
Abstract
Following ischemic stroke, the penumbra, at-risk neural tissue surrounding the core infarct, survives for a variable period of time before progressing to infarction. We investigated genetic determinants of the size of penumbra in mice subjected to middle cerebral artery occlusion (MCAO) using a genome-wide approach. 449 male mice from 33 inbred strains underwent MCAO for 6 hours (215 mice) or 24 hours (234 mice). A genome-wide association study using genetic data from the Mouse HapMap project was performed to examine the effects of genetic variants on the penumbra ratio, defined as the ratio of the infarct volume after 6 hours to the infarct volume after 24 hours of MCAO. Efficient mixed model analysis was used to account for strain interrelatedness. Penumbra ratio differed significantly by strain (F = 2.7, P < 0.001) and was associated with 18 significant SNPs, including 6 protein coding genes. We have identified 6 candidate genes for penumbra ratio: Clint1, Nbea, Smtnl2, Rin3, Dclk1, and Slc24a4.
Similar content being viewed by others


Introduction
Ischemic cerebrovascular disease is a leading cause of death globally, accounting for approximately 6.5 million deaths worldwide1, and is projected to increase in incidence over the coming decades2. Upon large cerebral artery occlusion, an area of core neural tissue infarcts within minutes while tissue surrounding the core, termed the penumbra, persists in an at-risk state. Within the penumbra, diverse and competing influences mediate whether this tissue will eventually infarct3. Recent large randomized clinical trials have demonstrated the success of intra-arterial clot removal within a specified time interval in rescuing not-yet-infarcted penumbra4,5,6. While this time window has increased in recent trials7,8, there are still restrictive requirements for thrombectomy, with only 7% of patients with ischemic stroke that were eligible for thrombectomy in 20169. Recently, the appreciation of fast and slow progressors after large vessel occlusion has prompted discussion of future trials using imaging data to determine who may benefit from treatment10. This heterogeneity in response to ischemia suggests the time windows used currently may not predict outcome accurately in all comers. Thus, understanding factors that affect the penumbra is important in determining in whom and at what time intervention would be beneficial after large artery occlusion.
Discrepancy in infarct volume between strains following middle cerebral artery occlusion (MCAO) is associated with both vascular and neural variation. In particular, differences in infarct size across mouse strains is associated with native arterial collateralization11,12,13,14, incomplete circle of Willis15,16, and susceptibility of neurons to ischemia17,18,19,20,21. However, the question of infarct volume is different from that of the proportion of penumbra to core infarct and is particularly pertinent in the era of endovascular stroke therapy. In this study, 449 male mice spanning 33 strains underwent middle cerebral artery occlusion (MCAO). Each strain contains a unique set of single nucleotide polymorphisms and by comparing the penumbra volume in each strain using a genome-wide analysis, we identified genetic polymorphisms associated with penumbral size.
Methods
Animal Use Statement
All animal care, housing, and experiments in this study were approved by and conducted in accordance with the PHS Policy on Humane Care and Use of Laboratory Animals from the Institutional Animal Care and Use Committee of the Harvard Medical Area Standing Committee on Animals. Experiments are reported in accordance with the ARRIVE guidelines.
Middle Cerebral Artery Occlusion Model
33 inbred strains from the Jackson Laboratory were included in this study. The mouse strains were chosen for genetic diversity22 and include mice that belong to 6 major groups: 1) Bagg albino (A/J, AKR/J, BALB/cJ, C3H/HeJ, CBA/J, CE/J, LG/J, MRL/MpJ, PL/J), 2) Swiss (BuB/BnJ, FVB/NJ, MA/MyJ, NOD/ShiLtJ, RIIIS/J, SJL/J, SWR/J), 3) Japanese and New Zealand (KK/HlJ, NON/ShiLtJ, NZO/HlLtJ, NZW/LacJ), 4) C57-related (C57BL/6J, C57BL/10J, C57BLKS/J, C57BR/cdJ, C57L/J), 5) Castle (129S1/SvImJ, BTBR T+ tf/J, LP/J), and 6) DBA (DBA/1J, DBA/2J, I/LnJ, P/J, SM/J) strains23. 449 mice underwent MCAO as previously described17. The number of mice excluded for each strain due to unsuccessful surgery or death for the 6-hour MCAO is shown in Supplementary Table 1. Animals excluded for the 24-hour model were previously reported17. Eight- to 10-week-old mice were used for the MCAO model. Briefly, mice were anesthetized with 1–3% isoflurane and a 7.0 monofilament threaded into the internal carotid artery to access the middle cerebral artery. Successful occlusion of the middle cerebral artery was defined as ≥80% decrease in cerebral blood flow (CBF) as measured by laser Doppler (Perimed, Jarfalla, Sweden) when comparing the CBF before and after MCAO. Animals were sacrificed by cervical dislocation under 1–3% isoflurane anesthesia. 215 were sacrificed 6 hours after MCAO, and the remaining 234 were sacrificed 24 hours after MCAO. The number of mice per strain at each time point along with age, weight, and mean arterial pressure are documented in Supplementary Table 2.
Evaluation of Infarct Volume
At the time of sacrifice, the brain was removed from the skull and sectioned into 1 mm coronal slices using a brain matrix. Slices were incubated in 2% triphenyltetrazolium (TTC, Sigma, St. Louis, MO) for 15 minutes at room temperature. Infarct volume was assessed by an investigator blinded to the genetics data at the time of measurement. Infarct volume was obtained by calculating the sum of infarct areas across all slices. Normalized infarct volumes were calculated using the indirect Swanson method: (contralateral volume – ipsilateral noninfarcted volume)/contralateral volume24. The penumbra ratio was defined as the ratio of normalized infarct volume at 6 hours to that of the mean normalized infarct volume at 24 hours across all mice in that particular strain. Data for the 24-hour time point were previously reported17. Strains with a penumbra ratio closer to one were considered to have a smaller penumbral volume as compared to strains with a lower ratio.
Association Between Circle of Willis Completeness and Penumbra Ratio
We have previously published the results of an analysis of circle of Willis variation between mouse strains25. Using these data, we calculated the average number of P1 segments per strain. We used the average P1 per strain value as a surrogate for completeness of the circle of Willis. A Spearman correlation was used to analyze the correlation between circle of Willis completeness with penumbra ratio.
Genome-Wide Association Analysis
Following volumetric assessment of MCA territory infarcts, two genome wide association analyses were performed using normalized infarct volume at 6 hours and penumbra ratio as the phenotypes. 132,285 SNPs from the NCBIM37 mouse genotype build were obtained from the UCLA and Broad Institute Mouse HapMap project (http://mouse.cs.ucla.edu/mousehapmap/). SNP data were converted to a numerical format with the R package GAPIT26. SNPs that were absent in greater than 10% of strains were excluded. Minor allele frequency (MAF) was calculated using Plink v1.927 (http://pngu.mgh.harvard.edu/purcell/plink/), with the major allele defined as the most frequent allele at each location amongst the strains included in this study. SNPs with a MAF of ≤0.05 were excluded. A total of 104,890 SNPs remained after filtering. The efficient mixed model association method was utilized, which accounts for inbred strain interrelatedness and population structure in mice, to generate a genome-wide association map (emma package in R28). Covariates were excluded on the basis of univariate regression P values > 0.1. These data were then visualized with Manhattan and QQ plots using the qqman package29. P values were adjusted for multiple testing using the Benjamini and Hochberg false discovery rate (FDR) algorithm30. FDR less than 0.05 were considered statistically significant. The genetic location of each SNP was obtained using the UCSC NCBIM37 (mm9) database31,32. Protein coding genes within 500 kbp of significant SNPs were identified using BiomaRt33 since the majority of enhancers fall within this window34. A network analysis was conducted on genes associated with significant SNPs using StringDB v10.535 (medium confidence threshold, all interaction sources, https://string-db.org/). Pathway overrepresentation analysis was performed using the package WebGestaltR36, with modifications, to search for pathways in KEGG37 (10/1/2016 release), Reactome38 (version 2016), and Wikipathway39 (5/10/18 release), for targets of miRNA and transcription factors from MSigDB40 (v6.0, 2017), and for protein-protein interaction networks from BioGRID41.
Power calculations
We performed simulations to analyze the statistical power using the package emmaPowerSim42 with modifications to account for replicates. The simulation assumes an average minor allele frequency of the causal SNP to be 0.3 and a background genetic effect size of 0.1. The simulation was performed in the 33 strains of mice used in this study for 100 causal SNPs, using 2, 4, 6, or 8 replicates per strain, and varying SNP effect size from the causal SNPs. The power analysis demonstrated that 6 replicates per strain are needed to achieve a power of 80% for a SNP effect of 0.4 (Supplementary Fig. 1). Furthermore, additional replicates only resulted in incremental increases in power. We therefore included a median of 6 technical replicates per strain.
Statistical Analysis
Statistical analysis was performed using R v3.443. Wilcoxon rank sum (Mann-Whitney U) test was used to assess differences in the infarct volume at 6 and 24 hours. ANOVA of univariate linear regression models were used to assess for inter-strain variability for each outcome. Univariate linear regression was used to screen covariates for inclusion in the mixed model. The ggrepel44, tidyr45, plyr46, dyplr47, broom48, data.table49, reshape250, RColorBrewer51, and ggplot252 packages were used for data preparation and visualization. Raw images of TTC stained brains were not modified with the exception of resizing of the whole image and cropping for purposes of organization into figures. Figures were assembled in Adobe Illustrator (Adobe Systems, San Jose, CA).
Results
Descriptive Analysis of Mouse Characteristics
A total of 449 male mice from 33 strains that underwent MCAO were included in this study, 215 of which were sacrificed at 6 hours and 234 of which at 24 hours (Table 1, stratified by strain in Supplementary Table 2). Mouse strains had significantly varied infarct volume at 6 hours (F = 9.74, P < 0.001), infarct volume at 24 hours (F = 8.54, P < 0.001), and penumbra ratio (F = 2.7, P < 0.001). Mean arterial pressure (MAP), weight, and age of mice sacrificed at 6 and 24 hours also varied between strains (Supplementary Table 3). However, these were not significantly associated with normalized infarct volume using univariate linear regression at their respective time points and were not significantly associated with penumbra ratio at 6 hours (Supplementary Table 3). Representative images of TTC staining in strains with relatively small (RIIIS/J) and large (BALB/cJ) penumbra ratios are presented in Fig. 1. Supplementary Figs 2A and 3A depict the distribution of normalized infarct volumes at 6 hours and penumbra ratio by strain, respectively.

Representative brain slices. (A) Graphical representations and (B) photographs of penumbral infarction variation in coronal mouse sections at 6 and 24 hours post MCAO stained with 2% triphenyltetrazolium (TTC). RIIIS/J mice have small and BALB/cJ mice have large penumbra ratios.
Correlation Between Circle of Willis Completeness and Penumbra Ratio
To assess the impact of variation in circle of Willis completeness, we investigated the association between the average number of P1 segments with penumbra ratio. P1 presence was previously reported by our group in 144 mice across the same 33 strains25. Using these data, the average number of P1s per strain did not correlate with penumbra ratio (ρ = 0.10, P = 0.56) (Supplementary Fig. 3). Lack of correlation between P1 presence, and hence circle of Willis completeness, enables analysis of the penumbra ratio with less concern for significant confounding from anatomic variation in the circle of Willis.
Genes Associated with Infarct Size at 6 hours
24 genome-wide significant SNPs were identified to be associated with the 6-hour normalized infarct volume (Supplementary Fig. 2B, Supplementary Table 5). QQ plot for the model demonstrated no notable deviation from expectation at high p values (Supplementary Fig. 2C). One of the significant SNPs, rs3677406, was previously reported by our group to be significant for the 24-hour normalized infarct volume17. Significant SNPs were located within four unique protein-coding genes – Tshz3, Zfp536, Ctif, And Clint1 – which did not have any associations in StringDB35 or pathway/target overrepresentation using WebGestaltR36.
Genes Associated with Penumbra Ratio
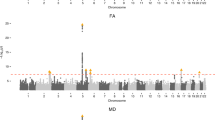
18 SNPs spanning four chromosomes and encompassing 6 protein coding genes were significantly associated with penumbra ratio following FDR correction (Fig. 2B, Table 2). QQ plot of the model demonstrated no notable deviation from expectation at high p values (Fig. 2C). The six protein coding genes – Clint1, Nbea, Smtnl2, Rin3, Dclk1, And Slc24a4 – did not have any interactions in StringDB35 or pathway/target overrepresentation using WebGestaltR36. However, 4 of the 6 significant genes (Dclk1, Slc24a4, Nbea, Smtnl2) are targets of the Myod1 transcription factor and 3 of the 6 significant genes (Dclk1, Clint1, and Slc24a4) are associated with miR-145. Two SNPs – rs31452396 and rs13472659 – were significantly associated with both infarct volume at 6 hours and penumbra ratio, one of which resides within Clint1 (rs13472659).

Summary of penumbra ratio by strain and results from genome-wide association analysis. (A) Box plot illustrating distribution of normalized infarct ratios sorted by strain. (B) Manhattan plot for normalized infarct ratio. (C) QQ plot for the penumbra ratio.
Discussion
In this study, we investigated the size of the ischemic penumbra at 6 hours by normalizing 6-hour infarct volumes to strain averaged infarct volumes at 24 hours, at which point the infarct has likely completed. Prior studies have identified SNPs associated with infarct volume at 24 hours and have linked those SNPs to genes potentially involved in stroke susceptibility17,19,20,21. Keum et al. used an inbred mouse genome wide approach to identify quantitative trait loci on chromosome 7, Civq1, as an important determinant of infarct volume19. The group subsequently identified Itgal120 and Il-21r21 as candidate genes associated with this region and has also reported additional loci contributing to variation in infarct volume amongst mice53. Our own group has previously reported an integrated analysis using inbred mice and human data to identify human ANGPT1 and ZBTB7C as candidate genes associated with middle cerebral artery infarct volume17.
However, the question of penumbra size has not previously been addressed. By dividing the normalized 6-hour infarct volume, which has previously been used to study penumbral gene expression54, by that of the average normalized volume at 24 hours, we generated a ratio of how much tissue had infarcted between 6 and 24 hours relative to the final infarct volume at 24 hours, which is representative of the penumbra. We utilized a proximal MCAO model that has the advantage of avoiding challenges with variations in distal MCA anatomy, but the final infarct volume may be affected by completeness of the circle of Willis15,16. To address this, we analyzed the association between P1 presence and penumbra ratio. The absence of a correlation suggests the penumbra volume calculated in this study is not dependent on circle of Willis completeness. The significant variation in the penumbra ratio across strains suggests that there is a genetic predisposition to penumbral size. This is, perhaps, not surprising, given the variation in infarct volume across strains11,12,13,14,15,16,17,18.
Of the 24 significant SNPs associated with infarct volume at 6 hours, one (rs3677406 on the Ctif gene) had been previously identified by our group to be associated with infarct volume at 24 hours17 whereas the remainder were unique to the 6-hour time point. It is plausible that the determinants of the size of the initial infarct core at 6 hours is not associated with that of the final infarct volume at 24 hours which is a function of both the core and the penumbra, therefore we would expect some of the associated SNPs to be unique to each time point. Of note, we also previously found the Ctif gene to be significantly associated with infarct volume in human stroke patients17. CTIF has been hypothesized to function as a translation initiation factor in the pioneer phase of translation, which appears to be the predominant mechanism by which mRNA is translated in the setting of hypoxia55. It follows that CTIF abnormalities could contribute to ischemic injury via decreased pioneer translation capacity.
To address the question of penumbral size, we performed a genome-wide association analysis with penumbra ratio as the outcome. The 18 SNPs significantly associated with penumbra volume were located within with 6 protein coding genes (Clint1, Dclk1, Slc24a4, Rin3, Smtnl2, And Nbea). The majority of known SNPs were found to be in introns, which suggests they may be involved in regulation of expression rather than in structural differences of the resulting protein. Two of the SNPs significant for penumbra volume were also significant at the 6-hour time point, rs31452396 and rs13472659, the latter of which is in Clint1. While rs13472659 is a synonymous coding SNP, it was found to be significant in two separate analyses is suggestive of its relevance to cerebral ischemia. CLINT1 (also called EPN4) is enriched in the brain and functions in the endocytosis of clathrin coated pits56. Clint1 mutant zebrafish have a phenotype mimicking psoriasis, suggesting it may function in mediating inflammation57. Genetic polymorphisms that result in dysregulated inflammatory responses to ischemia could potentiate the inflammatory penumbra, a concept proposed to account for secondary brain injury outside the initial ischemic territory58. Furthermore, zebrafish with Clint1 mutations have increased matrix metalloproteinase 9 (MMP-9) expression59. MMP-9, a proteolytic enzyme that normally functions in extracellular matrix remodeling that has been shown to function in blood brain barrier function, increases the degree of ischemic cerebral injury following stroke60,61,62,63. Increased permeability of the blood brain barrier enables immune cell extravasation and may exacerbate secondary brain injury. Another protein identified in this study, RIN3, a RAB5 guanine exchange factor, functions in early endocytosis64 and acts as a negative regulator in mast cells65. Mast cells in turn mediate early peri-infarct inflammation following MCAO in rats via blood brain barrier dysfunction and subsequent inflammatory cell extravasation66, potentially contributing to the inflammatory penumbra.
Clint1 expression has also been reported to increase in male, but not female, C57BL/6 mice following MCAO67, suggesting male and female mice might respond differently to ischemic injury. All mice included in this study were male, so our results are consistent with the study from Lusardi et al.67. Interestingly, expression of another gene associated with SNPs significantly associated with penumbra ratio, Dclk1, which encodes a kinase associated with microtubule polymerization68, has been shown to increase in ischemic female rats following the inhibition of let-7f, a microRNA that suppresses insulin-like growth factor 1 translation69. This effect was not observed in male or female rats following oophorectomy, suggesting this pathway is dependent on estrogen and hinting that DCLK1 activity in the setting of cerebral ischemia may have a protective role. Additionally, two long noncoding RNA (lncRNA) located within the Dclk1 locus are overexpressed following MCAO in rats, with a possible role in the epigenetic modifications in the setting of ischemia70. However, more research is necessary to better elucidate the function of these proteins following stroke.
Slc24a4 (also known as NCKX4) encodes a sodium/potassium/calcium exchanger and is immediately 5′ to Rin3 that is associated with lipid metabolism71. Slc24a4 knockout mice were found to be anorexic, with constitutively activated paraventricular nucleus neurons hypothesized to be secondary to elevated Ca2+ signal and melanocortin 4 receptor (MC4R) activity72. Melanocyte-stimulating hormone has been shown to have a dose-dependent neuroprotective effect via MC4R signaling following 10 minutes of bilateral carotid occlusion in gerbils via decreased TNF-α and IL-6, as well as decreased MAPK mediated apoptotic pathways73. However, this study specifically demonstrated neuroprotection in the hippocampus following global cerebral ischemia and did not investigate ischemic penumbra following stroke. Nevertheless, these findings illustrate a potential mechanism for how SLC24A4 mediates penumbra size. Additionally, SLC24A4 is part of a larger family of ion channels including NCKX2, which has been associated with ischemic brain injury74.
SMNTL2 is a downstream target of c-Jun-N-terminal kinases (JNK), predominantly in skeletal muscle although it is expressed in other tissue as well75. JNK isoforms facilitate neuron cell death in settings of ischemic stress76, and JNK inhibition is neuroprotective following MCAO77. However, the function of SMNTL2 is still largely uncharacterized and future study is needed to define its role, if any, in neuroprotection.
Finally, NBEA is a scaffolding protein with a Beige and Chediak-Higashi (BEACH) domain concentrated in neurons that functions in synaptic transmission at the neuromuscular junction78 as well as within the central nervous system79. Additionally, NBEA loss is associated with both autism and abnormal platelet morphology secondary to large dense core vesicle secretion80. Yet, the role of NBEA following stroke is not clear. Similar to SMNTL2, future research targeted at NBEA in stroke is needed.
To further investigate potential interaction amongst these candidate genes, we performed a pathway overrepresentation analysis. Myod1 (MyoD), a helix-loop-helix transcription factor involved in myocyte differentiation81, was identified as a regulator of the expression of 4 of the 6 significant genes—Dclk1, Slc24a4, Nbea, and Smtnl2. Myod1’s canonical role is to induce differentiation of fibroblasts to skeletal muscle cells, but it has also been shown to be expressed in the brain82 and may have a role in neural development83. Moreover, Dey et al. found loss of Myod1 catalyzed sonic hedgehog (Shh) driven neoplastic growth in medulloblastoma84. In the setting of ischemia, Shh activity increases in murine neural progenitor cells and neurons85. Specifically, the Shh pathway is active in cortex and striatum adjacent to the injured region86. Blocking the Shh pathway potentiates brain injury87 and Shh agonists have been found to neuroprotective after stroke88. The role, if any, of the 4 target genes identified here in Shh signaling is unclear.
Interestingly, the micro RNA miR-145 is associated with multiple proteins identified in the penumbra analysis. DCLK1 post-transcriptionally regulates miR-145 in pancreatic tumor zenografts and DCLK1 knockout mice were found to have increased expression of miR-14589. MiR-145 has also been implicated in ischemic stroke. Using an oxygen-glucose deprivation model with primary neuronal cultures, Zheng et al. found overexpression of miR-145 lead to decreased expression of Aqp4, the gene encoding aquaporin 4 (AQP4), in astrocytes resulting in a protective effect in the setting of ischemia90. MiR-145 was also found to be overexpressed in the cortex after transient (1 hour) MCAO and subsequent reperfusion in rats by Dharap et al. and antagonism of miR-145 led to increased levels of peri-infarct superoxide dismutase as well as decreased infarct volumes91. Moreover, miR-145 expression was found to be elevated within 24 hours of ischemic stroke in human peripheral blood and its expression correlated with the volume of the ischemic infarct as well as with NIH stroke scale score92. Furthermore, CLINT1 has also been shown to be inhibited by miR-145, resulting in cell death in a bladder cancer model93 and, using TargetScan94, SLC24A4 is a predicted target of miR-145 (http://www.targetscan.org/cgi-bin/targetscan/vert_70/targetscan.cgi?mirg = hsa-miR-145). The finding of multiple genes associated with penumbra ratio and miR-145 suggest genetic polymorphisms in these genes may be modifying a common pathway regulating the rate of penumbra infarction (Supplementary Fig. 4). However, further research is necessary to establish and better define the role of miR-145 in the rate of penumbra infarction.
Limitations
This study has important limitations warranting further discussion. First, we analyzed 6-hour infarct volumes in 215 mice from 33 unique strains, normalized to another 234 mice with 24-hour infarct volumes. Importantly, this method of estimating penumbra volume is a rough estimate as penumbra was not directly measured. These data should be therefore be considered carefully in the context of an estimated penumbra volume. The relatively small sample sizes may limit the statistical power of our study. The ratio of infarct volumes at six over 24 hours utilized approximate penumbra size but is not a direct measure of at-risk tissue and may result in an underestimation of the genetic effects. In addition, it is possible that other variations in blood vessel anatomy not examined in this study could impact the penumbra. It is also possible that serum pH, glucose, and other metabolites not measured in this study could affect penumbra ratio. Moreover, there are varying degrees of occlusion that can confound the results. In addition, the model used in this study, MCAO, may have limited applicability to human patients as it involves a single mechanism and does not capture the different stroke etiologies or the comorbidities commonly found in human patients presenting with ischemic stroke. Although we have minimized the number of animals necessary to achieve sufficient statistical power, a strain survey inherently involves a large number of animals. Nevertheless, these data provide an advantage over human GWAS in that a controlled experimental model, such as the MCAO, is possible whereas it is not in human subjects. Future mechanistic studies are required to investigate the role of the candidate genes identified on penumbral size.
Conclusions
The size of ischemic penumbra is paramount in determining the extent of salvageable brain following stroke. While prior studies have investigated genetic polymorphisms associated with infarct size, this is the first genome wide association study in mice specifically investigating penumbra size. We report 18 significant SNPs in 6 protein coding genes, including proteins potentially involved in the inflammatory penumbra and neuronal susceptibility to ischemia, which fundamentally make sense but require more research to better define. A better understanding of the genetic underpinnings of penumbral size could inform more personalized application of acute stroke treatment as well as provide the foundation for potential novel therapies.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Benjamin, E. J. et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation 135, e146–e603, https://doi.org/10.1161/cir.0000000000000485 (2017).
Ovbiagele, B. et al. Forecasting the future of stroke in the United States: a policy statement from the American Heart Association and American Stroke Association. Stroke 44, 2361–2375, https://doi.org/10.1161/STR.0b013e31829734f2 (2013).
Lo, E. H. A new penumbra: transitioning from injury into repair after stroke. Nature medicine 14, 497–500, https://doi.org/10.1038/nm1735 (2008).
Goyal, M. et al. Randomized assessment of rapid endovascular treatment of ischemic stroke. The New England journal of medicine 372, 1019–1030, https://doi.org/10.1056/NEJMoa1414905 (2015).
Berkhemer, O. A. et al. A randomized trial of intraarterial treatment for acute ischemic stroke. The New England journal of medicine 372, 11–20, https://doi.org/10.1056/NEJMoa1411587 (2015).
Campbell, B. C. et al. Endovascular therapy for ischemic stroke with perfusion-imaging selection. The New England journal of medicine 372, 1009–1018, https://doi.org/10.1056/NEJMoa1414792 (2015).
Nogueira, R. G. et al. Thrombectomy 6 to 24 Hours after Stroke with a Mismatch between Deficit and Infarct. The New England journal of medicine 378, 11–21, https://doi.org/10.1056/NEJMoa1706442 (2018).
Albers, G. W. et al. Thrombectomy for Stroke at 6 to 16 Hours with Selection by Perfusion Imaging. The New England journal of medicine 378, 708–718, https://doi.org/10.1056/NEJMoa1713973 (2018).
Chia, N. H., Leyden, J. M., Newbury, J., Jannes, J. & Kleinig, T. J. Determining the Number of Ischemic Strokes Potentially Eligible for Endovascular Thrombectomy: A Population-Based Study. Stroke 47, 1377–1380, https://doi.org/10.1161/strokeaha.116.013165 (2016).
Rocha, M. & Jovin, T. G. Fast Versus Slow Progressors of Infarct Growth in Large Vessel Occlusion Stroke: Clinical and Research Implications. Stroke 48, 2621–2627, https://doi.org/10.1161/strokeaha.117.017673 (2017).
Zhang, H., Prabhakar, P., Sealock, R. & Faber, J. E. Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism 30, 923–934, https://doi.org/10.1038/jcbfm.2010.10 (2010).
Sealock, R., Zhang, H., Lucitti, J. L., Moore, S. M. & Faber, J. E. Congenic fine-mapping identifies a major causal locus for variation in the native collateral circulation and ischemic injury in brain and lower extremity. Circulation research 114, 660–671, https://doi.org/10.1161/circresaha.114.302931 (2014).
Kao, Y. J., Oyarzabal, E. A., Zhang, H., Faber, J. E. & Shih, Y. I. Role of Genetic Variation in Collateral Circulation in the Evolution of Acute Stroke: A Multimodal Magnetic Resonance Imaging Study. Stroke 48, 754–761, https://doi.org/10.1161/strokeaha.116.015878 (2017).
Lucitti, J. L. et al. Variants of Rab GTPase-Effector Binding Protein-2 Cause Variation in the Collateral Circulation and Severity of Stroke. Stroke 47, 3022–3031, https://doi.org/10.1161/strokeaha.116.014160 (2016).
Kitagawa, K. et al. Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: evaluation of the patency of the posterior communicating artery. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism 18, 570–579, https://doi.org/10.1097/00004647-199805000-00012 (1998).
Barone, F. C., Knudsen, D. J., Nelson, A. H., Feuerstein, G. Z. & Willette, R. N. Mouse strain differences in susceptibility to cerebral ischemia are related to cerebral vascular anatomy. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism 13, 683–692, https://doi.org/10.1038/jcbfm.1993.87 (1993).
Du, R. et al. Integrative Mouse and Human Studies Implicate ANGPT1 and ZBTB7C as Susceptibility Genes to Ischemic Injury. Stroke 46, 3514–3522, https://doi.org/10.1161/strokeaha.115.010767 (2015).
Iadecola, C. et al. Reduced susceptibility to ischemic brain injury and N-methyl-D-aspartate-mediated neurotoxicity in cyclooxygenase-2-deficient mice. Proceedings of the National Academy of Sciences of the United States of America 98, 1294–1299, https://doi.org/10.1073/pnas.98.3.1294 (2001).
Keum, S. & Marchuk, D. A. A locus mapping to mouse chromosome 7 determines infarct volume in a mouse model of ischemic stroke. Circulation. Cardiovascular genetics 2, 591–598, https://doi.org/10.1161/circgenetics.109.883231 (2009).
Keum, S. et al. Natural genetic variation of integrin alpha L (Itgal) modulates ischemic brain injury in stroke. PLoS genetics 9, e1003807, https://doi.org/10.1371/journal.pgen.1003807 (2013).
Lee, H. K. et al. Natural allelic variation of the IL-21 receptor modulates ischemic stroke infarct volume. The Journal of clinical investigation 126, 2827–2838, https://doi.org/10.1172/jci84491 (2016).
Leme, A. S. et al. A survey of airway responsiveness in 36 inbred mouse strains facilitates gene mapping studies and identification of quantitative trait loci. Mol Genet Genomics 283, 317–326, https://doi.org/10.1007/s00438-010-0515-x (2010).
Petkov, P. M. et al. An efficient SNP system for mouse genome scanning and elucidating strain relationships. Genome research 14, 1806–1811, https://doi.org/10.1101/gr.2825804 (2004).
Swanson, R. A. & Sharp, F. R. Infarct measurement methodology. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism 14, 697–698, https://doi.org/10.1038/jcbfm.1994.88 (1994).
Qian, B., Rudy, R. F., Cai, T. & Du, R. Cerebral Artery Diameter in Inbred Mice Varies as a Function of Strain. Frontiers in neuroanatomy 12, 10, https://doi.org/10.3389/fnana.2018.00010 (2018).
Lipka, A. E. et al. GAPIT: genome association and prediction integrated tool. Bioinformatics (Oxford, England) 28, 2397–2399, https://doi.org/10.1093/bioinformatics/bts444 (2012).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. American journal of human genetics 81, 559–575, https://doi.org/10.1086/519795 (2007).
Kang, H. M. et al. Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723, https://doi.org/10.1534/genetics.107.080101 (2008).
qqman: Q-Q and Manhattan Plots for GWAS Data. R package version 0.1.4. (2017).
Benjamini, Y. & Hochberg, Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the royal statistical society. Series B (Methodological), 289–300 (1995).
Casper, J. et al. The UCSC Genome Browser database: 2018 update. Nucleic acids research 46, D762–d769, https://doi.org/10.1093/nar/gkx1020 (2018).
Kent, W. J. et al. The human genome browser at UCSC. Genome research 12, 996–1006, https://doi.org/10.1101/gr.229102 (2002).
Durinck, S., Spellman, P. T., Birney, E. & Huber, W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nature protocols 4, 1184–1191, https://doi.org/10.1038/nprot.2009.97 (2009).
Wang, K., Li, M. & Bucan, M. Pathway-based approaches for analysis of genomewide association studies. American journal of human genetics 81, 1278–1283, https://doi.org/10.1086/522374 (2007).
Szklarczyk, D. et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic acids research 45, D362–d368, https://doi.org/10.1093/nar/gkw937 (2017).
Wang, J., Duncan, D., Shi, Z. & Zhang, B. WEB-based GEne SeT AnaLysis Toolkit (WebGestalt): update 2013. Nucleic acids research 41, W77–83, https://doi.org/10.1093/nar/gkt439 (2013).
Kanehisa, M. et al. KEGG for linking genomes to life and the environment. Nucleic acids research 36, D480–484, https://doi.org/10.1093/nar/gkm882 (2008).
Fabregat, A. et al. The Reactome Pathway Knowledgebase. Nucleic acids research 46, D649–D655, https://doi.org/10.1093/nar/gkx1132 (2018).
Pico, A. R. et al. WikiPathways: pathway editing for the people. Plos Biol 6, e184, https://doi.org/10.1371/journal.pbio.0060184 (2008).
Liberzon, A. et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics (Oxford, England) 27, 1739–1740, https://doi.org/10.1093/bioinformatics/btr260 (2011).
Stark, C. et al. BioGRID: a general repository for interaction datasets. Nucleic acids research 34, D535–539, https://doi.org/10.1093/nar/gkj109 (2006).
emmaPowerSim: Power simulation experiments for efficient mixed model association. R package version 1.1 (2010).
R: A language and environment for statistical computing. Version 3.4 (R Foundation for Statistical Computing, Vienna, Austria, 2017).
ggrepel: Repulsive Text and Label Geoms for ‘ggplot2’. R package version 0.6.5 (2016).
tidyr: Easily Tidy Data with ‘spread()’ and ‘gather()’ Functions. R Package Version 0.6.3 (2017).
Wickham, H. The Split-Apply-Combine Strategy for Data Analysis. Journal of Statistical Software 40, 1–29 (2011).
dplyr: A Grammar of Data Manipulation. R package version 0.7.1 (2017).
broom: Convert Statistical Analysis Objects into Tidy Data Frames. R package version 0.4.2. (2017).
data.table: Extension of ‘data.frame‘. R package version 1.10.4 (2017).
Wickham, H. Reshaping Data with the reshape Package. Journal of Statistical Software 21, 1–20 (2007).
Neuwirth, E. RColorBrewer: ColorBrewer Palettes. R Package version 1, 1–2 (2014).
Wickham, H. ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag, New York, 2009).
Chu, P. L., Keum, S. & Marchuk, D. A. A novel genetic locus modulates infarct volume independently of the extent of collateral circulation. Physiological genomics 45, 751–763, https://doi.org/10.1152/physiolgenomics.00063.2013 (2013).
Hori, M. et al. Unraveling the Specific Ischemic Core and Penumbra Transcriptome in the Permanent Middle Cerebral Artery Occlusion Mouse Model Brain Treated with the Neuropeptide PACAP38. Microarrays (Basel, Switzerland) 4, 2–24, https://doi.org/10.3390/microarrays4010002 (2015).
Maquat, L. E., Tarn, W. Y. & Isken, O. The pioneer round of translation: features and functions. Cell 142, 368–374, https://doi.org/10.1016/j.cell.2010.07.022 (2010).
Miller, S. E., Collins, B. M., McCoy, A. J., Robinson, M. S. & Owen, D. J. A SNARE-adaptor interaction is a new mode of cargo recognition in clathrin-coated vesicles. Nature 450, 570–574, https://doi.org/10.1038/nature06353 (2007).
Dodd, M. E. et al. The ENTH domain protein Clint1 is required for epidermal homeostasis in zebrafish. Development (Cambridge, England) 136, 2591–2600, https://doi.org/10.1242/dev.038448 (2009).
Gauberti, M. et al. Ultra-sensitive molecular MRI of vascular cell adhesion molecule-1 reveals a dynamic inflammatory penumbra after strokes. Stroke 44, 1988–1996, https://doi.org/10.1161/strokeaha.111.000544 (2013).
LeBert, D. C. et al. Matrix metalloproteinase 9 modulates collagen matrices and wound repair. Development (Cambridge, England) 142, 2136–2146, https://doi.org/10.1242/dev.121160 (2015).
Asahi, M. et al. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism 20, 1681–1689, https://doi.org/10.1097/00004647-200012000-00007 (2000).
Asahi, M. et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. The Journal of neuroscience: the official journal of the Society for Neuroscience 21, 7724–7732 (2001).
Romanic, A. M., White, R. F., Arleth, A. J., Ohlstein, E. H. & Barone, F. C. Matrix metalloproteinase expression increases after cerebral focal ischemia in rats: inhibition of matrix metalloproteinase-9 reduces infarct size. Stroke 29, 1020–1030 (1998).
Rosell, A. et al. Increased brain expression of matrix metalloproteinase-9 after ischemic and hemorrhagic human stroke. Stroke 37, 1399–1406, https://doi.org/10.1161/01.STR.0000223001.06264.af (2006).
Kajiho, H. et al. RIN3: a novel Rab5 GEF interacting with amphiphysin II involved in the early endocytic pathway. Journal of cell science 116, 4159–4168, https://doi.org/10.1242/jcs.00718 (2003).
Janson, C., Kasahara, N., Prendergast, G. C. & Colicelli, J. RIN3 is a negative regulator of mast cell responses to SCF. PloS one 7, e49615, https://doi.org/10.1371/journal.pone.0049615 (2012).
Strbian, D., Karjalainen-Lindsberg, M. L., Tatlisumak, T. & Lindsberg, P. J. Cerebral mast cells regulate early ischemic brain swelling and neutrophil accumulation. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism 26, 605–612, https://doi.org/10.1038/sj.jcbfm.9600228 (2006).
Lusardi, T. A. et al. MicroRNA responses to focal cerebral ischemia in male and female mouse brain. Frontiers in molecular neuroscience 7, 11, https://doi.org/10.3389/fnmol.2014.00011 (2014).
Lin, P. T., Gleeson, J. G., Corbo, J. C., Flanagan, L. & Walsh, C. A. DCAMKL1 encodes a protein kinase with homology to doublecortin that regulates microtubule polymerization. The Journal of neuroscience: the official journal of the Society for Neuroscience 20, 9152–9161 (2000).
Selvamani, A., Sathyan, P., Miranda, R. C. & Sohrabji, F. An antagomir to microRNA Let7f promotes neuroprotection in an ischemic stroke model. PloS one 7, e32662, https://doi.org/10.1371/journal.pone.0032662 (2012).
Dharap, A., Pokrzywa, C. & Vemuganti, R. Increased binding of stroke-induced long non-coding RNAs to the transcriptional corepressors Sin3A and coREST. ASN neuro 5, 283–289, https://doi.org/10.1042/an20130029 (2013).
Kraja, A. T. et al. Genetic analysis of 16 NMR-lipoprotein fractions in humans, the GOLDN study. Lipids 48, 155–165, https://doi.org/10.1007/s11745-012-3740-8 (2013).
Li, X. F. & Lytton, J. An essential role for the K+−dependent Na+/Ca2+−exchanger, NCKX4, in melanocortin-4-receptor-dependent satiety. The Journal of biological chemistry 289, 25445–25459, https://doi.org/10.1074/jbc.M114.564450 (2014).
Giuliani, D. et al. Both early and delayed treatment with melanocortin 4 receptor-stimulating melanocortins produces neuroprotection in cerebral ischemia. Endocrinology 147, 1126–1135, https://doi.org/10.1210/en.2005-0692 (2006).
Cuomo, O. et al. A critical role for the potassium-dependent sodium-calcium exchanger NCKX2 in protection against focal ischemic brain damage. The Journal of neuroscience: the official journal of the Society for Neuroscience 28, 2053–2063, https://doi.org/10.1523/jneurosci.4912-07.2008 (2008).
Gordon, E. A. et al. Combining docking site and phosphosite predictions to find new substrates: identification of smoothelin-like-2 (SMTNL2) as a c-Jun N-terminal kinase (JNK) substrate. Cellular signalling 25, 2518–2529, https://doi.org/10.1016/j.cellsig.2013.08.004 (2013).
Vosler, P. S. & Chen, J. Potential molecular targets for translational stroke research. Stroke 40, S119–120, https://doi.org/10.1161/strokeaha.108.533109 (2009).
Borsello, T. et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nature medicine 9, 1180–1186, https://doi.org/10.1038/nm911 (2003).
Su, Y. et al. Neurobeachin is essential for neuromuscular synaptic transmission. The Journal of neuroscience: the official journal of the Society for Neuroscience 24, 3627–3636, https://doi.org/10.1523/jneurosci.4644-03.2004 (2004).
Medrihan, L. et al. Neurobeachin, a protein implicated in membrane protein traffic and autism, is required for the formation and functioning of central synapses. The Journal of physiology 587, 5095–5106, https://doi.org/10.1113/jphysiol.2009.178236 (2009).
Castermans, D. et al. SCAMP5, NBEA and AMISYN: three candidate genes for autism involved in secretion of large dense-core vesicles. Human molecular genetics 19, 1368–1378, https://doi.org/10.1093/hmg/ddq013 (2010).
Weintraub, H. et al. The myoD gene family: nodal point during specification of the muscle cell lineage. Science (New York, N.Y.) 251, 761–766 (1991).
Gerhart, J. et al. MyoD-positive myoblasts are present in mature fetal organs lacking skeletal muscle. The Journal of cell biology 155, 381–392, https://doi.org/10.1083/jcb.200105139 (2001).
Delfini, M. C. & Duprez, D. Ectopic Myf5 or MyoD prevents the neuronal differentiation program in addition to inducing skeletal muscle differentiation, in the chick neural tube. Development (Cambridge, England) 131, 713–723, https://doi.org/10.1242/dev.00967 (2004).
Dey, J. et al. MyoD is a tumor suppressor gene in medulloblastoma. Cancer research 73, 6828–6837, https://doi.org/10.1158/0008-5472.Can-13-0730-t (2013).
Sims, J. R. et al. Sonic hedgehog regulates ischemia/hypoxia-induced neural progenitor proliferation. Stroke 40, 3618–3626, https://doi.org/10.1161/strokeaha.109.561951 (2009).
Jin, Y. et al. The shh signaling pathway is upregulated in multiple cell types in cortical ischemia and influences the outcome of stroke in an animal model. PloS one 10, e0124657, https://doi.org/10.1371/journal.pone.0124657 (2015).
Ji, H. et al. Inhibition of sonic hedgehog signaling aggravates brain damage associated with the down-regulation of Gli1, Ptch1 and SOD1 expression in acute ischemic stroke. Neuroscience letters 506, 1–6, https://doi.org/10.1016/j.neulet.2011.11.027 (2012).
Jin, Y., Barnett, A., Zhang, Y., Yu, X. & Luo, Y. Poststroke Sonic Hedgehog Agonist Treatment Improves Functional Recovery by Enhancing Neurogenesis and Angiogenesis. Stroke 48, 1636–1645, https://doi.org/10.1161/strokeaha.117.016650 (2017).
Sureban, S. M. et al. DCLK1 regulates pluripotency and angiogenic factors via microRNA-dependent mechanisms in pancreatic cancer. PloS one 8, e73940, https://doi.org/10.1371/journal.pone.0073940 (2013).
Zheng, L. et al. Overexpression of MicroRNA-145 Ameliorates Astrocyte Injury by Targeting Aquaporin 4 in Cerebral Ischemic. Stroke. BioMed research international 2017, 9530951, https://doi.org/10.1155/2017/9530951 (2017).
Dharap, A., Bowen, K., Place, R., Li, L. C. & Vemuganti, R. Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism 29, 675–687, https://doi.org/10.1038/jcbfm.2008.157 (2009).
Jia, L., Hao, F., Wang, W. & Qu, Y. Circulating miR-145 is associated with plasma high-sensitivity C-reactive protein in acute ischemic stroke patients. Cell biochemistry and function 33, 314–319, https://doi.org/10.1002/cbf.3116 (2015).
Ostenfeld, M. S. et al. miR-145 induces caspase-dependent and -independent cell death in urothelial cancer cell lines with targeting of an expression signature present in Ta bladder tumors. Oncogene 29, 1073–1084, https://doi.org/10.1038/onc.2009.395 (2010).
Lewis, B. P., Burge, C. B. & Bartel, D. P. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20, https://doi.org/10.1016/j.cell.2004.12.035 (2005).
Acknowledgements
This work was supported by the National Institutes of Health grant K08NS067172 (RD).
Author information
Authors and Affiliations
Contributions
R.R.: performed analysis and drafted manuscript, N.C.: acquired data and critically revised manuscript, B.Q.: acquired data and critically revised manuscript, A.B.: interpreted data and critically revised manuscript, R.F.: interpreted data and critically revised manuscript, S.W.: interpreted data and critically revised manuscript, R.D.: conceived and designed experiments, performed analysis, supervised experiments and analysis, interpreted data, and critically revised manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rudy, R.F., Charoenvimolphan, N., Qian, B. et al. A Genome-Wide Analysis of the Penumbral Volume in Inbred Mice following Middle Cerebral Artery Occlusion. Sci Rep 9, 5070 (2019). https://doi.org/10.1038/s41598-019-41592-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-41592-5
This article is cited by
-
LncRNA SNHG1 promotes EMT process in gastric cancer cells through regulation of the miR-15b/DCLK1/Notch1 axis
BMC Gastroenterology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
