
Abstract
Retinitis pigmentosa (RP) is a common phenotype in multiple inherited retinal dystrophies (IRD). Disease gene identification can assist the clinical diagnosis of IRD patients for better clinical management, treatment and counseling. In this study, we aimed to delineate and characterize the disease-causing mutations in Chinese familial and sporadic patients with initial diagnosis of RP. Four unrelated Chinese families and 118 sporadic RP patients were recruited for whole exome sequencing analysis. A total of 5 reported and 3 novel USH2A mutations were identified in four Chinese probands. The probands and their family members showed typical RP features and mild to severe hearing impairment, confirming the diagnosis of Usher syndrome 2 (USH). Moreover, 11 sporadic RP patients were identified to carry the compound heterozygous mutations in the USH2A gene, confirming the diagnosis of USH2. The patients carried the truncating mutations had a younger age of first visit than the patients carried only the missense mutations (p = 0.017). In summary, this study revealed 8 novel USH2A variants in Chinese familial and sporadic RP patients, assuring that whole exome sequencing analysis is an adequate strategy to facilitate the clinical diagnosis of USH from the sporadic RP patients.
Similar content being viewed by others

Introduction
Usher syndrome (USH) is an autosomal recessive disorder characterized by retinitis pigmentosa (RP) and bilateral sensorineural hearing loss, with vestibular dysfunction in certain patients1. USH is a leading cause of hereditary deaf-blindness with an estimated prevalence ranging from 3.2 to 6.2 in 100,000 people worldwide2,3,4. On the basis of the severity, the progression of hearing impairment and RP as well as the presence of vestibular dysfunction, USH can be divided into three clinical subtypes5. USH1, the most severe subtype of USH, is featured by the congenital hearing loss and vestibular dysfunction as well as the pubertal onset of progressive RP6. USH2, the most common subtype of USH accounting for more than 50% of all USH patients, is classified by the moderate to severe deafness and the pubertal onset of progressive RP, but with normal vestibular functions. USH3 patients exhibits progressive hearing loss, but has variable onset of RP and vestibular dysfunction7. Despite no cure for USH, cochlear implants can help the USH2 patients for better hearing function8. Yet, there is still no effective treatment to restore the retinal function in USH patients although gene therapy has been proposed9.
USH is clinically and genetically heterogeneous with 16 identified USH-associated genes10. Three genes are responsible for USH2: USH2A (usherin)11, ADGRV1 (adhesion G protein-coupled receptor V1)12 and WHRN (whirlin)13. The USH2A gene is the major disease-causing gene for USH2; yet, it can also be relevant to non-syndromic RP or atypical USH11,14. The USH2A gene, located on chromosome 1q41, is coded for usherin, a transmembrane protein mainly produced in the photoreceptor layer of the retina, the hair cells in cochlea and the basement membranes of many tissues15. Different mutations in USH2A have been connected to non-syndromic autosomal recessive RP, apart from the typical USH216,17. Precise diagnosis can be difficult, especially with atypical fundus appearance18. Disease gene identification can greatly assist the clinical diagnosis of individual RP patient for better clinical management, treatment and counseling.
In this study, we applied the whole exome sequencing (WES) analysis to hunt for the disease-causing mutations in four unrelated Chinese families and 118 sporadic patients with initial diagnosis of RP. In addition, the clinical correlation of the identified mutations was also analyzed.
Results
Whole exome sequencing analysis in familial RP patients
Four unrelated Chinese families initially diagnosed with RP were recruited, following the recessive inheritance (Fig. 1). The proband of each recruited family was subjected to WES analysis. Each WES analysis resulted in a total of 70GB of sequence data, and 95.5% of sequence reads were originated from the exons, with a mean coverage of 100-fold. The total numbers of variants (SNPs and indels) of exons and splice sites identified were 23,774 for F1-II-6, 24,006 for F2-II-11, 23,499 for F3-II-6 and 23,911 for F4-II-15 (Supplementary Fig. 1). After filtering the synonymous, intergenic, intronic and common variants, the candidate variants of known RP genes were reduced to 2 for F1-II-6, 2 for F2-II-11, 5 for F3-II-6 and 3 for F4-II-15 (Table 1).

Pedigrees of the four recruited Chinese retinitis pigmentosa families. All the 4 recruited families initially diagnosed with RP followed the pattern of autosomal recessive inheritance. Squares and circles represent men and women respectively. The asterisk represents the participants’ blood were collected and the arrow indicates proband. The slash represents deceased person. Black: the affected members; White: the unaffected members.
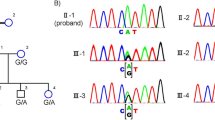
For the family F1, two heterozygous variants in USH2A gene were identified in all three patients (F1-II-2, F1-II-4 and the proband: F1-II-6): a nonsense variant c.187 C > T (p.R63*) in exon 2 and an insertion frameshift variant c.12086dupA (p.H4029Qfs*68) in exon 62. The identified variants has been confirmed by Sanger sequencing in these patients (Fig. 2). Their unaffected brother (F1-II-7) only carried the nonsense variants (c.187 C > T; p.R63*), whereas the three unaffected third-generation male subjects (F1-III-5, F1-III-8 and F1-III-9) only carried the insertion frameshift variant c.12086dupA (p.H4029Qfs*68), which was transmitted from the affected patients (Fig. 1 and Table 1). The c.187 C > T (p.R63*) variant has been previously reported19,20. The c.12086dupA (p.H4029Qfs*68) USH2A variant was neither found in 1000 genome, gnomAD and dbSNP nor previously reported, suggesting that it is a novel mutation. Furthermore, the c.187 C > T (p.R63*) and c.12086dupA (p.H4029Qfs*68) variants were also not found in 200 control subjects from our cohort. The c.187 C > T (p.R63*) and c.12086dupA (p.H4029Qfs*68) variants should be the causative mutations for the USH2 family F1.

Sanger sequencing confirmation of the identified USH2A mutations from the whole exome sequencing analysis in the recruited Chinese retinitis pigmentosa families. Sanger sequencing validation of the USH2A mutations (c.187 C > T and c.12086dupA) identified by the WES analysis in USH family F1.
For the family F2, two heterozygous variants in USH2A gene were identified in the two patients (F2-II-10 and the proband: F2-II-11), a non-synonymous variant c.5581 G > A (p.G1861S) in exon 28 and a splice site variant c.9570 + 1 G > A in intron 48 (Table 1). These two variants have been previously reported20,21. The c.5581 G > A (p.G1861S) and c.9570 + 1 G > A variants should be the causative mutations for the USH2 family F2.
For the family F3, two heterozygous variants in USH2A gene were identified in the proband (F3-II-6), a nonsense variant c.5079 G > A (p.W1693*) in exon 25 and a non-synonymous variant c.5168 G > A (p.G1723E) in exon 26 (Table 1), both of which are novel variants. The unaffected members (F3-II-4 and F3-II-8) only carried the nonsense variant c.5079 G > A (p.W1693*). In addition, non-synonymous variants in PRPH2 (c.367 C > T; p.R123W), EYS (c.2980 C > G; p.P994A) and KIAA1549 (c.5653 C > T; p.R1885W) genes were also found in the proband (F3-II-6); yet, these variants did not co-segregate with the phenotype in the pedigree. The c.5079 G > A (p.W1693*) and c.5168 G > A (p.G1723E) variants should be the causative mutations for the USH2 family F3.
For the family F4, two heterozygous variants in USH2A gene were identified in the proband (F4-II-15), a non-synonymous variant c.2802 T > G (p.C934W) in exon 13 and a splice site variant c.5858-1 G > A in intron 30 (Tables 1 and 2). Both variants have been previously reported16,17. The unaffected children (F4-III-34 and F4-III-35) only carried the splice site variant c.5858-1 G > A. Besides, non-synonymous variant in CNGB1 gene (c.367 C > T; p.R123W) was also identified in the proband (F4-II-15) and his unaffected son (F4-III-35), but the CNGB1 variant did not co-segregate with the phenotype in the pedigree. The c.2802 T > G (p.C934W) and c.5858-1 G > A variants should be the causative mutations for the USH2 family F4.
Clinical characteristics of the USH2 families
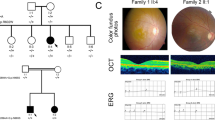
All the 4 recruited families initially diagnosed with RP followed the pattern of autosomal recessive inheritance (Fig. 1). For the four-generation family F1, the three affected patients (F1-II-2, F1-II-4 and the proband: F1-II-6) presented with the onset of night blindness at teenage and typical RP features. The fundus photographs showed the attenuation of retinal arteries, the bone-spicule pigmentation of the retina and the waxy optic disc (Fig. 3A). Patchy hypo-autofluorescence was observed in the mid-peripheral retina and hyper-autofluorescence in the macula by auto-fluorescence imaging (Fig. 3B). Moreover, optical coherence tomography (OCT) imaging displayed significant reduction in retinal thickness and extensive disruption in ellipsoid zone (Fig. 3C). The representative flattening in the rod and cone ERG response was detected (Fig. 3D). The pure-tone audiometry (PTA) results showed bilateral down-sloping severe sensorineural hearing loss (Fig. 3E), indicating the diagnosis of USH2. From the medical history, the affected members did not exhibit the delayed gait development, poor speech acquisition and balance disturbance or unstable walking. Their unaffected brother (F1-II-7) showed normal gross motor development, speech ability and eyesight; yet, he has bilateral mild sensorineural hearing loss and mild reduction in cone and rod response (Table 2). The remaining unaffected subjects (F1-III-5, F1-III-8, F1-III-9, F1-III-11) did not show any symptoms or abnormalities in the clinical examinations.

Clinical characteristics of the recruited Chinese retinitis pigmentosa family 1. (A) The fundus photographs of the affected and unaffected members of family F1 showed the attenuation of retinal arteries (arrow head), the bone-spicule pigmentation of the retina (arrows) and the waxy optic disc (double arrows). (B) Patchy hypo-autofluorescence (arrows) was observed in the mid-peripheral retina and hyper-autofluorescence in the macula (arrow head) by auto-fluorescence imaging. (C) OCT imaging displayed significant reduction in retinal thickness (arrow) and extensive disruption in ellipsoid zone (arrow head). (D) The representative flattening in the rod and cone ERG response was detected. (E) The PTA results showed bilateral down-sloping severe sensorineural hearing loss.
The three probands (F2-II-11, F3-II-6 and F4-II-15) from the other three recruited families showed progressive night blindness and mild to severe hearing impairment, which indicated the diagnosis of USH2. The fundus abnormalities, the flattening in the rod and cone responses ERG and the reduced photoreceptor layer thickness in OCT examination supported the typical RP features in these patients.
USH2A mutation analysis in sporadic RP patients
To further extend the USH2A mutation analysis, the USH2A gene variants was searched from 118 sporadic RP patients with the WES analysis. Total of 25 candidate variants were identified in 23 sporadic RP patients. Among these 23 sporadic RP patients, 13 of them carried only 1 USH2A variant, and 10 patients carried two or three variants in the USH2A gene. For these 10 sporadic RP patients, they showed progressive night blindness and mild to severe hearing impairment, indicating the diagnosis of USH2. For the 16 USH2A variants identified in the 10 sporadic RP (USH2) patients, 12 of them have been reported and 4 were novel (Table 3). Combining the heterozygous carrier and the compound heterozygous patients, 8 novel variants were discovered from 23 sporadic RP patients in this study. The novel variants have been verified by Sanger sequencing and were not found in our 200 control subjects.
Bioinformatics and clinical correlation of USH2A mutations
In this study, we identified 15 missense mutations from 14 USH2 and sporadic RP patients: c.2802 T > G (p.C934W), c.A4481A > T (p.N1494I), c.4576 G > A (p.G1526R), c.5168 G > A (p.G1723E), c.5398 T > A (p.W1800R), c.5581 G > A (p.G1861S), c.5953 G > A (p.E1985K), c.6998 T > C (p.V2333A), c.7100 G > A (p.G2367E), c.11233 T > C (p.Y3745H), c.11261 G > T (p.G3754V), c.13330 C > A (p.P4444T), c.13979 C > T (p.P4660L), c.14017 T > C (p.Y4673H) and c.15178 T > C (p.S5060P). All of the amino acids are conserved in USH2A protein across 8 different species (Fig. 4), except G3754 (partially conserved), indicating that the missense mutations could affect the USH2A protein function or structure.

Multiple sequence alignment of USH2A protein across different species. USH2A protein sequences of Homo sapiens, Pan troglodytes, Bos taurus, Rattus norvegicus, Mus musculus, Pteropus alecto, Numida meleagris, Xenopus laevis and Danio rerio were aligned by Clustal Omega. The conservative of the 15 missense mutations identified in USH and sporadic RP patients were displayed.
Compare to the missense mutations, the nonsense (c.187 C > T, p.R63*; c.2187 C > A, p.C729* and c.5079 G > A, p.W1693*), indel (c.12086dupA, p.H4029Qfs*68) and the splice site mutations (c.5858-1 G > A, c.8559-2 A > G and c.9570 + 1 G > A) would lead to drastic changes to the USH2A protein structure by creating premature stop codon (p.R63*: USH2A protein with 62 amino acids only, p.C729*: USH2A protein with 728 amino acids only, and p.W1693*: USH2A protein with 1692 amino acids only) or translational frameshifting (c.12086dupA: USH2A protein prematurely with 4097 amino acids, c.5858-1 G > A: affected mRNA processing for exon 30, c.8559-2 A > G: affected mRNA processing for exon 43 and c.9570 + 1 G > A: affected mRNA processing for exon 48). With references to the bioinformatics analyses, USH2A dysfunction is expected from a truncated protein or nonsense-mediated decay.
Based on the changes in protein properties by the mutations, we divided the 14 USH2 and sporadic RP patients into the missense mutation only group and the truncating mutation group. The patients carrying 1 or 2 nonsense, indel or splice site mutations in the truncating mutation group have a younger age of first visit (39.75 ± 16.34 years) than the patients only carrying the missense mutations (59.17 ± 10.76 years, p = 0.017; Fig. 5), implying that the nonsense, indel and splice site mutations could lead to earlier onset of disease or more severe disease progression compared to the missense mutations.

Age of first visit of USH2/RP patients with different properties of USH2A mutations. The 15 USH2/RP patients were divided into the missense mutation only group and the truncating mutation group (nonsense, indel or splice site mutations) based on the properties of USH2A mutations. The patients carried 1 or 2 nonsense, indel or splice site mutations in the protein truncational group have a younger age of first visit (39.75 ± 16.34 years) than the patients carried only the missense mutations (59.17 ± 10.76 years. *p < 0.05.
Discussion
USH is usually discovered first by RP in the initial screening, and ophthalmic examination alone might not easily discover other invisible dysfunctions. Genetic diagnosis, in this scenario, can facilitate the precise clinical diagnosis of complicated diseases for better management and counseling. In this study, we identified 10 sporadic RP patients, out of 118 recruited subjects, carrying at least 2 compound heterozygous USH2A mutations (Table 3), confirming the diagnosis of USH2. This indicates that 8.47% of sporadic RP patients could belong to USH in Southern Chinese population.
Previous studies in Chinese populations have reported the identification of USH2A and ADGRV1 mutations for USH2 in Chinese populations20,22,23,24. The USH2A gene, located on chromosome 1q41, is consisted of 72 exons, whereas the ADGRV1 gene, located on chromosome 5q14.3, is consisted of 90 exons. It is not efficient and time-consuming to screen the disease-causing mutations in these genes by traditional Sanger sequencing. Target exome sequencing has been applied to map the disease-causing gene for USH20,22,23,24. Yet, the WES analysis, based on the general enrichment and capturing platform, can efficiently delineate the variants in all genes with a reasonable price and processing time. It is a favorable sequencing analysis for the disease-causing mutation screening in the inherited retinal dystrophies20,25,26.
USH is an autosomal recessive disorder characterized by retinitis pigmentosa (RP) and bilateral sensorineural hearing loss. In family F1, the USH2A mutation carrier (F1-II-7; c.187 C > T p.R63*) had best corrected visual acuity of 1.0, showed normal gross motor development as well as speech ability and did not complained of nyctalopia; yet, he has bilateral mild sensorineural hearing loss and slightly reduction in rod and cone ERG responses (Fig. 3 and Table 2). The bilateral mild sensorineural hearing loss on PTA could probably be due to aging. However, whether the single nonsense mutation would lead to ERG response reduction or a milder symptoms of USH requires further investigations. Yet, we identified 21 sporadic RP patients carrying single USH2A variant, and whether carrying single USH2A variant contributes to the RP development is also unknown.
Usherin, coded by the USH2A gene, is a basement membrane protein with 5202 amino acids. The first 31 amino acids are responsible for the signal peptide, and the rest of the protein is composed of the N-terminal laminin domain, the laminin EGF-like domains, the laminin G-like domains, the fibronectin type-III domains, a transmembrane domain and a cytoplasmic tail. In this study, we identified 30 mutations in the USH2A gene, of which p.C934W and c.9570 + 1 G > A in 5 patients (18.52%) and p.S5060P in 4 patients (14.81%), indicating that these mutations have higher frequencies in Chinese populations and a potential of founder effect. In the 14 USH2 and sporadic RP patients, we identified 15 missense mutations from by the WES analysis (Tables 1 and 3). One mutation (p.C934W) is located in the laminin EGF-like domain, 3 mutations (p.G1526R, p.G1723E and p.W1800R) in the laminin G-like domains and 7 mutations (p.E1985K, p.V2333A, p.Y3745H, p.G3754V, p.P4444T, p.P4660L and p.Y4673H) in the fibronectin type-III domains and 3 mutations (p.N1494I, p.G1861S and p.S5060P) in the linker regions, indicating that they could induce structural or functional changes to the usherin protein. Moreover, the p.C934W mutation also belong to the site for disulphide bond formation. The amino acids of these 14 missense mutations, except p.G3754V (partially conserved), are highly conserved across 8 different species (Fig. 4), reassuring the importance of these amino acid residues for usherin protein function. Compared to the missense mutations, the nonsense (p.R63*, p.C729* and p.W1693*), indel (c.12086dupA) and the splice site mutations (c.5858-1 G > A, c.8559-2 A > G and c.9570 + 1 G > A) would lead to a truncated USH2A protein by the premature stop codon (p.R63*: USH2A protein with 62 amino acids only, p.C729*: USH2A protein with 728 amino acids only, and p.W1693*: USH2A protein with 1692 amino acids only), translational frameshifting (c.12086dupA: USH2A protein with 4097 amino acids, c.5858-1 G > A: affected mRNA processing for exon 30, c.8559-2 A > G: affected mRNA processing for exon 43 and c.9570 + 1 G > A: affected mRNA processing for exon 48) or reduced USH2A expression by nonsense-mediated decay. Accordingly, the patients carried the nonsense, indel or splice site mutations have a significant younger age of first visit (39.75 ± 16.34 years) than the patients carried only the missense mutations (59.17 ± 10.76 years; Fig. 5), implying that the nonsense, indel and splice site mutations could lead to earlier onset of disease or more severe disease progression compared to the missense mutations. Similarly, it was reported that USH2 patients carrying the p.Glu767Serfs*21 USH2A mutation can have earlier diagnosis of disease, onset of night blindness, onset of visual field loss, diagnosis of cataract and onset of hearing loss than those carrying the p.Cys759Phe mutation27. Moreover, patients with two truncating USH2A mutations develop significantly more severe hearing impairment throughout life than patients with two non-truncating mutations28. Yet, how these mutations cause visual defects and hearing impairment requires further functional and mechanism investigations.
In summary, this study revealed 11 novel variants in USH2A gene from four unrelated Chinese USH2 families and 23 sporadic RP patients, expanding the mutation spectrums of USH2A gene. Ten sporadic RP patients were diagnosed as USH2 after WES. Genetic diagnosis is an adequate strategy to aid the clinical diagnosis for better clinical management and counseling. It should be recommended as a routine examination for inherited retinal dystrophies.
Materials and Methods
Study subjects and clinical examinations
Four unrelated Chinese families initially diagnosed with RP were recruited from the Joint Shantou International Eye Center of Shantou University and the Chinese University of Hong Kong, including seven patients and nine unaffected family members (Fig. 1). The audiological criteria to consider a USH2 is mild to moderate sensorineural hearing loss29. Comprehensive ophthalmic examinations, including best-corrected visual acuity (BCVA), fundus photography, visual field tests, OCT, electroretinogram (ERG) and auto-fluorescence, were performed for all patients and some of the unaffected family members. The severity of hearing loss was evaluated by PTA and classified as mild (25–40 dB), moderate (41–70 dB), severe (71–90 dB), profound (>90 dB). Vestibular function was recorded from the detailed medical history, including motor development, speech acquisition, stability and balance.
For the validation analysis, 118 sporadic RP patients and 200 unrelated control subjects were also enrolled. The sporadic RP patients generally showed pubertal night blindness, restricted peripheral vision, progressive vision loss, overall bone-spicule pigmentation of the retina, attenuation of retinal vessels, and the flattening of the rod and cone ERG responses. The control subjects, aged 60 years or above, did not have any family history or symptom of inherited retinal diseases, hearing loss, or any other major eye diseases except mild senile cataracts and low myopia.
Peripheral blood was collected from the study subjects, and the genomic DNA was extracted by TIANGEN DNA blood Kit DP318 (TIANGEN, Beijing, China), and stored at −80 °C freezer before sequencing analysis.
This study protocol was approved by the Ethics Committee on Human Research at the Joint Shantou International Eye Center of Shantou University and the Chinese University of Hong Kong, which is in accordance with the tenets of the Declaration of Helsinki. Written informed consents were obtained from all study subjects or their guardians after explanation of the nature and possible consequences of the study.
Whole exome sequencing analysis
WES analysis was conducted for each proband from the recruited families and 118 sporadic RP patients. Briefly, genomic DNA was fragmented into 150–200 bp DNA libraries by S2/E210 Focused-ultrasonicator (Covaris, Woburn, MA). All coding exons were captured by and hybridized to a customized array (Agilent Technologies, Santa Clara, CA). The purified and enriched DNA libraries were sequenced using the HiSeq2500 platform (Illumina, San Diego, CA) to generate paired-end reads for 90 cycles per reads. The results were aligned to human genome reference (UCSC hg19) in the National Center for Biotechnology Information (NCBI) database using Burrows Wheeler Aligner Multi-Vision software package (BWA) for unique mapped reads. The single nucleotide polymorphisms (SNPs) and insertion/deletion variants (indels) were identified using the SAMtools and BCFtools. The detected variants were annotated with ANNOVAR with reference to the following databases for further variant filtering, including 1000 Genome Project (http://www.internationalgenome.org/), gnomAD (http://gnomad-old.broadinstitute.org/), EXAC (http://exac.broadinstitute.org/), and dbSNP (http://www3.ncbi.nlm.nih.gov/SNP/).
To ensure the accuracy and efficacy of the candidate mutations, the synonymous, intergenic and intronic variants were first filtered out. Those variants with minor allele frequency (MAF) lower than 1% or absent in East Asian population from gnomAD and EXAC were reserved. The candidate variants were compared to the reported RP causative genes in RentNet (http://sph.uth.edu/retnet/). The retained variants were predicted by the online bioinformatics software, including SIFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), mutationtaster (http://www.mutationtaster.org/), CADD (http://cadd.gs.washington.edu/), ExPASy (https://web.expasy.org/translate/) and Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/).
Sanger sequencing validation
The potential causative gene variants identified by WES in the Chinese RP families were validated by polymerase chain reaction (PCR) and Sanger sequencing with specific primers (AIJI Biotechnology Company, Guangdong, China; Supplementary Table 1) in the ABI 3730XL Genetic Analyzer (Applied Biosystems, USA) to confirm the co-segregation pattern. The sequencing results were analyzed by NOVOSNP (http://www.molgen.vib-ua.be/bioinfo/novosnp/) with the alignment to the reference DNA sequence. In addition, the identified variants were also screened in 200 control subjects.
Statistical analysis
Independent T-test was used to compare the means between the study groups with different properties of USH2A mutations. All statistical analyses were performed by the commercially available software (IBM SPSS Statistics 22; SPSS Inc., Chicago, IL). Significance was defined as p < 0.05.
References
Grøndahl, J. Estimation of prognosis and prevalence of retinitis pigmentosa and Usher syndrome in Norway. Clin Genet 31, 255–264 (1987).
Boughman, J. A., Vernon, M. & Shaver, K. A. Usher syndrome: definition and estimate of prevalence from two high-risk populations. J Chronic Dis 36, 595–603 (1983).
Rosenberg, T., Haim, M., Hauch, A. M. & Parving, A. The prevalence of Usher syndrome and other retinal dystrophy-hearing impairment associations. Clin Genet 51, 314–321 (1997).
Spandau, U. H. & Rohrschneider, K. Prevalence and geographical distribution of Usher syndrome in Germany. Graefes Arch Clin Exp Ophthalmol 240, 495–498 (2002).
Marazita, M. L. et al. Genetic epidemiological studies of early-onset deafness in the U.S.school-age population. Am J Med Genet 46, 486–491 (1993).
Tsilou, E. T. et al. Usher syndrome clinical types I and II: Could ocular symptoms and signs differentiate between the two types? Acta Ophthalmol Scan 80, 196–201 (2002).
Pakarinen, L., Karjalainen, S., Simola, K. O., Laippala, P. & Kaitalo, H. Usher’s syndrome type 3 in Finland. Laryngoscope 105, 613–617 (1995).
Jatana, K. R. et al. Usher syndrome: characteristics and outcomes of pediatric cochlear implant recipients. Otol Neurotol 34, 484–489 (2013).
Hashimoto, T. et al. Lentiviral gene replacement therapy of retinas in a mouse model for Usher syndrome type 1B. Gene Ther 14, 584–594 (2007).
Keats, B. J. & Savas, S. Genetic heterogeneity in Usher syndrome. Am J Med Genet 130, 13–16 (2004).
Eudy, J. D. et al. Mutation of a Gene Encoding a Protein with Extracellular Matrix Motifs in Usher Syndrome Type IIa. Science 280, 1753–1757 (1998).
Weston, M. D., Luijendijk, M. W., Humphrey, K. D., Möller, C. & Kimberling, W. J. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet 74, 357–366 (2004).
Ebermann, I. et al. A novel gene for Usher syndrome type 2: mutations in the long isoform of whirlin are associated with retinitis pigmentosa and sensorineural hearing loss. Hum Genet 121, 203–211 (2007).
Rivolta, C., Sweklo, E. A., Berson, E. L. & Dryja, T. P. Missense mutation in the USH2A gene: association with recessive retinitis pigmentosa without hearing loss. Am J Hum Genet 66, 1975–1978 (2000).
Liu, X. et al. Usherin is required for maintenance of retinal photoreceptors and normal development of cochlear hair cells. Proc Natl Acad Sci USA 104, 4413–4418 (2007).
Xu, W. et al. Seven novel mutations in the long isoform of the USH2A gene in Chinese families with nonsyndromic retinitis pigmentosa and Usher syndrome Type II. Mol Vis 17, 1537–1552 (2011).
Lenassi, E. et al. A detailed clinical and molecular survey of subjects with nonsyndromic USH2A retinopathy reveals an allelic hierarchy of disease-causing variants. Eur J Hum Genet 23, 1318–1327 (2015).
Verbakel, S. K. et al. Non-syndromic retinitis pigmentosa. Prog Retin Eye Res 66, 157–186 (2018).
Dreyer, B. et al. Spectrum of USH2A mutations in Scandinavian patients with Usher syndrome type II. Hum Mutat 29, 451 (2008).
Huang, X. F. et al. Targeted exome sequencing identified novel USH2A mutations in Usher syndrome families. PLoS One 8, e63832 (2013).
Yang, T., Wei, X., Chai, Y., Li, L. & Wu, H. Genetic etiology study of the non-syndromic deafness in Chinese Hans by targeted next-generation sequencing. Orphanet J Rare Dis 8, 85 (2013).
Chen, X. et al. Targeted next-generation sequencing reveals novel USH2A mutations associated with diverse disease phenotypes: implications for clinical and molecular diagnosis. PLoS One 9, e105439 (2014).
Wang, X. et al. Genetic analysis of a Chinese family with members affected with Usher syndrome type II and Waardenburg syndrome type IV. Int J Pediatr Otorhinolaryngol 102, 114–118 (2017).
Sun, T. et al. Comprehensive Molecular Screening in Chinese Usher Syndrome Patients. Invest Ophthalmol Vis Sci 59, 1229–1237 (2018).
Timal, S. et al. Gene identification in the congenital disorders of glycosylation type I by whole-exome sequencing. Hum Mol Genet 21, 4151–4161 (2012).
Corton, M., Nishiguchi, K. M. & Avila-Fernández, A. Exome Sequencing of Index Patients with Retinal Dystrophies as a Tool for Molecular Diagnosis. PLoS One 11, e0153121 (2016).
Blanco-Kelly, F. et al. Clinical aspects of Usher syndrome and the USH2A gene in a cohort of 433 patients. JAMA Ophthalmol 133, 157–164 (2015).
Hartel, B. P. et al. A combination of two truncating mutations in USH2A causes more severe and progressive hearing impairment in Usher syndrome type IIa. Hear Res 339, 60–68 (2016).
Bernal, S. et al. Clinical and genetic studies in Spanish patients with Usher syndrome type II: description of new mutations and evidence for a lack of genotype–phenotype correlation. Clin Genet 68, 204–214 (2005).
Acknowledgements
We are grateful to the participants in this study. This work was supported by the National Nature Science Foundation of China (30901646 and 81170853), YangFan Program, and TeZhi Program of Guangdong Province, China.
Author information
Authors and Affiliations
Contributions
H.C. conception and design. X.X. and H.C. financial support. H.C. provision of study materials or patients. W.T., Y.C., S.C. and Y.Z. collection and/or assembly of data. W.T., Y.C., T.K.N. and H.C. data analysis and interpretation. W.T. and T.K.N. manuscript writing. T.K.N. and H.C. final approval of manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ng, T.K., Tang, W., Cao, Y. et al. Whole exome sequencing identifies novel USH2A mutations and confirms Usher syndrome 2 diagnosis in Chinese retinitis pigmentosa patients. Sci Rep 9, 5628 (2019). https://doi.org/10.1038/s41598-019-42105-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-42105-0
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
