
Abstract
Benign prostatic hyperplasia (BPH) results in a significant public health burden due to the morbidity caused by the disease and many of the available remedies. As much as 70% of men over 70 will develop BPH. Few studies have been conducted to discover the genetic determinants of BPH risk. Understanding the biological basis for this condition may provide necessary insight for development of novel pharmaceutical therapies or risk prediction. We have evaluated SNP-based heritability of BPH in two cohorts and conducted a genome-wide association study (GWAS) of BPH risk using 2,656 cases and 7,763 controls identified from the Electronic Medical Records and Genomics (eMERGE) network. SNP-based heritability estimates suggest that roughly 60% of the phenotypic variation in BPH is accounted for by genetic factors. We used logistic regression to model BPH risk as a function of principal components of ancestry, age, and imputed genotype data, with meta-analysis performed using METAL. The top result was on chromosome 22 in SYN3 at rs2710383 (p-value = 4.6 × 10−7; Odds Ratio = 0.69, 95% confidence interval = 0.55–0.83). Other suggestive signals were near genes GLGC, UNCA13, SORCS1 and between BTBD3 and SPTLC3. We also evaluated genetically-predicted gene expression in prostate tissue. The most significant result was with increasing predicted expression of ETV4 (chr17; p-value = 0.0015). Overexpression of this gene has been associated with poor prognosis in prostate cancer. In conclusion, although there were no genome-wide significant variants identified for BPH susceptibility, we present evidence supporting the heritability of this phenotype, have identified suggestive signals, and evaluated the association between BPH and genetically-predicted gene expression in prostate.
Similar content being viewed by others


Introduction
Benign prostatic hyperplasia (BPH) is characterized by an enlarged prostate that affects a moderate proportion of middle-aged men and a large proportion of elderly men and can result in significant discomfort and reproductive and urinary tract dysfunction1,2,3,4. Lower urinary tract symptoms (LUTS) are commonly attributed to BPH in the absence of other causes5,6,7. Very severe cases can result in urinary tract infections and bleeding, bladder stones, and kidney damage from failing to void7,8,9,10. Pharmaceutical treatments for BPH include alpha blockers to relax muscles and treat some LUTS symptoms, and 5-alpha reductase inhibitors which can shrink the prostate in some patients but may increase risk for prostate cancer11,12,13,14,15,16. Additionally, the available surgical remedies can present additional risks and have considerable potential consequences for reproductive and urinary tract health17,18,19,20,21.
Heritability of LUTS scores in twins has been estimated at 20–40%22, with some estimates as high as 83%23, while heritability of benign prostate disease has been estimated at 49% from twin studies24. The presence of racial disparities also supports a genetic contribution to BPH risk25,26. Evaluation of the SNP-based additive genetic heritability has not yet been published.
The genetic factors underlying BPH risk remain unclear. To date there have been many BPH candidate gene studies, often evaluating the effect of prostate cancer susceptibility variants, with mixed success27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46,47. Three larger-scale studies have been performed, a MetaboChip analysis of prostate volume48 and two recent genome-wide association studies (GWAS) of BPH49,50. In the present study, we evaluated genetic heritability of clinically reported BPH and conducted a GWAS using cases and controls identified from the Electronic Medical Records and Genomic (eMERGE) network, and evaluated the contribution of the genetic associations to gene expression in prostate tissue.
Results
Heritability
SNP-based additive heritability among common variants was assessed in the 5 sites of the eMERGE-1 network and one of the Geisinger datasets (CoreExome) as they had the largest number of cases assessed on a common genotyping array (Table 1). After stringent filters to remove residual population stratification, there were 755 cases and 899 controls included from eMERGE-1 and 423 cases and 1278 controls included from Geisinger CoreExome. Heritability results were consistent between the two groups, with an estimated heritability of 0.65 (±0.30) in eMERGE-1 (p-value = 0.011) and 0.56 (±0.38) in Geisinger (p-value = 0.070) (Table 2). Results were largely consistent across inclusion of increasing PCs (Supplementary Table 1). Results across chromosomes varied substantially (Supplementary Fig. 1), however, chromosomes 6 and 7 were present among the top 5 results for both eMERGE-1 and Geisinger, suggesting the likelihood of one or more BPH-susceptibility loci being located on these chromosomes.
Genome-wide association
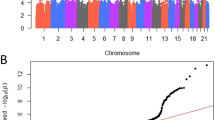
In total, 2,656 cases and 7,763 controls were included across eight eMERGE sites (including the data used in the heritability analysis; Table 1) for the analysis of common (minor allele frequency [MAF] > 0.05) genetic association at 10,973,920 SNPs. Overall, the samples were predominantly identified as white (85%) and cases were slightly older than controls (average age of 68.88 years in cases vs 61.45 years in controls). The most statistically significant results from single SNP GWAS analyses was on chromosome 22 in synpasin 3 (SYN3) at rs2710383 (allele frequency = 0.12, p-value = 4.56 × 10−7; Odds Ratio [OR] = 0.69, 95% confidence interval [CI] = 0.55–0.83; Table 3; Fig. 1). Other suggestive signals were within genes glutamate-cysteine ligase catalytic subunit (GCLC; chromosome 6), unc-13 homolog A (UNC13A; chromosome 19), and ELOVL [elongation of very long chain fatty acids] fatty acid elongase 6 (ELOVL6; chromosome 4), near the long intergenic non-protein coding RNA 1919 (LINC01919; chromosome 18), and in an intergenic region on chromosome 20 between BTB domain containing 3 (BTBD3) and serine palmitoyltransferase long chain base subunit 3 (SPTLC3). Secondary analysis restricting to whites yielded consistent results for top SNPs (Table 3), but identified the top variants as rs10786938 in SORCS1 (p-value = 3.84 × 10−7, OR = 1.23; Supplementary Table 2).

Genome-wide genetically-predicted gene expression and SNP association meta-analysis results with BPH.
We also evaluated the previously identified variants from recent GWAS to determine whether variants replicated across studies (Table 4)49,50. None of the variants reported in those studies were significant or suggestively associated with BPH in this analysis, although effect estimates were largely consistent in both direction and magnitude. Restricting to whites only to more closely match the papers49,50 did not yield significant results.
Gene expression
We also evaluated genetically predicted gene expression (GPGE) in prostate tissue using S-PrediXcan51 and models constructed in GTEx samples52 (Table 5; Fig. 1). The top result did not reach statistical significance (Bonferroni threshold for number of genes, p-value < 1.93 × 10−5) was with increased predicted expression of ETS variant 4 (ETV4; chromosome 17; p-value = 0.0015). Other nominally significant genes were identified on chromosomes 6, 20, 3, 14, 1, and 7. It is noteworthy that neither of the genes on chromosome 6 (histone cluster 1 H3 family member e [HIST1H3E]) and 20 (gonadotropin-releasing hormone 2 [GNRH2], were near the top signals implicated in the GWAS results (GLGC and the intergenic region between BTBD3 and SPTLC3), instead these suggestive GPGE results arose from secondary signals in other regions.
Discussion
We have performed the first SNP-based heritability assessments of BPH followed by a trans-ethnic GWAS and evaluation of genetically predicted gene expression in prostate tissue. Our results indicate that BPH is likely to be substantially heritable, with consistent point estimates near 60% across two comparable EMR-based cohorts, which is somewhat higher than the 49% reported previously from twin studies24. The LUTS symptom score heritability however has been reported to be variable, with estimates ranging from 20 to 83%. The cases in this study likely have overt symptoms of BPH that lead to their clinical diagnoses and treatments, and may represent a more severe phenotype than from some cohort studies.
In this first GWAS of EHR-assessed BPH, we identified previously unreported suggestive SNPs. The gene containing the top SNP from the GWAS, SYN3 is a neuronal protein53,54 which has been implicated in GWAS of many diverse phenotypes including age-related macular degeneration55,56,57, height58,59,60, and uric acid levels61. Expression of SYN3 in GTEx is highest in testis, followed by several brain regions, but is low in prostate and no predictive model was constructed for SYN3 expression in that tissue52,62. Another neuronal protein UNC13A (unc-13 homolog A) was also implicated from these GWAS results. Variants near this gene have been consistently associated with amyotrophic lateral sclerosis in several genome-wide studies63,64,65.
The second suggestive signal from GWAS, in the gene GCLC is also interesting, due in part to the localization of the SNP-based heritability on chromosome 6. Also relevant is the finding of modest association with the lead variant (rs534957) from our study which also demonstrated a weak association with prostatitis in the UK Biobank data (p-value = 6 × 10−3; as viewed in the Global Biobank Engine66 [https://biobankengine.stanford.edu/]). This suggests a consistent finding with another EHR-defined data set despite differences in case/control classification. Additionally, the identification of suggestive GPGE on chromosome 6 apart from the GCLC locus provides modest support for the heritability analysis, suggesting that the relevant SNPs have yet to be detected, perhaps due to a lack of power in the present studies.
One of the more biologically interesting candidates identified in this study is GNRH2 (gonadotropin-releasing hormone 2). GPGE analysis indicated that reduced expression of this gene in the prostate was associated with increased risk of BPH (p-value = 0.021). GNRH2 is expressed in the prostate67,68,69,70 and its expression is regulated by several reproductive hormones71. Both gonadotropin releasing hormone (GnRH) antagonists and agonists have been investigated as treatments for BPH and prostate cancer12,16,72,73,74,75,76,77, however, the side effects have made many of these impractical as therapeutic options. There is currently a Phase 3 trial underway to evaluate whether a GnRH antagonist in combination with radiation can improve progression of prostate cancer. This therapeutic was previously part of a phase 2 trial for efficacy in BPH, however the trial was stopped early due to not meeting primary efficacy endpoints. This is potentially consistent with the results observed here in which reduced expression levels of GNRH2 are associated with increased risk of BPH. A genetic variant in GNRH2 (rs6051545) was observed to impact testosterone levels during androgen deprivation therapy to treat metastatic prostate cancer78. It has been suggested that this may lead to a negative effect of the therapy on prognosis78.
Of the 11 genes included in Table 5, more than half of them have been previously reported such that expression changes have been associated in prostate tissue, often with various stages of prostate cancer. The top result from the GPGE analysis, ETV4 (ETS variant 4) has been previously found in studies of prostate cancer to have significantly higher relative expression in the tumor tissues than in benign samples79, as well as an association with poor prognosis80,81. We found that increased predicted expression of ETV4 is associated with increased risk of BPH in this study (p-value = 0.0015). Laminin subunit beta 2 (LAMB2) has been identified as being downregulated in the transition from prostate intraepithelial neoplasia to invasive prostate cancer from differential expression analysis82. Our results suggest that increasing LAMB2 expression is associated with increased risk of BPH. SCAP, which encodes SREBP cleavage-activating protein, has also been identified to show expression changes in prostate cancer83,84, and has been specifically noted to be regulated by androgens83,85. Recently, TIGIT expression has been implicated in failures of prostate cancer checkpoint inhibition86,87. Together, these results suggest that though these results did not achieve statistical significance, germline genetic associations with BPH may alter gene expression in prostate tissue, and that those genes without a presently documented role may yet be identified as important in studies of prostate gene expression implicated in disease.
There have been two recent GWAS of BPH in whites which have identified many significant and suggestive associations, though none were identified by both studies. Evaluation of these reported signals in the eMERGE data revealed modest associations at only five loci, including BCL11A, TERT, CLPTM1L, GATA6, and FGFR2 (Table 4). It is notable, that although none of the variants reported were significant in this study, effect estimates were largely consistent in both direction and magnitude. The lack of replication may be due in part to differences across studies in disease definition (varied use of IPSS scores, prostate volume, history of transurethral resection of the prostate, etc), participant recruitment from clinical trials, community cohorts, and hospital-based populations, or differences in age. Evaluation of associated variation reported in candidate gene analyses of BPH28,30,31,32,34,37,38,88 and an evaluation of prostate volume48 did not yield any suggestive results in the present study (Supplementary Table 3).
Previous studies have shown adequate positive and negative predictive values based on electronic diagnoses (International Classification of Diseases, Ninth Revision (ICD9) codes and problem list) for BPH89, however, the phenotyping of BPH in the medical record likely reflects the presence of symptoms. Studies of care-seeking behavior with respect to BPH and LUTS have consistently shown that those seeking medical care tend to have higher symptom scores/more severe symptoms, but that reasons for not seeking treatment include diverse social and treatment concerns, even among those experiencing symptoms90,91,92,93,94. This suggests the possibility that some portion of the controls in our study may have experienced (or will experience in the future) symptoms of BPH but have not (yet) sought treatment for the condition. This is a limitation of the present study.
Based on these results, wherein BPH was shown to be heritable but no significant susceptibility loci were detected, it seems that BPH is a complex disease made up of many physiological symptoms and the genetic underpinnings of this trait are likely to consist of a multitude of variants of small effect. This makes large sample sizes crucial for detecting genetic loci associated with BPH as has been demonstrated50. In conclusion, we have shown that BPH is heritable, identified suggestive association signals, and are the first to evaluate the association between BPH and genetically-predicted gene expression in prostate.
Methods
Study Populations
The eMERGE Network is a consortium of several EHR-linked biorepositories formed with the goal of developing approaches for the use of the EHR in genomic research95,96. Consortium membership has evolved over eMERGE’s 11 year history, with many sites contributing data including Group Health/University of Washington, Marshfield Clinic, Mayo Clinic, Northwestern University, Vanderbilt University (Phase 1 sites), Children’s Hospital of Philadelphia (CHOP), Boston Children’s Hospital (BCH), Cincinnati Children’s Hospital Medical Center (CCHMC), Geisinger Health System, Mount Sinai School of Medicine (sites added in Phase 2), Harvard University and Columbia University (sites added in Phase 3). The eMERGE study was approved by the Ethical Committee/Institutional Review Board at each site (Vanderbilt University Medical Center, Group Health/University of Washington, Marshfield Clinic, Mayo Clinic, Northwestern University, Children’s Hospital of Philadelphia, Boston Children’s Hospital, Cincinnati Children’s Hospital Medical Center, Geisinger Health System, Mount Sinai School of Medicine, Harvard University and Columbia University) and all methods were performed in accordance with the relevant guidelines and regulations. In this study of BPH, data from the eMERGE pediatric study sites (CHOP, BCH, CCHMC) were not included. Participants at all study sites provided written, informed consent, and for participants under the age of 18 years (who were not included in the analyses presented herein), informed consent was obtained from a parent and/or legal guardian.
Phenotyping
Among men of at least age 40, without prostate or bladder cancers (defined via ICD9 codes [233.4, 233.7 or 233.9], tumor registries [Primary site = C619] and problem lists [containing keywords e.g. “prostate cancer”, “malignant tumor of the prostate”, “bladder cancer”, “bladder CA”), we included all cases of BPH with at least two ICD9 codes indicating a BPH diagnosis (600, 600.0, 600.0*, 600.2, 600.2*, 600.9, 600.9*), in addition to either receiving medications for the treatment of BPH or 1 or more procedure (Current Procedural Terminology [CPT]) codes for BPH-related surgeries (52450, 52601, 52630, 52648, 53850, 53852). Controls were males of at least age 40, with at least 3 outpatient visits within any 2-year period after the age of 40, without prostate or bladder cancer or instances of BPH ICD9 codes, medications or BPH-related surgical CPT codes. The algorithm is available on PheKB.org (https://phekb.org/phenotype/216).
Genotyping and Quality Control
Genotyping was performed for eMERGE-1 study sites using one of two Illumina arrays across two genotyping centers. Individuals of self-identified or administratively-assigned European-descent were genotyped on the Illumina 660W-Quad, while individuals of self-identified or administratively-assigned African-descent were genotyped on the Illumina 1 M. For the majority of patients, genotyping was performed at one of two centers: the Center for Inherited Disease Research (CIDR) at Johns Hopkins University and the Center for Genotyping and Analysis at the Broad Institute as previously described95,96. Existing genotype data available for eMERGE-2 and -3 study sites included data from the Illumina 550, Illumina 610, Illumina HumanOmni Express, Illumina MultiEthnic Genotyping Array, Illumina CoreExome, and Affymetrix 6.0 arrays.
Genotype quality control (QC) was performed within each study population, and a uniform protocol was implemented. QC for all studies was performed using PLINK97, including a 95% single nucleotide polymorphism (SNP) and individual call rate threshold, removal of first-degree related individuals, sex checks, alignment of alleles to the genomic ‘+’ strand. Visualization of ancestry by principal components analysis was performed by study using either Eigenstrat98 or flashPCA99.
Statistical Analysis
Restricted maximum likelihood estimation as implemented in GCTA100 was used to determine the proportion of phenotypic variance explained by common additive genetic variants in two cohorts, eMERGE-1 and Geisinger CoreExome. Data was filtered to include only common variants (MAF > 0.05), and samples with IBD probabilities <0.025, as well as restricted using principal components to retain only EA samples. Disease prevalence was based on average age within cohort and set at 0.67 for eMERGE-1 and 0.63 for Geisinger CoreExome.
Genotype data was imputed from the 1000 Genomes Project haplotypes using SHAPEIT2101 and IMPUTE2102 by site or study and analyzed for associations separately. We used logistic regression to model BPH risk as a function of genotype, age, and principal components of ancestry with the SNPTEST software package, with subsequent meta-analysis performed using METAL103. There was no substantial genomic inflation observed, with a meta-analysis lambda of 1.017 (Supplementary Fig. 2).
To further evaluate the genetic association results in the context of gene expression, we employed the novel method S-PrediXcan51, an extension of the PrediXcan method62. PrediXcan conducts a test of association between phenotypes and gene expression levels predicted by genetic variants in a library of tissues from the Genotype-Tissue Expression (GTEx) project52,104. S-PrediXcan is a meta-analysis approach that conducts the PrediXcan test using genotype association summary statistics, rather than performing the tests in individual-level data. We utilized covariance matrices built for prostate tissue from GTEx to annotate SNP association signals as well as to provide information about likely tissue expression patterns and relevant biological information.
Data Availability
The datasets generated during and/or analysed during the current study are available in the eMERGE dbGAP repository (accession phs000888.v1.p1; https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000888.v1.p1).
References
Lee, S. W. H., Chan, E. M. C. & Lai, Y. K. The global burden of lower urinary tract symptoms suggestive of benign prostatic hyperplasia: A systematic review and meta-analysis. Scientific reports 7, 7984, https://doi.org/10.1038/s41598-017-06628-8 (2017).
Foster, S. A., Shortridge, E. F., DiBonaventura, M. & Viktrup, L. Predictors of self-reported benign prostatic hyperplasia in European men: analysis of the European National Health and Wellness Survey. World journal of urology 33, 639–647, https://doi.org/10.1007/s00345-014-1366-6 (2015).
Chokkalingam, A. P. et al. Prevalence of BPH and lower urinary tract symptoms in West Africans. Prostate cancer and prostatic diseases 15, 170–176, https://doi.org/10.1038/pcan.2011.43 (2012).
Foo, K. T. Pathophysiology of clinical benign prostatic hyperplasia. Asian journal of urology 4, 152–157, https://doi.org/10.1016/j.ajur.2017.06.003 (2017).
Egan, K. B. The Epidemiology of Benign Prostatic Hyperplasia Associated with Lower Urinary Tract Symptoms: Prevalence and Incident Rates. The Urologic clinics of North America 43, 289–297, https://doi.org/10.1016/j.ucl.2016.04.001 (2016).
Speakman, M., Kirby, R., Doyle, S. & Ioannou, C. Burden of male lower urinary tract symptoms (LUTS) suggestive of benign prostatic hyperplasia (BPH) - focus on the UK. BJU international 115, 508–519, https://doi.org/10.1111/bju.12745 (2015).
Rodriguez-Nieves, J. A. & Macoska, J. A. Prostatic fibrosis, lower urinary tract symptoms, and BPH. Nature reviews. Urology 10, 546–550, https://doi.org/10.1038/nrurol.2013.149 (2013).
Jung, J. H. et al. The association of benign prostatic hyperplasia with lower urinary tract stones in adult men: A retrospective multicenter study. Asian journal of urology 5, 118–121, https://doi.org/10.1016/j.ajur.2017.06.008 (2018).
Huang, W. et al. Risk factors for bladder calculi in patients with benign prostatic hyperplasia. Medicine 96, e7728, https://doi.org/10.1097/md.0000000000007728 (2017).
Ko, Y. H. et al. Clinical Implications of Residual Urine in Korean Benign Prostatic Hyperplasia (BPH) Patients: A Prognostic Factor for BPH-Related Clinical Events. International neurourology journal 14, 238–244, https://doi.org/10.5213/inj.2010.14.4.238 (2010).
Bishr, M. et al. Medical management of benign prostatic hyperplasia: Results from a population-based study. Canadian Urological Association journal = Journal de l’Association des urologues du Canada 10, 55–59, https://doi.org/10.5489/cuaj.3058 (2016).
Kumar, R., Malla, P. & Kumar, M. Advances in the design and discovery of drugs for the treatment of prostatic hyperplasia. Expert opinion on drug discovery 8, 1013–1027, https://doi.org/10.1517/17460441.2013.797960 (2013).
Kim, E. H., Brockman, J. A. & Andriole, G. L. The use of 5-alpha reductase inhibitors in the treatment of benign prostatic hyperplasia. Asian journal of urology 5, 28–32, https://doi.org/10.1016/j.ajur.2017.11.005 (2018).
Wallner, L. P. et al. 5-Alpha Reductase Inhibitors and the Risk of Prostate Cancer Mortality in Men Treated for Benign Prostatic Hyperplasia. Mayo Clinic proceedings 91, 1717–1726, https://doi.org/10.1016/j.mayocp.2016.07.023 (2016).
Hirshburg, J. M., Kelsey, P. A., Therrien, C. A., Gavino, A. C. & Reichenberg, J. S. Adverse Effects and Safety of 5-alpha Reductase Inhibitors (Finasteride, Dutasteride): A Systematic Review. The Journal of clinical and aesthetic dermatology 9, 56–62 (2016).
Russo, A. et al. Latest pharmacotherapy options for benign prostatic hyperplasia. Expert opinion on pharmacotherapy 15, 2319–2328, https://doi.org/10.1517/14656566.2014.955470 (2014).
Malaeb, B. S., Yu, X., McBean, A. M. & Elliott, S. P. National trends in surgical therapy for benign prostatic hyperplasia in the United States (2000-2008). Urology 79, 1111–1116, https://doi.org/10.1016/j.urology.2011.11.084 (2012).
Rieken, M., Antunes-Lopes, T., Geavlete, B. & Marcelissen, T. What Is New with Sexual Side Effects After Transurethral Male Lower Urinary Tract Symptom Surgery? European urology focus 4, 43–45, https://doi.org/10.1016/j.euf.2018.05.001 (2018).
Gacci, M. et al. Best practice in the management of storage symptoms in male lower urinary tract symptoms: a review of the evidence base. Therapeutic advances in urology 10, 79–92, https://doi.org/10.1177/1756287217742837 (2018).
Moreira, A. M. et al. A Review of Adverse Events Related to Prostatic Artery Embolization for Treatment of Bladder Outlet Obstruction Due to BPH. Cardiovascular and interventional radiology 40, 1490–1500, https://doi.org/10.1007/s00270-017-1765-3 (2017).
Qian, X. et al. Functional outcomes and complications following B-TURP versus HoLEP for the treatment of benign prostatic hyperplasia: a review of the literature and Meta-analysis. The aging male: the official journal of the International Society for the Study of the Aging Male 20, 184–191, https://doi.org/10.1080/13685538.2017.1295436 (2017).
Afari, N. et al. Heritability of Lower Urinary Tract Symptoms in Men: A Twin Study. The Journal of urology 196, 1486–1492, https://doi.org/10.1016/j.juro.2016.06.018 (2016).
Meikle, A. W., Bansal, A., Murray, D. K., Stephenson, R. A. & Middleton, R. G. Heritability of the symptoms of benign prostatic hyperplasia and the roles of age and zonal prostate volumes in twins. Urology 53, 701–706 (1999).
Partin, A. W. et al. Concordance rates for benign prostatic disease among twins suggest hereditary influence. Urology 44, 646–650 (1994).
Hoke, G. P. & McWilliams, G. W. Epidemiology of benign prostatic hyperplasia and comorbidities in racial and ethnic minority populations. The American journal of medicine 121, S3–10, https://doi.org/10.1016/j.amjmed.2008.05.021 (2008).
Colon, I. & Payne, R. E. Benign prostatic hyperplasia and lower urinary tract symptoms in African Americans and Latinos: treatment in the context of common comorbidities. The American journal of medicine 121, S18–26, https://doi.org/10.1016/j.amjmed.2008.05.023 (2008).
Lee, C. L., Lee, J., Na, Y. G. & Song, K. H. Combined effect of polymorphisms in type III 5-alpha reductase and androgen receptor gene with the risk of benign prostatic hyperplasia in Korea. Journal of exercise rehabilitation 12, 504–508, https://doi.org/10.12965/jer.1632802.401 (2016).
Kim, S. K. et al. Association between polymorphisms of estrogen receptor 2 and benign prostatic hyperplasia. Experimental and therapeutic medicine 10, 1990–1994, https://doi.org/10.3892/etm.2015.2755 (2015).
Choubey, V. K. et al. SRD5A2 gene polymorphisms and the risk of benign prostatic hyperplasia but not prostate cancer. Asian Pacific journal of cancer prevention: APJCP 16, 1033–1036 (2015).
Ruan, L. et al. Association between single nucleotide polymorphism of vitamin D receptor gene FokI polymorphism and clinical progress of benign prostatic hyperplasia. TheScientificWorldJournal 2015, 235895, https://doi.org/10.1155/2015/235895 (2015).
Winchester, D. et al. SPINK1 Promoter Variants Are Associated with Prostate Cancer Predisposing Alterations in Benign Prostatic Hyperplasia Patients. Anticancer research 35, 3811–3819 (2015).
Ban, J. Y. & Yoo, K. H. Promoter Polymorphism (rs12770170, -184C/T) of Microseminoprotein, Beta as a Risk Factor for Benign Prostatic Hyperplasia in Korean Population. International neurourology journal 18, 63–67, https://doi.org/10.5213/inj.2014.18.2.63 (2014).
Kumar, V. et al. Association of CYP1A1, CYP1B1 and CYP17 gene polymorphisms and organochlorine pesticides with benign prostatic hyperplasia. Chemosphere 108, 40–45, https://doi.org/10.1016/j.chemosphere.2014.02.081 (2014).
Seok, H., Yoo, K. H., Kim, Y. O. & Chung, J. H. Association of a Missense ALDH2 Single Nucleotide Polymorphism (Glu504Lys) With Benign Prostate Hyperplasia in a Korean Population. International neurourology journal 17, 168–173, https://doi.org/10.5213/inj.2013.17.4.168 (2013).
Karatzas, A. et al. Lack of association between the UDP-glucuronosyltransferase 1A1 (UGT1A1) gene polymorphism and the risk of benign prostatic hyperplasia in Caucasian men. Molecular biology reports 40, 6665–6669, https://doi.org/10.1007/s11033-013-2781-2 (2013).
Zambra, F. M., Biolchi, V., Brum, I. S. & Chies, J. A. CCR2 and CCR5 genes polymorphisms in benign prostatic hyperplasia and prostate cancer. Human immunology 74, 1003–1008, https://doi.org/10.1016/j.humimm.2013.04.031 (2013).
Qi, J. et al. Genetic variants in 2q31 and 5p15 are associated with aggressive benign prostatic hyperplasia in a Chinese population. Prostate 73, 1182–1190, https://doi.org/10.1002/pros.22666 (2013).
Jiao, Y. et al. LILRA3 is associated with benign prostatic hyperplasia risk in a Chinese Population. International journal of molecular sciences 14, 8832–8840, https://doi.org/10.3390/ijms14058832 (2013).
Biolchi, V. et al. Androgen receptor GGC polymorphism and testosterone levels associated with high risk of prostate cancer and benign prostatic hyperplasia. Molecular biology reports 40, 2749–2756, https://doi.org/10.1007/s11033-012-2293-5 (2013).
Choubey, V. K. et al. Null genotypes at the GSTM1 and GSTT1 genes and the risk of benign prostatic hyperplasia: a case-control study and a meta-analysis. Prostate 73, 146–152, https://doi.org/10.1002/pros.22549 (2013).
Biolchi, V., Silva Neto, B., Koff, W. & Brum, I. S. Androgen receptor CAG polymorphism and the risk of benign prostatic hyperplasia in a Brazilian population. International braz j urol: official journal of the Brazilian Society of Urology 38, 373–379 (2012).
Izmirli, M., Arikan, B., Bayazit, Y. & Alptekin, D. Associations of polymorphisms in HPC2/ELAC2 and SRD5A2 genes with benign prostate hyperplasia in Turkish men. Asian Pacific journal of cancer prevention: APJCP 12, 731–733 (2011).
Konwar, R. et al. Glutathione S-transferase (GST) gene variants and risk of benign prostatic hyperplasia: a report in a North Indian population. Asian Pacific journal of cancer prevention: APJCP 11, 1067–1072 (2010).
Mittal, R. D., Kesarwani, P., Singh, R., Ahirwar, D. & Mandhani, A. GSTM1, GSTM3 and GSTT1 gene variants and risk of benign prostate hyperplasia in North India. Disease markers 26, 85–91, https://doi.org/10.3233/dma-2009-0611 (2009).
Ma, Z. et al. Polymorphisms of fibroblast growth factor receptor 4 have association with the development of prostate cancer and benign prostatic hyperplasia and the progression of prostate cancer in a Japanese population. International journal of cancer. Journal international du cancer 123, 2574–2579, https://doi.org/10.1002/ijc.23578 (2008).
Das, K. et al. Shorter CAG repeats in androgen receptor and non-GG genotypes in prostate-specific antigen loci are associated with decreased risk of benign prostatic hyperplasia and prostate cancer. Cancer Lett 268, 340–347, https://doi.org/10.1016/j.canlet.2008.04.009 (2008).
Cartwright, R. et al. Systematic review and meta-analysis of candidate gene association studies of lower urinary tract symptoms in men. European urology 66, 752–768, https://doi.org/10.1016/j.eururo.2014.01.007 (2014).
Giri, A., Edwards, T. L., Motley, S. S., Byerly, S. H. & Fowke, J. H. Genetic Determinants of Metabolism and Benign Prostate Enlargement: Associations with Prostate Volume. PLoS One 10, e0132028, https://doi.org/10.1371/journal.pone.0132028 (2015).
Na, R. et al. A genetic variant near GATA3 implicated in inherited susceptibility and etiology of benign prostatic hyperplasia (BPH) and lower urinary tract symptoms (LUTS). Prostate 77, 1213–1220, https://doi.org/10.1002/pros.23380 (2017).
Gudmundsson, J. et al. Genome-wide associations for benign prostatic hyperplasia reveal a genetic correlation with serum levels of PSA. Nature communications 9, 4568, https://doi.org/10.1038/s41467-018-06920-9 (2018).
Barbeira, A. N. et al. Exploring the phenotypic consequences of tissue specific gene expression variation inferred from GWAS summary statistics. Nature communications 9, 1825, https://doi.org/10.1038/s41467-018-03621-1 (2018).
The Genotype-Tissue Expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science (New York, N.Y.) 348, 648–660, https://doi.org/10.1126/science.1262110 (2015).
Kao, H. T. et al. A third member of the synapsin gene family. Proc Natl Acad Sci USA 95, 4667–4672 (1998).
Hosaka, M. & Sudhof, T. C. Synapsin III, a novel synapsin with an unusual regulation by Ca2+. J Biol Chem 273, 13371–13374 (1998).
Fritsche, L. G. et al. Seven new loci associated with age-related macular degeneration. Nat Genet 45(433–439), 439e431–432, https://doi.org/10.1038/ng.2578 (2013).
Neale, B. M. et al. Genome-wide association study of advanced age-related macular degeneration identifies a role of the hepatic lipase gene (LIPC). Proc Natl Acad Sci USA 107, 7395–7400, https://doi.org/10.1073/pnas.0912019107 (2010).
Chen, W. et al. Genetic variants near TIMP3 and high-density lipoprotein-associated loci influence susceptibility to age-related macular degeneration. Proc Natl Acad Sci USA 107, 7401–7406, https://doi.org/10.1073/pnas.0912702107 (2010).
Wood, A. R. et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nat Genet 46, 1173–1186, https://doi.org/10.1038/ng.3097 (2014).
He, M. et al. Meta-analysis of genome-wide association studies of adult height in East Asians identifies 17 novel loci. Hum Mol Genet 24, 1791–1800, https://doi.org/10.1093/hmg/ddu583 (2015).
Lango Allen, H. et al. Hundreds of variants clustered in genomic loci and biological pathways affect human height. Nature 467, 832–838, https://doi.org/10.1038/nature09410 (2010).
Charles, B. A. et al. A genome-wide association study of serum uric acid in African Americans. BMC medical genomics 4, 17, https://doi.org/10.1186/1755-8794-4-17 (2011).
Gamazon, E. R. et al. A gene-based association method for mapping traits using reference transcriptome data. Nat Genet 47, 1091–1098, https://doi.org/10.1038/ng.3367 (2015).
van Es, M. A. et al. Genome-wide association study identifies 19p13.3 (UNC13A) and 9p21.2 as susceptibility loci for sporadic amyotrophic lateral sclerosis. Nat Genet 41, 1083–1087, https://doi.org/10.1038/ng.442 (2009).
Benyamin, B. et al. Cross-ethnic meta-analysis identifies association of the GPX3-TNIP1 locus with amyotrophic lateral sclerosis. Nature communications 8, 611, https://doi.org/10.1038/s41467-017-00471-1 (2017).
van Rheenen, W. et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet 48, 1043–1048, https://doi.org/10.1038/ng.3622 (2016).
McInnes, G. et al. Global Biobank Engine: enabling genotype-phenotype browsing for biobank summary statistics. bioRxiv, https://doi.org/10.1101/304188 (2018).
Desaulniers, A. T., Cederberg, R. A., Lents, C. A. & White, B. R. Expression and Role of Gonadotropin-Releasing Hormone 2 and Its Receptor in Mammals. Frontiers in endocrinology 8, 269, https://doi.org/10.3389/fendo.2017.00269 (2017).
Millar, R. et al. A novel mammalian receptor for the evolutionarily conserved type II GnRH. Proc Natl Acad Sci USA 98, 9636–9641, https://doi.org/10.1073/pnas.141048498 (2001).
Neill, J. D., Duck, L. W., Sellers, J. C. & Musgrove, L. C. A gonadotropin-releasing hormone (GnRH) receptor specific for GnRH II in primates. Biochemical and biophysical research communications 282, 1012–1018, https://doi.org/10.1006/bbrc.2001.4678 (2001).
White, R. B., Eisen, J. A., Kasten, T. L. & Fernald, R. D. Second gene for gonadotropin-releasing hormone in humans. Proc Natl Acad Sci USA 95, 305–309 (1998).
Darby, S. et al. Expression of GnRH type II is regulated by the androgen receptor in prostate cancer. Endocrine-related cancer 14, 613–624, https://doi.org/10.1677/erc-07-0041 (2007).
Sakai, M., Elhilali, M. & Papadopoulos, V. The GnRH Antagonist Degarelix Directly Inhibits Benign Prostate Hyperplasia Cell Growth. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme 47, 925–931, https://doi.org/10.1055/s-0035-1555899 (2015).
Raja, A., Hori, S. & Armitage, J. N. Hormonal manipulation of lower urinary tract symptoms secondary to benign prostatic obstruction. Indian journal of urology: IJU: journal of the Urological Society of India 30, 189–193, https://doi.org/10.4103/0970-1591.126904 (2014).
Rick, F. G. et al. Hormonal manipulation of benign prostatic hyperplasia. Current opinion in urology 23, 17–24, https://doi.org/10.1097/MOU.0b013e32835abd18 (2013).
Polisca, A. et al. Clinical efficacy of the GnRH agonist (deslorelin) in dogs affected by benign prostatic hyperplasia and evaluation of prostatic blood flow by Doppler ultrasound. Reproduction in domestic animals = Zuchthygiene 48, 673–680, https://doi.org/10.1111/rda.12143 (2013).
Soler, R. et al. Future direction in pharmacotherapy for non-neurogenic male lower urinary tract symptoms. European urology 64, 610–621, https://doi.org/10.1016/j.eururo.2013.04.042 (2013).
Lepor, H. The role of gonadotropin-releasing hormone antagonists for the treatment of benign prostatic hyperplasia. Reviews in urology 8, 183–189 (2006).
Shiota, M. et al. The Association of Polymorphisms in the Gene Encoding Gonadotropin-Releasing Hormone with Serum Testosterone Level during Androgen Deprivation Therapy and Prognosis of Metastatic Prostate Cancer. The Journal of urology, https://doi.org/10.1016/j.juro.2017.09.076 (2017).
Cavazzola, L. R. et al. Relative mRNA expression of prostate-derived E-twenty-six factor and E-twenty-six variant 4 transcription factors, and of uridine phosphorylase-1 and thymidine phosphorylase enzymes, in benign and malignant prostatic tissue. Oncology letters 9, 2886–2894, https://doi.org/10.3892/ol.2015.3093 (2015).
Qi, M. et al. Overexpression of ETV4 is associated with poor prognosis in prostate cancer: involvement of uPA/uPAR and MMPs. Tumour biology: the journal of the International Society for Oncodevelopmental Biology and Medicine 36, 3565–3572, https://doi.org/10.1007/s13277-014-2993-7 (2015).
Mesquita, D. et al. Specific and redundant activities of ETV1 and ETV4 in prostate cancer aggressiveness revealed by co-overexpression cellular contexts. Oncotarget 6, 5217–5236, https://doi.org/10.18632/oncotarget.2847 (2015).
Ashida, S. et al. Molecular features of the transition from prostatic intraepithelial neoplasia (PIN) to prostate cancer: genome-wide gene-expression profiles of prostate cancers and PINs. Cancer research 64, 5963–5972, https://doi.org/10.1158/0008-5472.can-04-0020 (2004).
Heemers, H. et al. Androgens stimulate lipogenic gene expression in prostate cancer cells by activation of the sterol regulatory element-binding protein cleavage activating protein/sterol regulatory element-binding protein pathway. Molecular endocrinology (Baltimore, Md.) 15, 1817–1828, https://doi.org/10.1210/mend.15.10.0703 (2001).
Prabhu, A. V., Krycer, J. R. & Brown, A. J. Overexpression of a key regulator of lipid homeostasis, Scap, promotes respiration in prostate cancer cells. FEBS letters 587, 983–988, https://doi.org/10.1016/j.febslet.2013.02.040 (2013).
Heemers, H. et al. Identification of an androgen response element in intron 8 of the sterol regulatory element-binding protein cleavage-activating protein gene allowing direct regulation by the androgen receptor. J Biol Chem 279, 30880–30887, https://doi.org/10.1074/jbc.M401615200 (2004).
Papanicolau-Sengos, A. et al. Identification of targets for prostate cancer immunotherapy. Prostate 79, 498–505, https://doi.org/10.1002/pros.23756 (2019).
Nava Rodrigues, D. et al. Immunogenomic analyses associate immunological alterations with mismatch repair defects in prostate cancer. J Clin Invest 128, 4441–4453, https://doi.org/10.1172/jci121924 (2018).
Gu, X. et al. SRD5A1 and SRD5A2 are associated with treatment for benign prostatic hyperplasia with the combination of 5alpha-reductase inhibitors and alpha-adrenergic receptor antagonists. The Journal of urology 190, 615–619, https://doi.org/10.1016/j.juro.2013.03.024 (2013).
Szeto, H. C., Coleman, R. K., Gholami, P., Hoffman, B. B. & Goldstein, M. K. Accuracy of computerized outpatient diagnoses in a Veterans Affairs general medicine clinic. The American journal of managed care 8, 37–43 (2002).
Kok, E. T. et al. Simple case definition of clinical benign prostatic hyperplasia, based on International Prostate Symptom Score, predicts general practitioner consultation rates. Urology 68, 784–789, https://doi.org/10.1016/j.urology.2006.04.008 (2006).
Roberts, R. O. et al. Natural history of prostatism: worry and embarrassment from urinary symptoms and health care-seeking behavior. Urology 43, 621–628 (1994).
Jacobsen, S. J. et al. Natural history of prostatism: factors associated with discordance between frequency and bother of urinary symptoms. Urology 42, 663–671 (1993).
Sarma, A. V., Wallner, L., Jacobsen, S. J., Dunn, R. L. & Wei, J. T. Health seeking behavior for lower urinary tract symptoms in black men. The Journal of urology 180, 227–232, https://doi.org/10.1016/j.juro.2008.03.032 (2008).
Wolters, R., Wensing, M., van Weel, C., van der Wilt, G. J. & Grol, R. P. Lower urinary tract symptoms: social influence is more important than symptoms in seeking medical care. BJU international 90, 655–661 (2002).
Gottesman, O. et al. The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and. future. Genetics in medicine: official journal of the American College of Medical Genetics 15, 761–771, https://doi.org/10.1038/gim.2013.72 (2013).
McCarty, C. A. et al. The eMERGE. Network: a consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC medical genomics 4, 13, https://doi.org/10.1186/1755-8794-4-13 (2011).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559–575, https://doi.org/10.1086/519795 (2007).
Price, A. L. et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 38, 904–909, https://doi.org/10.1038/ng1847 (2006).
Abraham, G. & Inouye, M. Fast principal component analysis of large-scale genome-wide data. PLoS One 9, e93766, https://doi.org/10.1371/journal.pone.0093766 (2014).
Yang, J., Lee, S. H., Goddard, M. E. & Visscher, P. M. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88, 76–82, https://doi.org/10.1016/j.ajhg.2010.11.011 (2011).
Delaneau, O., Marchini, J. & Zagury, J. F. A linear complexity phasing method for thousands of genomes. Nat Methods 9, 179–181, https://doi.org/10.1038/nmeth.1785 (2012).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5, e1000529, https://doi.org/10.1371/journal.pgen.1000529 (2009).
Willer, C. J., Li, Y. & Abecasis, G. R. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191, https://doi.org/10.1093/bioinformatics/btq340 (2010).
Mele, M. et al. Human genomics. The human transcriptome across tissues and individuals. Science (New York, N.Y.) 348, 660–665, https://doi.org/10.1126/science.aaa0355 (2015).
Acknowledgements
J.N.H. was supported by T32CA160056 (P.I. X-O Shu). The eMERGE Network was initiated and funded by the NHGRI through the following grants: U01HG8657 (Kaiser Washington/University of Washington); U01HG8685 (Brigham and Women’s Hospital); U01HG8672 (Vanderbilt University Medical Center); U01HG8666 (Cincinnati Children’s Hospital Medical Center); U01HG6379 (Mayo Clinic); U01HG8679 (Geisinger Clinic); U01HG8680 (Columbia University Health Sciences); U01HG8684 (Children’s Hospital of Philadelphia); U01HG8673 (Northwestern University); U01HG8701 (Vanderbilt University Medical Center serving as the Coordinating Center); U01HG8676 (Partners Healthcare/Broad Institute); and U01HG8664 (Baylor College of Medicine). Some of the dataset(s) used for the analyses described were obtained from Vanderbilt University Medical Center’s BioVU which is supported by institutional funding, the 1S10RR025141-01 instrumentation award, and by the CTSA grant UL1TR000445 from NCATS/NIH. The authors would also like to acknowledge NCATS grant UL1TR002373 (P.I. Marc Drezner).
Author information
Authors and Affiliations
Contributions
Study design/conception: D.M.R., J.C.D., T.L.E.; Data collection/Sample recruitment: K.M.B., D.M.R., J.C.D., M.B., J.G.L., G.J., N.S., G.H., D.C., A.G., P.D., P.L.P., P.A.M.S., H.H.; Data analysis: J.N.H., E.S.T., T.L.E.; Methods/Algorithm Development: J.C.D., R.C., S.S.; Manuscript writing: J.N.H., D.R.V.E., T.L.E.; Manuscript editing/comments: M.D.R., S.S.V., D.M.R., J.C.D., J.G.L.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hellwege, J.N., Stallings, S., Torstenson, E.S. et al. Heritability and genome-wide association study of benign prostatic hyperplasia (BPH) in the eMERGE network. Sci Rep 9, 6077 (2019). https://doi.org/10.1038/s41598-019-42427-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42427-z
This article is cited by
-
Polygenic risk score predicting susceptibility and outcome of benign prostatic hyperplasia in the Han Chinese
Human Genomics (2024)
-
Association of PSA variability with prostate cancer development using large-scale medical information data: a retrospective cohort study
Genes and Environment (2023)
-
Genetic, Genomic, and Heritable Components of Benign Prostatic Hyperplasia
Current Bladder Dysfunction Reports (2023)
-
TNF is a potential therapeutic target to suppress prostatic inflammation and hyperplasia in autoimmune disease
Nature Communications (2022)
-
Genome-wide association study identifies a role for the progesterone receptor in benign prostatic hyperplasia risk
Prostate Cancer and Prostatic Diseases (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
