the ideas; P.M.R.G., A.A. and C.G. designed the methodology; P.M.R.G., A.A. and T.M.H. collected the data; P.M.R.G., C.G., T.M.H., J.B.G., D.M., F.S. and J.M. analysed the data; P.M.R.G. and C.G. led the writing of the manuscript. All authors contributed critically to the drafts and gave final approval for publication.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rodríguez-González, P.M., García, C., Albuquerque, A. et al. A spatial stream-network approach assists in managing the remnant genetic diversity of riparian forests. Sci Rep 9, 6741 (2019). https://doi.org/10.1038/s41598-019-43132-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-43132-7
This article is cited by
-
Analysing the distance decay of community similarity in river networks using Bayesian methods
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
