
Abstract
A fundamental understanding of the phonon transport mechanism is important for optimizing the efficiency of thermoelectric devices. In this study, we investigate the thermal transport properties of the oxidized form of phosphorene called phosphorene oxide (PO) by solving phonon Boltzmann transport equation based on first-principles density functional theory. We reveal that PO exhibits a much lower thermal conductivity (2.42–7.08 W/mK at 300 K) than its pristine counterpart as well as other two-dimensional materials. To comprehend the physical origin of such low thermal conductivity, we scrutinize the contribution of each phonon branch to the thermal conductivity by evaluating various mode-dependent quantities including Grüneisen parameters, anharmonic three-phonon scattering rate, and phase space of three-phonon scattering processes. Our results show that its flexible puckered structure of PO leads to smaller sound velocities; its broken-mirror symmetry allows more ZA phonon scattering; and the relatively-free vibration of dangling oxygen atoms in PO gives rise to additional scattering resulting in further reduction in the phonon lifetime. These results can be verified by the fact that PO has larger phase space for three-phonon processes than phosphorene. Furthermore we show that the thermal conductivity of PO can be optimized by controlling its size or its phonon mean free path, indicating that PO can be a promising candidate for low-dimensional thermoelectric devices.
Similar content being viewed by others
Introduction
The thermoelectric effect is one of the promising next-generation energy harvesting technologies and has been studied extensively in recent decades1,2. The efficiency of a thermoelectric device can be expressed using a dimensionless quantity, ZT defined as
where S, σ, T, λ, and κ represent the Seebeck coefficient, electrical conductivity, absolute temperature, electron and lattice thermal conductivities, respectively. In order to increase ZT value, we need to increase the power factor S2σ, and at the same time to decrease thermal conductivities. Seebeck coefficient S is usually much higher in semiconductor than in metal, since S is strongly related to differential total electron density of state (DOS), which is maximized near band gaps, where DOS changes abruptly. On the other hand, it is, unfortunately, impossible to adjust σ high, and λ low simultaneously to enhance the thermoelectricity, because they are coupled to each other and governed by the Wiedemann-Franz law. In semiconductors, the phononic contribution of the thermal conductivity (κ) is much larger than its electronic contribution (λ). Moreover, κ is partially decoupled from σ, and thus its reduction is a useful strategy for high-performance thermoelectric materials. Recently, a few studies proposed that introduction of particular defect structures increases σ/(λ + κ) since the thermal conductivity decreases much more than its electrical counterpart, although the defects usually reduce both3,4. Another interesting research showed that making low-dimensional structures enhances thermoelectric power. In the low-dimensional materials, S can be further increased due to its unique properties, such as sudden changes in the DOS5, and quantum confinement effects6,7. Moreover, such structural redesign may affect thermal conductivity. κ can be lowered more while keeping σ by forming superlattice or nanocrystal structures8,9, because the mean free path of phonon is longer than that of electron. To realize state-of-the-art thermoelectric devices, therefore, one should design a quasi low-dimensional or superlattice structure with a semiconductor, which has a low lattice thermal conductivity. It is, for example, known that the best thermoelectric materials are Bi2 Sb3/Sb2 Te3 superlattice10 and layered SnSe structure11, which exhibit the highest ZT values.
Phosphorene, which has recently attracted attentions in various fields related to materials research12,13, would fit in a category satisfying conditions mentioned above. Due to its puckered structure representing strong anisotropy, its mechanical, electrical and thermal properties shows remarkably different directional behavior, especially along the armchair and zigzag directions14,15,16,17,18,19. There have been several reports showing that phosphorene has a high ZT value along the armchair direction20,21.
However, the poor stability of phosphorene under ambient air conditions has been a crucial problem for its various applications including thermoelectric devices. Earlier studies reported that phosphorene can be easily oxidized in air because of the lone-pair electrons in each phosphorus atom22,23,24,25. Although there were a few proposals to prevent phosphorene from being oxidized26, they are not easily applicable, and thus it is still difficult to use phosphorene as a thermoelectric device. Another way to overcome the poor air stability of phosphorene is to use the oxidized structure itself as a device. It was reported that a particular structure of phosphorene oxide (PO) is a semiconductor with a direct band gap of 0.6~0.88 eV23,24,25, which is slightly smaller than that of its pristine counterpart, phosphorene. It was also reported that PO has a relatively small electron effective mass of 0.18 me24, which is similar to that of phosphorene along the armchair direction and much smaller than along the zigzag direction. In addition, phosphorene oxide exhibits some interesting physical properties such as non-symmorphic phase protected band structure25.
Moreover, PO satisfies the conditions mentioned above for good thermoelectric materials. In this paper, we report our investigation on the thermal transport properties of the oxidized structure of phosphorene by solving the Boltzmann transport equation based on first-principles density functional theory. PO shows significantly lower lattice thermal conductivity than phosphorene. Furthermore, we present our analysis of why such low lattice thermal conductivity was observed in PO. Our analysis includes the evaluation of the phonon relaxation time, Grüneisen parameters, and phase space of the three-phonon processes, all of which enable us to understand the fundamental origin of the low thermal conductivity of PO.
Results and Discussion
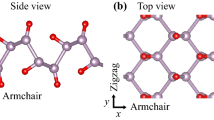
In an earlier study25, we found the equilibrium structure of PO, as shown in Fig. 1. Similar to phosphorene, PO has a puckered configuration, composed of four phosphorus and four oxygen atoms in a primitive unit cell. Every P atom is connected to three other P atoms and one O atom, whereas every O atom to one P atom and has three lone pairs of electrons. The optimized lattice constants along the armchair and zigzag directions were calculated to be 5.12 and 3.66 Å, respectively, in good agreement with previous studies of PO23,24. We expanded this equilibrium structure to 4 × 4 × 1 super cell to investigate the thermal properties.

Equilibrium structure of PO shown in side (a) and top (b) views. Phosphorus and oxygen atoms are depicted by blue and red spheres.
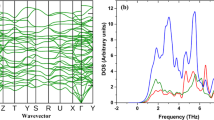
Figure 2 shows our calculated phonon dispersion of PO plotted along the special lines shown in its inset in (a). We focused more on three acoustic branches (LA: longitudinal, TA: transverse, ZA: flexural modes), which are main contribution to the thermal conductivity. As shown in Fig. 2(b), which is zoomed in below ω ≤ 200 from (a), the acoustic modes exhibit anisotropic dispersion along the Γ-Y (zigzag) and Γ-X (armchair) directions. To clearly describe such an anisotropic behaviors in the dispersion, we computed the group velocity vg of the three acoustic branches along these two directions. As shown in Fig. 3(a,b), the sound velocities of LA and TA modes are in the range of 2.9–5.2 km/s, which are slightly smaller than those of other 2D materials such as silicene (5.4–8.8 km/s)27, MoS2 (4.2–6.8 km/s)28, graphene (3.7–6.0 km/s)29, and phosphorene (4.0–7.8 km/s)16. We found that vg of the LA phonon mode along the zigzag direction is larger than that along the armchair direction, as we easily understand since the LA mode depends strongly on the elastic modulus of the material. Due to the puckered structure shown in Fig. 1(a), PO can be more easily stretched or compressed along the armchair direction compared to along their zigzag counterpart, similar to phosphorene. On the other hand, the group velocities of the TA mode do not show strong directional dependence, and so do those of the ZA mode exhibiting the quadratic behaviors as in planar graphene. Therefore, the LA mode should be responsible for the anisotropicity in thermal transport property of PO.

Phonon dispersion of PO in (a) the whole frequency range and in (b) the low frequency region (ω ≤ 200 cm−1). The three acoustic branches (LA: longitudinal, TA: transverse, ZA: flexural modes) are represented respectively by sky blue, green, and purple colors. The BZ of PO and the special path and points are shown in the inset in (a).

Absolute values of phonon group velocities of PO for the three acoustic branches along (a) the Γ-Y and (b) the Γ-X directions. (c) Grüneisen parameters calculated for the three acoustic modes in the first BZ. These calculated values for three modes LA, TA, and ZA are depicted by sky blue, green and purple colors, respectively. To compensate for the imaginary frequencies found in the ZA mode, which should exhibit the quadratic behavior near Γ, its group velocity along the Γ-Y direction was fitted using a polynomial function.
In a carrier transport phenomenon in a crystal solid, not only the group velocity but the carrier lifetime or scattering rate also play a crucial role in determining its transport properties. The evaluation of the carrier lifetime requires anharmonic phonon-phonon scattering or Umklapp process evolving three phonons, since the momentum-conserved phonon scattering process described by only two phonons is not relevant to the phonon lifetime.
We noticed that the Grüneisen parameter (GP), which is usually considered for the thermal expansion behaviors, gives us a useful information on the anharmonic phonon scattering rate. The mode-dependent GP is defined as
Figure 3(c) shows the GP values evaluated for the three acoustic phonon branches. The ZA-mode GP γZA becomes very large in the long wavelength region near the Γ point as in other 2D materials30 as described by membrane effect31. It is also clear that the GPs are largely anisotropic and discontinuous at the Γ point, reflecting its structural features similar to the sound velocity. It was derived that the mode-dependent anharmonic phonon lifetime is inversely proportional to the mode-dependent GP squared or \({\tau }_{{\rm{U}}}(j,{\bf{q}})\propto \frac{1}{|{\gamma }_{j}({\bf{q}}{)|}^{2}}\)32,33,34,35. Thus LA mode of PO, which has smaller GP values than the TA or ZA, may exhibit longer phonon lifetime than the other acoustic modes. Therefore, the LA mode plays an important role in PO’s thermal transport behavior as we will later describe in detail.
To investigate the temperature dependence of the lattice thermal conductivity κPO(T) of PO, we solved phonon Boltzmann equation using the iterative approach mentioned in the Method section. We also applied the same method to evaluate the lattice thermal conductivity κP(T) of phosphorene for comparison. Figure 4 shows \({\kappa }_{{\rm{PO}}}^{{\rm{A}}}(T)\), \({\kappa }_{{\rm{PO}}}^{{\rm{Z}}}(T)\), \({\kappa }_{{\rm{P}}}^{{\rm{A}}}(T)\), and \({\kappa }_{{\rm{P}}}^{{\rm{Z}}}(T)\), where A and Z indicate the armchair and zigzag directions along which the thermal conductivities were calculated. These calculated values were almost perfectly fitted with an inverse function a/T with a single parameter a, which is expected from the three-phonon anharmonic process36. Our predicted room-temperature thermal conductivity values of phosphorene, which are 146 W/mK and 65 W/mK along the zigzag and armchair directions, respectively, agree reasonably well with previously reported values ranging from 110 to 153 W/mK and from 33 to 64 W/mK along the respective directions18,19,37. Both phosphorene and PO exhibit that their lattice thermal conductivities along their armchair directions are approximately 2.5 times smaller than those along the zigzag directions, owing to the structural anisotropy mentioned above. Although PO is structurally similar to phosphorene, we observed that the thermal conductivity values of PO are much lower than those of phosphorene over the whole temperature range regardless of the transport directions. For example, at room temperature (T = 300 K), we found \({\kappa }_{{\rm{PO}}}^{{\rm{A}}}\) to be 2.42 W/mK, which is lower than the values not only of phosphorene (\({\kappa }_{{\rm{P}}}^{{\rm{A}}}=65\) W/mK), but also of other 2D materials such as silicene (26 W/mK)38, MoS2 (23.2~34.5 W/mK)39,40,41, stanene (11.6 W/mK)42, graphene (3000~6000 W/mK)43,44, and hexagonal boron nitride (hBN) (350~600 W/mK)44,45. Low thermal conductivity is one of sufficient conditions for high-performance thermoelectric materials. In this sense, PO can be a new candidate for 2D thermoelectric materials.

Calculated lattice thermal conductivities of phosphorene (red symbols) and PO (blue symbols) along zigzag (squares) and armchair (triangles) directions as a function of temperature ranging from 200 K to 800 K. The data points in each category were well fitted with an inverse function a/T with a single parameter a, plotted with the solid or dashed line.
To explore what causes such a low lattice thermal conductivity in PO, we first estimated the contributions of various phonon modes to the κPO and summarized in Table 1. It was revealed that the ZA phonon mode has a significantly long lifetime in a completely flat 2D material because its mirror symmetry limits the phase space for phonon-phonon scattering of the ZA phonon mode46. For example, in graphene and hBN, whose room-temperature thermal conductivities have been reported to be a few thousands43,46 and a few hundreds44,45 in W/mK, their ZA modes contribute to their thermal conductivities by about 75%44,46. We found, on the other hand, that the contribution of the ZA mode in PO becomes less significant to be only 15~17%, whereas the contribution from the other modes including the LA and TA modes becomes dominant. This is because PO is not a perfect 2D structure, but a puckered one, allowing more phonon-phonon scattering of the ZA mode. Thus, puckered structures would be advantageous for a low-dimensional thermoelectric application.
To further analyze the low lattice thermal conductivity of PO, we evaluated the relaxation times of phosphorene and PO as a function of frequency. In general, there are various sources giving rise to phonon scattering, for example, anharmonic phonon-phonon or Umklapp (U) scattering, phonon-electron scattering, impurity effect, boundary effect, isotope effect, and so on. Among these scattering sources, we took only the phonon-phonon scattering into account, since the other sources were much smaller in PO. Therefore, we simply replaced the total scattering rate 1/τ with the Umklapp scattering rate 1/τU. Figure 5 shows our calculated mode-dependent anharmonic phonon relaxation time of phosphorene and PO for three acoustic modes (ZA, TA, and LA) and the other higher modes. The phonon lifetime of PO is more than one-tenth smaller than that of phosphorene as shown in Fig. 5. We assigned this reduction to two effects of the oxygen atoms in PO. One is the trivial P-O composite effect, and the other is due to the flexibility of oxygen atoms. Thus, oxidation occuring spontaneously on the phosphorene surface, becomes an additional advantage leading to low thermal conductivity. Each oxygen atom, which is connected only to one phosphrous atoms, is a kind of a dangling atom, whereas each phosphorous atom has four tetrahedral bonds with neighboring atoms. Thus, oxygen atoms may participate not only into their optical P − O vibration, but also vibrate along the in-plane directions together with phosphorous atoms contributing to the acoustic modes. This contribution may be responsible for the acoustic phonon softening leading to the reduction in the thermal conductivity of PO. To verify the effect of existence of such dangling atoms on the acoustic phonon modes, we devised a model structure mimicking the PO system. See Supplementary Information for our model calculation.

The mode-dependent anharmonic phonon relaxation time of (a) phosphorene and (b) PO for three acoustic modes, ZA (purple), TA (green), LA (blue), and all the other modes (black). The relaxation time is given in a logarithmic scale.
To discuss the physical phenomena of phonon scattering in more detail, we analyzed the allowed phase space P3 for the anharmonic three-phonon scattering process introduced by Lindsay and Broido47. The three-phonon scattering is allowed only when the energy and momentum conservation conditions are satisfied. Thus, the phonon scattering rate of each phonon state is proportional to the number of available final states. In other words, the phonon lifetime of each mode ωj(q) is inversely correlated with the mode-dependent phase space P3(ωj(q)) defined by48
where ΩBZ is the volume of BZ; \({D}_{j}^{(\pm )}({\bf{q}})\) are two-phonon densities of states49 for absorption (+) and emission (−) processes. Difference of a factor two between two coefficients was introduced to avoid double counting. \({D}_{j}^{(\pm )}({\bf{q}})\) can be evaluated by
Here the momentum conservation was already imposed to replace q″ with q ± q′ − G, where G vectors are the reciprocal lattice vectors to describe Umklapp processes. Figure 6 shows P3 values as a function of frequency ωj evaluated over the Brillouin zone. It is clear that PO has a much larger P3 value than phosphorene, indicating that more scattering processes occur in PO than in phosphorene. This explains why PO has a much shorter relaxation time than phosphorene.

Allowed mode-dependent phase spaces P3(ωj(q)) of three-phonon scattering processes evaluated in (a) phosphorene and (b) PO for three acoustic modes ZA (purple), TA (green), LA (blue) and all the other modes (black). P3 values are given in a logarithmic scale.
To design high-performance thermoelectric devices, most important strategy is to reduce the thermal conductivity while remaining its electronic counterpart. Usually the lattice thermal conductivity of a system can be decreased by reducing the size of the system, since the size reduction results in shortening of its phonon MFP. On the other hand, electron MFP is much smaller than that of phonon especially in semiconductors. For example, the electron MFP of phosphorene is known to be around only a few nm at 300 K20. Therefore, optimization of grain size in nanocrystalline structures or nanowire approach can be an effective way to enhance the thermoelectric figure of merit. To explore the size dependence of thermal conductivity of PO, we evaluated the cumulative thermal conductivity κ(l) by summing up all the contributions from phonon modes with MFP smaller than l. Figure 7 shows the computed κ(l) as a function of phonon MFP along both zigzag and armchair directions at various temperatures. As shown in the figure, κ(l) curves resemble a logistic function when MFP is given in a logarithmic scale, and thus we fit our calculated data to a single parametric function given as50
where l0 is the fitting parameter determining the characteristic value of the MFP, yielding κ(l = l0) = κ/2. The l0 values are fitted to be 40.26 (39.98), 15.30 (16.46), 9.44 (10.48), and 6.71 (7.64) nm at 200, 400, 600, and 800 K, respectively, for the zigzag (armchair) direction.

Cumulative lattice thermal conductivities (solid lines) of PO along (a) zigzag and (b) armchair directions with respect to the phonon mean free path (MFP) at various temperatures. These conductivities were fitted by the function defined in the text, plotted with dotted lines.
Summary and Conclusions
We presented our investigation on the thermal transport properties of phosphorene oxide (PO) as well as phosphorene using the first-principles calculations combining with the semiclassical Boltzmann transport theory. We found that the thermal conductivity of PO is much lower than those of other two-dimensional materials including phosphorene, revealing that oxidation is responsible for the reduction in the thermal conductivity. The thermal conductivity of PO depends strongly on its transport directions, and were calculated to be 2.42 and 7.08 W/mK along the armchair and the zigzag directions at 300 K. Similar to phosphorene, PO is structurally characterized by the flexible puckered structure, which leads to lower-frequency acoustic phonon modes and smaller sound velocities than genuine 2D flat materials. In addition, PO possesses no mirror symmetry allowing more ZA phonon scattering. Furthermore, we identified that nearly-free vibration of dangling oxygen atoms gives rise to additional scattering resulting in further reduction in the phonon lifetime. Spontaneous oxidation of phosphorene greatly reduces its thermal conductivity, which can be additionally optimized by controlling the size, and thus PO can be a promising candidate for use in low-dimensional thermoelectric devices.
Methods
To investigate the thermal property of PO, we performed first-principles calculations based on density functional theory51 as implemented in the Vienna ab initio simulation package (VASP)52,53. We employed the projector augmented wave potentials54,55 to describe the valence electrons, and treated exchange-correlation functional within the generalized gradient approximation of Perdew-Burke-Ernzerhof 56. The plane-wave kinetic energy cutoff was selected to be 500 eV, and c = 20 Å was chosen for the lattice constant of the direction perpendicular to the plane to minimize the interlayer interaction. The Brillouin zone (BZ) of each structure was sampled using a 11 × 11 × 1 k-point grid for the primitive unit cells of phosphorene and PO.
To precisely evaluate the temperature dependence of the lattice thermal conductivity κPO(T) of PO, we solved the phonon Boltzmann transport equation using an iterative approach proposed by Omini et al.57, which implemented in the ShengBTE code50. The thermal conductivity tensor καβ, where α and β denote x, y or z, can be obtained from
where kB is the Boltzmann constant, Ω the unit cell volume, and N the Γ-centered q-point grids. In the summand, f0 is the Bose-Einstein distribution function; and ωj,q and vj,q are respectively the phonon frequency and group velocity of the phonon mode with the branch index j and wavevector q. Lj,q was introduced to compensate the phonon distribution deviated from f0 in the presence of the temperature gradient. This quantity with a dimension of length can be expressed as
where \({\tau }_{j,{\bf{q}}}^{0}\) is the single-mode relaxation time (SMRT) estimated from the imaginary part of self energy obtained by many body perturbation theory including the anharmonic three phonon scattering. Here, uj,q is the correction to the SMRT approach due to the deviation of the phonon population at the specific mode with j and q from the Bose-Einstein statistics. Since it is nonlinearly coupled with Lj,q, we solved Eq. (6) iteratively to evaluate uj,q and thus Lj,q. For more complete expressions of \({\tau }_{j,{\bf{q}}}^{0}\), Lj,q, and uj,q, and their evaluation approach, see the reference [ShengBTE_2014]. The phonon dispersion relations of phosphorene and PO were calculated by applying the finite displacement method58 to their respective 6 × 6 × 1 supercells. We also computed their third-order interatomic force constants and phonon relaxation times to evaluate their correpsonding lattice thermal conductivities50,59 using 4 × 4 × 1 supercell. We took up to the fourth nearest neighboring interaction and the corresponding BZ was sampled by a 2 × 2 × 1 k-point grid. Note that the cross-section area should be determined in order to evaluate the thermal conductivity. As usually done in two dimensional cases, the thicknesses of phosphorene and PO were set to 5.5 and 8.0 Å,respectively, approximately corresponding to interlayer distances in their bulk configurations.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Snyder, G. J. & Toberer, E. S. Complex thermoelectric materials. Nat. Mater. 7, 105–114, https://doi.org/10.1038/nmat2090 (2008).
Curtarolo, S. et al. The high-throughput highway to computational materials design. Nat. Mater. 12, 191–201, https://doi.org/10.1038/nmat3568 (2013).
Kim, J. Y. & Grossman, J. C. High-efficiency thermoelectrics with functionalized graphene. Nano Lett. 15, 2830–2835, https://doi.org/10.1021/nl504257q (2015).
Anno, Y., Imakita, Y., Takei, K., Akita, S. & Arie, T. Enhancement of graphene thermoelectric performance through defect engineering. 2D Mater. 4, 025019, https://doi.org/10.1088/2053-1583/aa57fc (2017).
Mahan, G. D. & Sofo, J. O. The best thermoelectric. Proc. Natl. Acad. Sci. 93, 7436–7439, https://doi.org/10.1073/pnas.93.15.7436 (1996).
Hicks, L. D. & Dresselhaus, M. S. Effect of quantum-well structures on the thermomagnetic figure of merit. Phys. Rev. B 47, 12727–12731, https://doi.org/10.1103/PhysRevB.47.12727 (1993).
Hicks, L. D. & Dresselhaus, M. S. Thermoelectric figure of merit of a one-dimensional conductor. Phys. Rev. B 47, 16631–16634, https://doi.org/10.1103/PhysRevB.47.16631 (1993).
Boukai, A. I. et al. Silicon nanowires as efficient thermoelectric materials. Nat. 451, 168–171, https://doi.org/10.1038/nature06458 (2008).
Majumdar, A. Thermoelectricity in Semiconductor Nanostructures. Sci. 303, 777–778, https://doi.org/10.1126/science.1093164 (2004).
Venkatasubramanian, R., Siivola, E., Colpitts, T. & O’Quinn, B. Thin-film thermoelectric devices with high roomtemperature figures of merit. Nat. 413, 597–602, https://doi.org/10.1038/35098012 (2001).
Zhao, L.-D. et al. Ultralow thermal conductivity and high thermoelectric figure of merit in SnSe crystals. Nat. 508, 373–377, https://doi.org/10.1038/nature13184 (2014).
Liu, H. et al. Phosphorene: An Unexplored 2D Semiconductor with a High Hole Mobility. ACS Nano 8, 4033–4041, https://doi.org/10.1021/nn501226z (2014).
Akhtar, M. et al. Recent advances in synthesis, properties, and applications of phosphorene. npj 2D Mater. Appl. 1, 5, https://doi.org/10.1038/s41699-017-0007-5 and see references therein (2017).
Cai, Y. et al. Giant Phononic Anisotropy and Unusual Anharmonicity of Phosphorene: Interlayer Coupling and Strain Engineering. Adv. Func. Mater. 25, 2230–2236, https://doi.org/10.1002/adfm.201404294 (2015).
Ong, Z.-Y., Cai, Y., Zhang, G. & Zhang, Y.-W. Strong Thermal Transport Anisotropy and Strain Modulation in Single-Layer Phosphorene. J. Phys. Chem. C 118, 25272–25277, https://doi.org/10.1021/jp5079357 (2014).
Qin, G. et al. Anisotropic intrinsic lattice thermal conductivity of phosphorene from first principles. Phys. Chem. Chem. Phys. 17, 4854–4858, https://doi.org/10.1039/C4CP04858J (2015).
Zhu, L., Zhang, G. & Li, B. Coexistence of size-dependent and size-independent thermal conductivities in phosphorene. Phys. Rev. B 90, 214302, https://doi.org/10.1103/PhysRevB.90.214302 (2014).
Jain, A. & McGaughey, A. J. H. Strongly anisotropic in-plane thermal transport in single-layer black phosphorene. Sci. Rep. 5, 8501, https://doi.org/10.1038/srep08501 (2015).
Xu, W., Zhu, L., Cai, Y., Zhang, G. & Li, B. Direction dependent thermal conductivity of monolayer phosphorene: Parameterization of Stillinger-Weber potential and molecular dynamics study. J. Appl. Phys. 117, 214308, https://doi.org/10.1063/1.4922118 (2015).
Liao, B., Zhou, J., Qiu, B., Dresselhaus, M. S. & Chen, G. Ab initio study of electron-phonon interaction in phosphorene. Phys. Rev. B 91, 235419, https://doi.org/10.1103/PhysRevB.91.235419 (2015).
Fei, R. et al. Enhanced Thermoelectric Efficiency via Orthogonal Electrical and Thermal Conductances in Phosphorene. Nano Lett. 14, 6393–6399, https://doi.org/10.1021/nl502865s (2014).
Koenig, S. P., Doganov, R. A., Schmidt, H., Castro Neto, A. H. & Zyilmaz, B. Electric field effect in ultrathin black phosphorus. Appl. Phys. Lett. 104, 103106, https://doi.org/10.1063/1.4868132 (2014).
Ziletti, A. et al. Phosphorene oxides: Bandgap engineering of phosphorene by oxidation. Phys. Rev. B 91, 085407, https://doi.org/10.1103/PhysRevB.91.085407 (2015).
Wang, G. X., Pandey, R. & Karna, S. P. Phosphorene oxide: stability and electronic properties of a novel two-dimensional material. Nanoscale 7, 524–531, https://doi.org/10.1039/c4nr05384b (2015).
Kang, S.-H., Park, J., Woo, S. & Kwon, Y.-K. Symmetry-protected degeneracies in the electronic band structure of oxidized black phosphorous. unpublished (2019).
Wood, J. D. et al. Effective passivation of exfoliated black phosphorus transistors against ambient degradation. Nano Lett. 14, 6964–6970, https://doi.org/10.1021/nl5032293 (2014).
Li, X. et al. Intrinsic electrical transport properties of monolayer silicene and MoS2 from first principles. Phys. Rev. B 87, 115418, https://doi.org/10.1103/PhysRevB.87.115418 (2013).
Liu, T.-H., Chen, Y.-C., Pao, C.-W. & Chang, C.-C. Anisotropic thermal conductivity of MoS2 nanoribbons: Chirality and edge effects. Appl. Phys. Lett. 104, 201909, https://doi.org/10.1063/1.4878395 (2014).
Ong, Z. Y. & Pop, E. Effect of substrate modes on thermal transport in supported graphene. Phys. Rev. B 84, 075471, https://doi.org/10.1103/PhysRevB.84.075471 (2011).
Sevik, C. Assessment on lattice thermal properties of two-dimensional honeycomb structures: Graphene, h-BN, h-MoS2, and h-MoSe2. Phys. Rev. B 89, 035422, https://doi.org/10.1103/PhysRevB.89.035422 (2014).
Lifshitz, I. M. Thermal properties of chain and layered structures at low temperatures. Zh. Eksp. Teor. Fiz. 22, 475 (1952).
Klemens, P. G. Thermal conductivity and lattice vibrational modes. vol. 7 of Solid State Physics, 1–98, https://doi.org/10.1016/S0081-1947(08)60551-2 (Academic Press, 1958).
Klemens, P. G. Theory of Thermal Conductivity of Nanophase Materials. In Chemistry and Physics of Nanostructures and Related Non-Equilibrium Materials, 97–104 (Minerals, Metals, and Materials Society, 1997).
Zou, J. & Balandin, A. Phonon heat conduction in a semiconductor nanowire. J. Appl. Phys. 89, 2932–2938, https://doi.org/10.1063/1.1345515 (2001).
Nika, D. L., Pokatilov, E. P., Askerov, A. S. & Balandin, A. Phonon thermal conduction in graphene: Role of Umklapp and edge roughness scattering. Phys. Rev. B 79, 155413, https://doi.org/10.1103/PhysRevB.79.155413 (2009).
Grimvall, G. Thermophysical properties of materials. (Elsevier Science, 1999).
Hong, Y., Zhang, J., Huang, X. & Zeng, X. C. Thermal Conductivity of Phosphorene Nanoribbon: A Comparative Study with Graphene. Nanoscale 7, 18716–18724, https://doi.org/10.1039/C5NR03577E (2015).
Gu, X. & Yang, R. First-principles prediction of phononic thermal conductivity of silicene: A comparison with graphene. J. Appl. Phys. 117, 025102, https://doi.org/10.1063/1.4905540 (2015).
Cai, Y., Lan, J., Zhang, G. & Zhang, Y.-W. Lattice vibrational modes and phonon thermal conductivity of monolayer MoS2. Phys. Rev. B 89, 035438, https://doi.org/10.1103/PhysRevB.89.035438 (2014).
Peng, B. et al. Thermal conductivity of monolayer MoS2, MoSe2, and WS2: interplay of mass effect, interatomic bonding and anharmonicity. RSC Adv. 6, 5767–5773, https://doi.org/10.1039/C5RA19747C (2016).
Yan, R. et al. Thermal conductivity of monolayer molybdenum disulfide obtained from temperature-dependent Raman spectroscopy. ACS Nano 8, 986–993, https://doi.org/10.1021/nn405826k (2014).
Peng, B. et al. Low lattice thermal conductivity of stanene. Sci. Rep. 6, 20225, https://doi.org/10.1038/srep20225 (2016).
Berber, S., Kwon, Y.-K. & Tománek, D. Unusually High Thermal Conductivity of Carbon Nanotubes. Phys. Rev. Lett. 84, 4613–4616, https://doi.org/10.1103/PhysRevLett.84.4613 (2000).
Lindsay, L. & Broido, D. A. Enhanced thermal conductivity and isotope effect in single-layer hexagonal boron nitride. Phys. Rev. B 84, 155421, https://doi.org/10.1103/PhysRevB.84.155421 (2011).
Sevik, C., Kinaci, A., Haskins, J. B. & Çağin, T. Characterization of thermal transport in low-dimensional boron nitride nanostructures. Phys. Rev. B 84, 085409, https://doi.org/10.1103/PhysRevB.84.085409 (2011).
Lindsay, L., Broido, D. A. & Mingo, N. Flexural phonons and thermal transport in graphene. Phys. Rev. B 82, 115427, https://doi.org/10.1103/PhysRevB.82.115427 (2010).
Lindsay, L. & Broido, D. A. Three-phonon phase space and lattice thermal conductivity in semiconductors. J. Phys.: Condens. Matter 20, 165209, https://doi.org/10.1088/0953-8984/20/16/165209 (2008).
Wu, X. et al. Hydrogenation of penta-graphene leads to unexpected large improvement in thermal conductivity. Nano Lett. 16, 3925–3935, https://doi.org/10.1021/acs.nanolett.6b01536 (2016).
Okubo, K. & Tamura, S. Two-phonon density of states and anharmonic decay of large-wave-vector LA phonons. Phys. Rev. B 28, 4847–4850, https://doi.org/10.1103/PhysRevB.28.4847 (1983).
Li, W., Carrete, J., Katcho, N. A. & Mingo, N. ShengBTE: a solver of the Boltzmann transport equation for phonons. Comp. Phys. Commun. 185, 1747–1758, https://doi.org/10.1016/j.cpc.2014.02.015 (2014).
Kohn, W. & Sham, L. J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 140, A1133–A1138, https://doi.org/10.1103/PhysRev.140.A1133 (1965).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186, https://doi.org/10.1103/PhysRevB.54.11169 (1996).
Kresse, G. & Hafner, J. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561, https://doi.org/10.1103/PhysRevB.47.558 (1993).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979, https://doi.org/10.1103/PhysRevB.50.17953 (1994).
Kresse, G. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775, https://doi.org/10.1103/PhysRevB.59.1758 (1999).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868, https://doi.org/10.1103/PhysRevLett.77.3865 (1996).
Omini, M. & Sparavigna, A. An iterative approach to the phonon Boltzmann equation in the theory of thermal conductivity. Phys. B 212, 101–112, https://doi.org/10.1016/0921-4526(95)00016-3 (1995).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5, https://doi.org/10.1016/j.scriptamat.2015.07.021 (2015).
Li, W., Lindsay, L., Broido, D. A., Stewart, D. A. & Mingo, N. Thermal conductivity of bulk and nanowire Mg2SixSn1−x alloys from first principles. Phys. Rev. B 86, 174307, https://doi.org/10.1103/PhysRevB.86.174307 (2012).
Acknowledgements
We gratefully acknowledge financial support from the Korean government (MSIT) through the National Research Foundation (NRF) of Korea (2015R1A2A2A01006204, 2019R1A2C1005417). A portion of our computational work was done using the resources of the KISTI Supercomputing Center (KSC-2018-CHA-0052, KSC-2018-C2-0033).
Author information
Authors and Affiliations
Contributions
S.L. performed all the calculations and analyzed the data with help from S.-H.K. and Y.-K.K. S.L. and Y.-K.K. wrote the manuscript, on which S.-H.K. commented. All authors reviewed the manuscript. Y.-K.K. supervised the whole project.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lee, S., Kang, SH. & Kwon, YK. Low Lattice Thermal Conductivity of a Two-Dimensional Phosphorene Oxide. Sci Rep 9, 5149 (2019). https://doi.org/10.1038/s41598-019-41696-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41696-y
This article is cited by
-
Oxidation-enhanced thermoelectric efficiency in a two-dimensional phosphorene oxide
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
