
Abstract
Vancomycin-resistant enterococci (VRE) can rapidly spread through hospitals. Therefore, our hospital employs a screening program whereby rectal swabs are screened for the presence of vanA and vanB, and only PCR-positive broths are cultured on VRE selection agar. Early November 2016, a clinical vanA-/vanB-negative VRE isolate was detected in a vanA/vanB-screening-negative patient, giving the possibility that an undetected VRE might be spreading within our hospital. Whole-genome-sequencing of the isolate showed that resistance was vanD-mediated and core genome multilocus sequence typing showed it was a rare type: ST17/CT154. To determine the prevalence of vanA/B/C/D-carrying enterococci, we designed a real-time PCR for vanC1/2/3 and vanD and screened rectal swabs from 360 patients. vanD was found in 27.8% of the patients, yet culture demonstrated only E. faecium from vanA-positive broths and E. gallinarum from vanC1-positive broths. No vanD-positive VRE were found, limiting the possibility of nosocomial spread of this VRE. Moreover, the high prevalence of non-VRE vanD in rectal swabs makes it unfeasible to include the vanD PCR in our VRE screening. However, having validated the vanC1/2/3 and vanD PCRs allows us to rapidly check future vanA/B-negative VRE for the presence of vanC and vanD genes.
Similar content being viewed by others


Introduction
Vancomycin-resistant enterococci (VRE) may be considered as an important healthcare-associated pathogen, especially in immunocompromised patients1,2. Acquisition of vancomycin resistance is mediated by several van gene clusters3. The most commonly encountered vancomycin resistance genes are vanA and vanB, which confer either high level resistance to vancomycin (MIC > 128 mg/L) or more moderate levels (16 to 64 mg/L), respectively3,4. Moreover, the localization of these vanA- and vanB-containing pathogenicity islands on mobile genetic elements allow this type of resistance to spread clonally and laterally. Therefore, these resistance genes are highly transferable between enterococci and thus pose a risk for nosocomial spread5. Consequently, several public health institutes, including our hospital, now screen for the carriage of vanA- and vanB-positive VRE in hospitalized patients6.
In contrast to vanA and vanB, vanC and vanD in VRE are chromosomally associated, i.e. much less transferable7. vanC generally presents with low-level resistance against vancomycin (MIC 2–32 mg/L) and teicoplanin (≤1 mg/L)3. Moreover, VRE containing vanC are typically E. gallinarum or E. casseliflavus/E. flavescens, and associated with low risk of mortality and rarely cause nosocomial outbreaks8,9,10,11. Hence, vanC-positive patients typically are not isolated, contrary to patients with vanA- or vanB-positive VRE9. The vanD gene, generally, confers moderate-level resistance against vancomycin (MIC 64–128 mg/L) and teicoplanin (MIC 4–64 mg/L) and is constitutively expressed3,12. Until recently, vanD was mostly found in anaerobic commensals of the human gut microbiome13 and was seldom found in enterococci. In Europe, individual cases with vanD-positive enterococci have been reported in patients from France (2005)14 and Sweden (2007, 2016)15,16. Yet, recently six clinical vanD-positive E. faecium isolates were reported from the Netherlands3,17. These isolates presented with MIC values of 8–256 mg/L (vancomycin) and <0.5–4 mg/L (teicoplanin)17. Overall, vanD-positive enterococci are relatively unknown and, as far as we know, are not included in surveillance screenings. Thus, VRE surveillance programs might miss the presence of vanD-positive enterococci within a hospital.
Screening for VRE carriage can be useful to identify unrecognized cases to prevent nosocomial transmission of VRE and reduce the subsequent risk of VRE infection18. Therefore, the Maastricht University Medical Centre (MUMC+) adopted a monthly point prevalence screening of all admitted patients in all wards of the hospital. As part of the screening, a rectal swab is taken from a patient, cultured in TSB broth, and subsequently screened with vanA/vanB PCR. PCR-positive broths are then inoculated on VRE selection agar to determine whether a vancomycin-resistant Enterococcus faecium or Enterococcus faecalis is present. Parallel to the screening described above, high-risk wards (e.g. Intensive Care, gastroenterology) also submit rectal swabs to screen for ESBL and VRE by culture on selection agars without prior vanA/vanB PCR.
The first VRE isolate of each patient is typed by conventional multilocus sequence typing (MLST). Epidemiological data and typing results are combined to assess whether nosocomial transmission is likely. In case of expected nosocomial transmission, patients are isolated, isolates are whole-genome-sequenced, specific cleaning procedures are implemented and screening of the ward is intensified.
Case Description
Within our hospital, a 59 year-old male presented with acute, severe necrotizing pancreatitis, following heavy alcohol consumption. Computed tomography scan revealed a necrotizing pancreatitis and intra-abdominal air, without a known cause. Although initially clinically stable, the patient was transferred to the Intensive Care Unit due to respiratory failure and progressive pancreatic necrosis. Over the course of four months, various isolates were cultured from aspirations and drainages (Fig. 1). The isolation of Serratia marcescens and E. faecium from transgastric fluid and blood cultures prompted an antibiotic switch to meropenem and vancomycin.

The time line of the patient, who initially presented with acute severe necrotizing pancreatitis on day 0 (admission). Over time, various microorganisms were isolated from clinical samples: E. coli on day 21 from fine needle aspirate (sensitive to all β-lactam antibiotics), Serratia marcescens (pip/tazo resistant) and Enterococcus faecium (amoxicillin resistant, vancomycin susceptible) on day 35 from transgastric drainage fluid. Serratia marcescens was also isolated from blood cultures on day 37. From day 65 on, a vancomycin resistant E. faecium was also isolated from rectal swabs. Earlier rectal swabs did not produce vancomycin resistant enterococci. Abbreviations: Amoxi-Clav (Amoxicillin-clavulanic acid), Pip/Tazo (piperacillin-tazobactam).
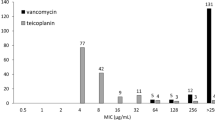
Moreover, the patient was routinely screened twice-weekly for carriage of VRE, by directly inoculating the rectal swabs on VRE selection agar without PCR-based selection. During the first one and half month, thirteen swabs were taken and none yielded VRE. Yet, on day 65 of admission, a vancomycin-resistant E. faecium was isolated which had MIC values of 16 mg/L for vancomycin, 1 mg/L for teicoplanin, and 2 mg/L for linezolid (vanD-65). The isolate was analyzed by PCR and proved to be negative for vanA and vanB. Hereafter, more VRE were isolated from rectal swabs of this patient (Fig. 1). The last known VRE isolate was found on day 107 of admission (vanD-107), which had MIC values of 24 mg/L for vancomycin, 1 mg/L for teicoplanin, and ≥8 mg/L for linezolid (Table 1). On the 116th day of admission, antibiotic therapy was discontinued. In total, the patient had received meropenem and vancomycin for 73 days. Follow-up testing was performed at one year after discharge and the patient was asked to submit ten rectal swabs taken at least 12 h apart (i.e. spanning ≥5 days). None of the swabs yielded VRE.
Results
Vancomycin-resistant isolate
The vancomycin-resistant E. faecium isolated on hospital day 65 (vanD-65) was negative for vanA and vanB by PCR. The species identity was confirmed by biochemical properties (resistant for furazolidone, sensistive for both mupirocine and tellurite), MALDI-TOF and 16S sequencing. To determine the origin of the vancomycin resistance, whole genome sequencing (WGS) was performed at the Statens Serum Institut (SSI) in Denmark.
Assembly resulted in 246 contigs, genome size 3102488 bp. The resulting genome was analyzed for resistance genes using ResFinder 2.119, which identified a full vanD gene cluster, located on position 198734..104387 of contig 1 (contig length = 192.330 bp; Hit length 5654 bp) with a 99,94% identity to the vanD cluster in the species Enterococcus raffinosus (Strain GV5; GenBank: AB242319.1). Additional resistance genes were identified; three coding for aminoglycoside resistance and two for macrolide resistance, respectively (Supplement A1).
Interestingly, both the left and right flank of contig 1 showed >99% identity to fully sequenced E. faecium chromosomes such as Aus0085 (Genbank: CP006620.1), while a large internal fragment of contig 1 (approximately 168.900 bp containing the vanD gene cluster) showed very little similarity to any sequences at NCBI except from some DNA fragments ranging between <100 bp and 21892 bp with 88–97% identity to a Blautia (previously Clostridium) coccoides genome (Strain YL58; GenBank NZ CP022713) and the vanD gene cluster of E. raffinosus mentioned above.
Subsequent MLST and cgMLST analysis at the SSI showed the isolate to be MLST type ST17, clonal type CT154 (ST17/CT154), a strain rarely found in the cgMLST database. The cgMLST database contained six other ST17/CT154 VRE. However, none of these contained a vanD gene cluster, and contained various other additional resistance genes (Supplement A1).
Retrospectively, all vanA-/vanB-negative VRE isolates of the patient (N = 7) were subjected to WGS at the Maastricht University Medical Center (MUMC+). All seven isolates had vanD-mediated resistance to vancomycin (Table 1), belonged to ST17/CT154 and whole-genome-MLST showed a maximum of one locus difference between the seven isolates. The vanD gene belonged to vanD4, as did three out of six of the previous Dutch vanD-positive isolates (Fig. 2, Top et al.17). Comparison of the genetic environment of the vanD locus in these four Dutch vanD-positive VRE revealed >99.9% homology (Supplement A2A,B). However, our isolate belonged to a different ST/CT and is thus not clonally related to previously described Dutch vanD-positive VRE (Table 1, Supplement A2C). The patient also had a vancomycin susceptible E. faecium, which had been isolated on day 35. Sadly, this isolate could not be retrieved for analysis. Therefore, no clonal relatedness can be inferred between this isolate and the subsequent VRE isolates.

Phylogenetic neighbour-joining (NJ) tree of the vanD-positive E. faecium described in this report and the sequenced vanD-PCR fragments (~150 nt). NJ tree was generated and bootstrapped (1000 repetitions) from ClustalX2 multiple alignment of trimmed DNA sequences (Supplement A6). The type of vancomycin-resistance gene is denoted on the right-hand side of the figure, based on the most similar reported vanD gene. Further included isolates include the six Dutch vanD-positive isolates recently reported17 and reference genomes. Accession numbers are given in the figure. The tree was rooted using a randomly scrambled E. faecium fragment. Bar represents substitutions per site.
VRE screening strategy
Upon encountering the vanD-positive VRE, the prevalence of vanD-positive VRE within our hospital was determined. For this goal, PCRs for vanC1/2/3 and vanD (covering all currently known vanD1 – vanD5 sequences) were designed and validated using clinical isolates and transformants (Supplement A3). Next, using these validated PCRs, the prevalence of the vanA, vanB, vanC1, vanC2/3 and vanD genes within our hospital was assessed. In the period of four days, rectal swabs were taken from 360 patients on 12 different wards. In principle, each patient had only one swab taken as part of this monthly point prevalence screening. Each swab was shaken in a TSB broth. Subsequently, the broths were incubated overnight prior to being subjected to PCR analysis for vancomycin-resistance genes (Table 2 below). The prevalence is calculated as the number of PCR-positive broths divided by the total number of broths.
Secondly, to determine whether these genes represented VRE carriage, 47 of the PCR-positive broths were sub-cultured on VRE chromogenic agar to determine VRE carriage (100% of vanA-positive (2 out of 2), 100% of vanB-positive (38 out of 38), 17.6% of vanC-positive (9 out of 51), and 32% of vanD-positive (32/100) broths), which resulted in 2 vanA-positive E. faecium (Supplement A5).
Since the VRE-selective agar inhibits growth of vanC-containing E. gallinarum and E. casseliflavus, another strategy was employed to identify VRE; an additional 16 TSB broths were selected based on their low Ct values for vanC1, vanC2/3, and vanD (i.e. cultures with the highest load); vanC1 Ct 21–29, vanC2/3 Ct 17–33, and vanD Ct 26–39. These TSB broths were inoculated on both CNA agar and Columbia 5% sheep blood agar, on which a 5 μg vancomycin disc was placed. The rationale to select broths with the lowest Ct values was based on our validation showing that broths with a Ct value over 35.0 for either vanA or vanB do not result in culturable VRE’s (data not shown).
After 24 h and after 48 h of incubation at 37 °C, colonies that grew within the zone of inhibition were selected and characterized by PCR, MALDI-ToF and antibiotic susceptibility testing (Supplement A5). This resulted in 8 agar plates with growth: 5 vancomycin resistant vanC1-positive E. gallinarum (Ct 21–29, MIC 4 to 16 mg/L), a vancomycin susceptible vanC2/3-positive E. casseliflavus (Ct 17, MIC 1 mg/L) and a vancomycine susceptible Enterococcus faecalis negative for vanA/B/C/D (MIC 2 mg/L). Additionally, a vancomycin-susceptible Staphylococcus haemolyticus was isolated. No vanD-positive enterococci were identified.
Given the multitude of broths positive for vanD by PCR (100 out of 360; 27.8%), eleven samples were randomly selected among the high-positive PCRs (i.e. low Ct value), sequenced to confirm the specificity of amplification, and aligned with known vanD-sequences (Fig. 2 below, Supplement A6). Alignment and BLAST analysis revealed high similarity to the vanD sequence of our isolate and to other vanD sequences previously found in E. faecium and E. raffinosus. One sequence (26-5_E12) is most similar to a vanD sequence found in Ruminococcus gauvreauii, an anaerobic species.
Lastly, the results of this screening were combined with existing data, enabling calculation of the positive predictive value (PPV) of our vanA, vanB, and vanD PCRs for VRE in a non-outbreak situation in our hospital. Here, the positive predictive value of the PCR indicates which fraction of PCR-positive broths yield VRE when cultured on VRE-selective agar. This resulted in; vanA 82% PPV (N = 65), vanB 8% PPV (N = 426), vanD 0% PPV (N = 100). The PPV of vanC1/2/3 PCR was not calculated given the low clinical relevance of the microorganisms typically harbouring these resistance genes, and the fact that these microorganisms do not grow on the VRE-selective agars.
Discussion
In response to the isolation of a vanD-positive VRE, PCRs for vanC1/2/3 and vanD were devised and validated to determine the prevalence of vanC/D-positive enterococci within our hospital. A hospital-wide screening, comprising 360 patients, did not show any relevant vanC- or vanD-positive enterococci. The observed prevalence of the vanB gene (10.6%) and the vanD gene (27.8%) in our hospital are similar to those in literature13, whereas the prevalence of the vanA gene is lower (0.6% versus 9.3%). For the vanC genes, the prevalence of 7.2% (vanC1) and 7.8% (vanC2/3) in our hospital is higher than in literature10,20, as will be discussed below. Culture of vanC-positive broths yielded 5 vanC1-positive and 1 vanC2/3-positive enterococci (success rate 6/14: ~43%). Yet, these vanC-positive enterococci have sub-clinical vancomycin MIC values (range: 1–16 mg/L), values which are well achievable in serum21.
Previous studies into the prevalence of vancomycin-resistant enterococci used 6 mg/L vancomycin during the enrichment step and this likely has affected the observed prevalence of vanC. For example, in Hungary, clinical samples were first grown on VRE selection agars, resulting in a prevalence of 0.19% and 0.3% for vanC1 and vanC2, respectively, among all enterococci-positive samples10. In Nigeria, rectal swabs were inoculated in broth containing 6 mg/L vancomycin prior to screening, resulting in a prevalence of 2.8% for vanC1- and 0.3% for vanC2-positive enterococci20. We used broths without any vancomycin and noted a prevalence of 7.8% and 7.2% for vanC1 and vanC2/3, respectively (Table 2). The VRE-selective agar used in this study contains 8 mg/L vancomycin, besides other chemicals that inhibit growth of vanC-positive E. gallinarum and E. casseliflavus (data not shown). When CNA agar with a disk of vancomycin was used for culture, vanC-positive enterococci were isolated in 43% of the tested PCR-positive broths indicating that these enterococci could readily be isolated in case of PCR-positivity. Furthermore, given the relatively low MIC values conferred by vanC genes, the majority of vanC-containing enterococci possibly will be missed if selection is applied prior to screening. Yet, this is currently not seen as a flaw given the low clinical relevance of vanC-positive E. gallinarum, E. casseliflavus/E. flavescens8,9,10,11.
In contrast, the prevalence of vanD apparently is less affected by the use of selective broths since we found a prevalence of 27.8% (the Netherlands) without selective broth and others found 43.8% (Montreal, Canada) and 26.7% (Boston, USA) after enrichment in selective broth containing 6 mg/L vancomycin and 60 mg/L aztreonam13. These prevalences are quite similar despite the geographic distance and the presence or absence of selection. Despite the high prevalence, and the fact that the sequenced vanD amplicons were most similar to vanD genes associated with enterococci (Fig. 2), neither we nor other authors13 were able to grow vanD-positive enterococci. The fact that the attempts to culture vanD-positive enterococci were futile suggests that the vanD gene is more present in our commensal microbiota than previously appraised. Furthermore, the fact that the isolated VRE had chromosomally integrated vanD suggests that the gene is not readily transferable between bacteria. Hence, where did this vanD-positive VRE originate?
One hypothesis is that the patient became colonized within the hospital. However, as described above, we could not isolate further vanD-positive VRE from other patients despite multiple attempts and approaches. Moreover, our isolated vanD-positive VRE was studied by WGS, revealing it to be an MLST ST17/CT154, a rare type that is currently predominantly encountered in Germany22. However, these ST17/CT154 VRE are vanA-positive and have different antibiotic gene repertoires suggesting that there is no direct connection between the German isolates and our isolate (Supplement A1).
Thus, it is more likely that the patient had the vanD gene present in his gut, either already in an E. faecium or in other microorganisms. In case of the first, the initial load of VRE was perhaps too low for efficient culture during the first 1.5 month of hospitalization. Vancomycin-based therapy then enabled the VRE to expand while the other fecal microbiota diminished23,24. The alternative hypothesis is that the E. faecium of the patient had acquired the vanD gene cluster from neighbouring anaerobic microorganisms, e.g. Clostridium spp, Blautia spp, or Ruminococcus spp13,25. Since anaerobic microorganisms are not expected to grow under aerobic conditions, i.e. under our screening protocol, the presence of the vancomycin resistance genes could be detected by PCR in a growth-independent manner while the subsequent screening on agar would not produce any growth. In line with the possibility of vanD gene cluster transferal from neighbouring anaerobic microorganisms, the vanD gene cluster in our VRE isolate seems to have integrated along with fragments of Blautia spp.-related genomic DNA.
Yet, given the high homology of the vanD gene cluster in our isolate and three other, older, Dutch vanD-positive VRE17, this vanD gene cluster may once have crossed from Blautia spp. into an enterococ and thereafter have spread horizontally to other ST of E. faecium (Supplement A2A–C). Furthermore, the high homology in these Dutch VRE suggests that our patient acquired a VRE that was already vanD-positive. More importantly, if the vanD gene cluster is indeed derived from Blautia spp., this implies that new resistance mechanisms can be introduced from gut microbiota into enterococci, especially after prolonged treatment with antibiotics (this report)16.
Since many clinical laboratories focus on pre-screening for the presence of vanA and vnB genes by PCR, this could lead to underestimation of non-vanA/non-vanB VRE. Therefore, in response to this vanD-positive VRE, a PCR for vanC1/2/3 and vanD was designed and validated to determine the prevalence of vanC/D-positive enterococci within our hospital. Yet, the screening did not yield any relevant vanC- or vanD-positive enterococci.
We conclude that the occurrence of a vanD-positive VRE is currently still very rare in the clinical setting. Yet, this report and others16,17 indicate that vanD-positive VRE certainly can be found in patients and, being a VRE, pose a risk for nosocomial spread. Given the high prevalence of vanD in rectal swabs within our hospitalized population, and the low PPV of vanD-positive PCR, it would not be cost-effective to include the vanD PCR in our VRE screening program. However, having validated these vanC1/2/3 and vanD PCRs in our hospital, future vanA/B-negative VREs can now be rapidly checked for the presence of a vanC and vanD gene.
Materials and Methods
Surveillance
As part of the routine screening for the presence of vancomycin-resistant enterococci (VRE) within the MUMC+, 360 patients from 12 different wards were screened by taking an rectal swab, which were subsequently brought to the department of Medical Microbiology for analysis. Here, the swabs were suspended in Tryptic soy broth (TSB) medium (Tritium microbiologie, the Netherlands) and then cultured overnight at 37 °C prior to DNA extraction and analysis.
DNA extraction
200 μL of overnight cultures was extracted on a MagNA Pure 96 system using MagNA Pure LC 96 DNA and viral NA small volume kit (Roche Diagnostics, the Netherlands). Total nucleic acids were eluted in 100 μL eluate.
Molecular detection of vancomycin-resistance genes
Primers and probes sequences to detect vanA and vanB genes were obtained from Erik van Hannen (St Antonius Hospital, Nieuwegein, the Netherlands). Primers and probes to detect vanC and vanD genes were designed in-house for this study. All available sequences of vanC and vanD genes were obtained from NCBI’s GenBank (Supplement A3), after which all unique sequences were aligned using Clustal Omega. Next, using IDT’s PrimerQuest and OligoAnalyzer tools, primers and a TaqMan probe were designed to match all allele variants of the vanC1, vanC2/3 and vanD genes. The primers and probes were analyzed in-silico to avoid cross-annealing.
Absence of cross-annealing was validated in-vitro against strains harboring either vanA, vanB, vanC1, vanC2/3 or vanD (Supplement A4). The specificity of the PCRs was further validated by testing against; I) reference positive and negative strains and plasmids, II) known negative clinical isolates, and by sequencing PCR products of vanC1/2/3- and vanD-positive PCRs (Fig. 2). All primer/probe sequences are displayed in Table 3 below.
Controls and template plasmids
A plasmid containing murine CMV glycoprotein B (mCMV-gb) was used as internal spike to check for efficient DNA extraction and PCR-inhibitory compounds. Control plasmids for vanC1 and vanC2/3 were constructed by cloning the corresponding PCR amplicon into a pGEM-T easy vector (Promega Corporation, Madison, WI, USA). The constructs were transformed into E. coli TOP10 and used as positive control for DNA extraction and PCR detection. Clinical isolates of enterococci containing vanA, vanB and vanD were used as positive controls.
Amplification and detection
PCR of vanA, vanB and the internal control mCMVgb was performed in a multiplex reaction. Similarly vanC1 and vanC2/3 were combined in a multiplex reaction, whereas vanD was a simplex reaction. Primer-probe concentrations for vanA and vanB were 800 nM of each primer and 200 nM probe. The PCR of mCMVgb, vanC1, vanC2/3 and vanD contained 300 nM of each primer and 200 nM probe. Each PCR was performed in a total volume of 25 μL consisting of 12.5 μL ABsolute QPCR ROX Mix (Thermo Scientific, Waltham, MA, USA), the primer/probe mixture at the concentrations given above and 5 μL of DNA eluate. Amplification was performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) under the following conditions: 15 minutes at 95 °C, followed by 42 cycles of 15 seconds at 95 °C and 1 minute at 60 °C.
Isolation and culture of vancomycin-resistant microorganisms
Based on the PCR screening, all TSB cultures that were positive for vanA or vanB were inoculated onto VRE-selective chromogenic agar (bioMérieux, Marcy-l′Etoile, France). In addition, a selection of the vanC and vanD PCR-positive TSB cultures were streaked on VRE-selective agar to check for presence of VRE. TSB cultures positive for vanC or vanD were streaked onto CNA agar (colistin, nalidixic acid) and blood agar (both from Bection-Dickinson, USA) on which a 5 μg vancomycin disc (Rosco diagnostica, Danmark) was placed. All agar plates were incubated for at least 48 h and checked after 24 h and 48 h of incubation for growth. Colonies within the inhibition zone were further cultured and characterized by screening axenic microbial cultures by PCR for the presence of vanC1, vanC2/3, or vanD. Isolates positive for a vancomycin-resistance gene were identified using VITEK MS (MALDI-TOF, bioMérieux). Antibiotic resistance was determined using VITEK II and E-test (bioMérieux), according to manufacturer’ instructions. As controls, agar plates were inoculated with a 0.5mcFarland suspension of fresh grown bacterial isolates. These controls were prepared by mixing a VSE with a VRE in the ratio of 95:5 volume/volume %, respectively. For E. faecalis, this was ATCC 29212 (VSE) and ATCC 51299 (vanB-positive VRE with low-level resistance). For E. faecium, clinical isolates were used (one each for vanA, vanB and vanD).
Whole genome sequencing
Genomic DNA was purified using NucliSENS easyMAG (bioMérieux) following the manufacturer’s instructions, and libraries were prepared using a Nextera XT Kit (Illumina, Little Chesterford, UK). Illumina MiSeq 250 bp paired end sequencing was applied. Genome assembly of E. faecium was performed with SPAdes genome assembler, version 3.10.026. The assembled genome was submitted to ResFinder 2.1 (http://cge.cbs.dtu.dk/services/ResFinder/), using default settings (90% sequence similarity and ≥60% coverage length) which identifies acquired antimicrobial resistance genes19.
cgMLST and wgMLST analysis
cgMLST was performed using the official E. faecium cgMLST scheme provided by Ridom SeqSphere+ (v5.1.0). Similar ST17/CT154 genomes were identified at cgMLST.org and downloaded as draft genomes. wgMLST was performed using the BioNumerics (v7.6.3) WGS plugin with wgMLST E. faecium scheme.
Amplicon sequencing
Sequencing of PCR products was performed as described before27 using the PCR primers and an ABI BigDye Terminator v1.1 cycle sequencing kit (Applied Biosystems, Foster City, CA, USA). Sequencing data were obtained by using an ABI 3730 DNA Analyzer (Applied Biosystems) and were analyzed using BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). 16S sequencing was performed as previously described28.
Mauve analysis
Mauve v20150222 build 10 was used to compare the DNA contig carrying vanD gene cluster in VRE to the most similar contigs carrying vanD gene available in Genbank and published by Top et al.17. DNA similarity between these vanD-carrying contigs was analyzed by performing pairwise BLASTn analysis.
Phylogenetic analysis of contigs carrying the vanD gene clusters
The same DNA contigs from the MAUVE analysis were also subjected to phylogenetic analysis based on variations in single Nucleotide Polymorphisms (SNPs) to determine their genetic relatedness. The 192 kb contig 1 from strain vanD-65 was used as reference and the phylogenetic distance between the selected contigs were calculated using CSIPhylogeny (https://cge.cbs.dtu.dk/services/CSIPhylogeny/) with a pruning distance of 100 bp.
Ethics statement
All materials were derived from routine clinical diagnostics, and were de-identified prior to analysis. Neither information nor samples have been collected specifically for this study. All data were analyzed anonymously.
Data Availability
The Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under bioproject PRJDB7459, Biosamples: SAMD00141057 – SAMD00141063.
References
Kim, Y. J. et al. Risk factors for vancomycin-resistant enterococci infection and mortality in colonized patients on intensive care unit admission. Am J Infect Control 40, 1018–1019, https://doi.org/10.1016/j.ajic.2012.01.009 (2012).
Jiang, H. L. et al. The Risk Factors, Costs, and Survival Analysis of Invasive VRE Infections at a Medical Center in Eastern Taiwan. Int J Infect Dis 54, 18–24, https://doi.org/10.1016/j.ijid.2016.11.005 (2017).
Werner, G. et al. Emergence and spread of vancomycin resistance among enterococci in Europe. Euro Surveill 13 (2008).
Naas, T. et al. First nosocomial outbreak of vancomycin-resistant Enterococcus faecium expressing a VanD-like phenotype associated with a vanA genotype. J Clin Microbiol 43, 3642–3649, https://doi.org/10.1128/JCM.43.8.3642-3649.2005 (2005).
Willems, R. J. et al. Global spread of vancomycin-resistant Enterococcus faecium from distinct nosocomial genetic complex. Emerg Infect Dis 11, 821–828, https://doi.org/10.3201/eid1106.041204 (2005).
Frakking, F. N. J. et al. Recommendations for the successful control of a large outbreak of vancomycin-resistant Enterococcus faecium in a non-endemic hospital setting. J Hosp Infect, https://doi.org/10.1016/j.jhin.2018.02.016 (2018).
Cetinkaya, Y., Falk, P. & Mayhall, C. G. Vancomycin-resistant enterococci. Clin Microbiol Rev 13, 686–707 (2000).
Choi, S. H. et al. Clinical features and outcomes of bacteremia caused by Enterococcus casseliflavus and Enterococcus gallinarum: analysis of 56 cases. Clin Infect Dis 38, 53–61, https://doi.org/10.1086/380452 (2004).
Tschudin Sutter, S. et al. Not all patients with vancomycin-resistant enterococci need to be isolated. Clin Infect Dis 51, 678–683, https://doi.org/10.1086/655824 (2010).
Dombradi, Z. et al. Prevalence of vanC vancomycin-resistant enterococci in the teaching hospitals of the University of Debrecen, Hungary. Microb Drug Resist 18, 47–51, https://doi.org/10.1089/mdr.2011.0014 (2012).
Faron, M. L., Ledeboer, N. A. & Buchan, B. W. Resistance Mechanisms, Epidemiology, and Approaches to Screening for Vancomycin-Resistant Enterococcus in the Health Care Setting. J Clin Microbiol 54, 2436–2447, https://doi.org/10.1128/JCM.00211-16 (2016).
Casadewall, B., Reynolds, P. E. & Courvalin, P. Regulation of expression of the vanD glycopeptide resistance gene cluster from Enterococcus faecium BM4339. J Bacteriol 183, 3436–3446, https://doi.org/10.1128/JB.183.11.3436-3446.2001 (2001).
Domingo, M. C. et al. High prevalence of glycopeptide resistance genes vanB, vanD, and vanG not associated with enterococci in human fecal flora. Antimicrob Agents Chemother 49, 4784–4786, https://doi.org/10.1128/AAC.49.11.4784-4786.2005 (2005).
Lavigne, J. P., Marchandin, H., Bouziges, N. & Sotto, A. First infection with VanD-type glycopeptide-resistant Enterococcus faecium in Europe. J Clin Microbiol 43, 3512–3515, https://doi.org/10.1128/JCM.43.7.3512-3515.2005 (2005).
Fang, H., Hedin, G., Telander, B., Li, G. & Nord, C. E. Emergence of VanD-type vancomycin-resistant Enterococcus faecium in Stockholm, Sweden. Clin Microbiol Infect 13, 106–108, https://doi.org/10.1111/j.1469-0691.2006.01569.x (2007).
Starlander, G., Tellgren-Roth, C. & Melhus, A. Fatal acquisition of vanD gene during vancomycin treatment of septicaemia caused by Enterococcus faecium. J Hosp Infect 92, 409–410, https://doi.org/10.1016/j.jhin.2016.01.002 (2016).
Top, J. et al. Identification of a novel genomic island associated with vanD-type vancomycin-resistance in six Dutch vancomycin-resistant Enterococcus faecium. Antimicrob Agents Chemother, https://doi.org/10.1128/AAC.01793-17 (2018).
Kampmeier, S. et al. Weekly screening supports terminating nosocomial transmissions of vancomycin-resistant enterococci on an oncologic ward - a retrospective analysis. Antimicrob Resist Infect Control 6, 48, https://doi.org/10.1186/s13756-017-0206-z (2017).
Zankari, E. et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67, 2640–2644, https://doi.org/10.1093/jac/dks261 (2012).
Ekuma, A. E., Oduyebo, O. O., Efunshile, A. M. & Konig, B. Surveillance for Vancomycin Resistant Enterococci in a Tertiary Institution in South Western Nigeria. Afr J Infect Dis 10, 121–126, https://doi.org/10.21010/ajid.v10i2.8 (2016).
Genuini, M. et al. Achievement of Therapeutic Vancomycin Exposure With Continuous Infusion in Critically Ill Children. Pediatr Crit Care Med, https://doi.org/10.1097/PCC.0000000000001474 (2018).
cgMLST_database, http://www.cgmlst.org/ncs/schema/991893/searchstrain/?f=ct&v=154 (2017).
Isaac, S. et al. Short- and long-term effects of oral vancomycin on the human intestinal microbiota. J Antimicrob Chemother 72, 128–136, https://doi.org/10.1093/jac/dkw383 (2017).
Van der Auwera, P., Pensart, N., Korten, V., Murray, B. E. & Leclercq, R. Influence of oral glycopeptides on the fecal flora of human volunteers: selection of highly glycopeptide-resistant enterococci. J Infect Dis 173, 1129–1136 (1996).
Domingo, M. C., Huletsky, A., Giroux, R., Picard, F. J. & Bergeron, M. G. vanD and vanG-like gene clusters in a Ruminococcus species isolated from human bowel flora. Antimicrob Agents Chemother 51, 4111–4117, https://doi.org/10.1128/AAC.00584-07 (2007).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455–477, https://doi.org/10.1089/cmb.2012.0021 (2012).
von Wintersdorff, C. J. et al. High rates of antimicrobial drug resistance gene acquisition after international travel, The Netherlands. Emerg Infect Dis 20, 649–657, https://doi.org/10.3201/eid.2004.131718 (2014).
Vliegen, I., Jacobs, J. A., Beuken, E., Bruggeman, C. A. & Vink, C. Rapid identification of bacteria by real-time amplification and sequencing of the 16S rRNA gene. J Microbiol Methods 66, 156–164, https://doi.org/10.1016/j.mimet.2005.11.005 (2006).
Acknowledgements
The authors would like to thank Dr. Erik van Hannen (St. Antonius Hospital, Utrecht, the Netherlands) for providing the vanA and vanB primer sequences. Furthermore, we thank Dr. Ir. Petra F.G. Wolffs and Selma B. Herngreen for implementing and validating the vanA/B PCR within our diagnostic setting. Lastly, we are grateful to the technicians of the sections Bacteriology and Molecular Diagnostics for their work involving the samples and analyses of this study.
Author information
Authors and Affiliations
Contributions
J.F. & C.v.W. designed and conducted the screenings. C.J., H.H. and L.v.A. conducted and analyzed the Whole-Genome Sequencing. J.F. & L.v.A. wrote the main manuscript text. J.F., C.v.W., J.v.N., C.J., F.v.T., H.H. and L.v.A. analyzed the data and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Flipse, J., von Wintersdorff, C.J., van Niekerk, J.M. et al. Appearance of vanD-positive Enterococcus faecium in a tertiary hospital in the Netherlands: prevalence of vanC and vanD in hospitalized patients. Sci Rep 9, 6949 (2019). https://doi.org/10.1038/s41598-019-42824-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42824-4
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
