
Abstract
Perivascular space (PVS) burden is an emerging, poorly understood, magnetic resonance imaging marker of cerebral small vessel disease, a leading cause of stroke and dementia. Genome-wide association studies in up to 40,095 participants (18 population-based cohorts, 66.3 ± 8.6 yr, 96.9% European ancestry) revealed 24 genome-wide significant PVS risk loci, mainly in the white matter. These were associated with white matter PVS already in young adults (N = 1,748; 22.1 ± 2.3 yr) and were enriched in early-onset leukodystrophy genes and genes expressed in fetal brain endothelial cells, suggesting early-life mechanisms. In total, 53% of white matter PVS risk loci showed nominally significant associations (27% after multiple-testing correction) in a Japanese population-based cohort (N = 2,862; 68.3 ± 5.3 yr). Mendelian randomization supported causal associations of high blood pressure with basal ganglia and hippocampal PVS, and of basal ganglia PVS and hippocampal PVS with stroke, accounting for blood pressure. Our findings provide insight into the biology of PVS and cerebral small vessel disease, pointing to pathways involving extracellular matrix, membrane transport and developmental processes, and the potential for genetically informed prioritization of drug targets.
Similar content being viewed by others


Main
PVS are physiological spaces surrounding small vessel walls as they run from the subarachnoid space through the brain parenchyma1,2,3. Dilation of PVS observed on brain magnetic resonance imaging (MRI) is thought to be a marker of PVS dysfunction and, speculated from preclinical studies, to reflect impairment of brain fluid and waste clearance2,4.
PVS increase in number with age and vascular risk factors, especially hypertension2. They are associated with white matter hyperintensities (WMH) of presumed vascular origin, lacunes and cerebral microbleeds2, all MRI features of cerebral small vessel disease (cSVD), a leading cause of stroke and dementia with no specific mechanistic treatment to date5,6. PVS are detected on brain MRI much earlier than WMH, lacunes or cerebral microbleeds7, and are described as the earliest stage of cSVD lesions on neuropathology8. Their pathophysiology is poorly understood6,9.
In experimental models, PVS appear to be important conduits for substrate delivery, flushing interstitial fluid, clearing metabolic waste (for example, beta-amyloid peptide) and brain fluid regulation, as part of the ‘glymphatic system’4,7. These processes were described to increase during sleep2,4,7. Mounting evidence suggests a major role of PVS in cerebral injury. Several studies suggested associations of PVS burden (number of visible PVS on brain MRI) with stroke2,6,10, Alzheimer’s disease pathology2 and cerebral amyloid angiopathy (CAA)11,12,13. Post-stroke edema has been linked to post-stroke PVS enlargement14, and in amyotrophic lateral sclerosis PVS dilation was observed and perivascular fibroblast proteins were associated with survival15.
PVS burden is highly heritable16. Identifying genetic risk variants for PVS could be a powerful tool to decipher underlying biological pathways. We conducted genome-wide association study (GWAS) meta-analyses and whole-exome/whole-genome sequencing (WES/WGS) studies of extensive PVS burden in up to 40,095 and 19,010 older community participants, respectively. Given differential associations with risk factors and neurological traits2,10,17 and anatomical differences18, we ran analyses separately for white matter (WM)-PVS, basal ganglia (BG)-PVS and hippocampal (HIP)-PVS. We followed up identified risk loci in independent samples of young healthy adults and older Japanese community participants and examined shared genetic determinants with other vascular and neurological traits. Leveraging tissue and cell-specific gene expression databases and drug target libraries, we conducted extensive bioinformatics exploration of identified PVS risk loci.
Results
Genetic discovery
Twenty-one population-based cohorts were included, of which 18 were for GWAS and 8 for whole-exome association studies (Supplementary Table 1 and Methods). We tested associations of extensive PVS burden with ~8 million single-nucleotide polymorphisms (SNPs) (minor allele frequency (MAF) ≥ 1%) in GWAS meta-analyses, gathering up to 40,095 participants (66.3 ± 8.6 yr, 51.7% female, 66.7% with hypertension; Supplementary Tables 1–3). We dichotomized PVS burden based on cut-offs closest to the top quartile of PVS distribution to account for differences in PVS quantification methods, image acquisition and participant characteristics (Supplementary Tables 1 and 2 and Methods). In total, 9,607 of 39,822, 9,189 of 40,000, and 9,339 of 40,095 participants had extensive PVS burden in WM, BG and hippocampus.
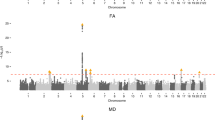
The GWAS meta-analysis comprised 17 cohorts from the Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium (N ≤ 11,511)19, with PVS quantification primarily on visual rating scales, and UK Biobank (UKB, N ≤ 28,655), with computational PVS quantification (Methods). Participants were of European (N = 38,871), Hispanic (N = 717), East-Asian (N = 339) and African-American (N = 168) ancestry. We identified 22 independent genome-wide significant risk loci for extensive PVS burden (WM-PVS: 19; BG-PVS: 2; HIP-PVS: 3 (2 shared with WM-PVS)) and two additional risk loci for WM-PVS in Europeans only, leading to 24 independent signals (Table 1, Fig. 1, Extended Data Fig. 1 and Supplementary Fig. 1). There was no systematic inflation of association statistics (Supplementary Table 4 and Extended Data Fig. 1).

a, Extensive PVS burden (arrows) in WM (top, WM-PVS), BG (middle, BG-PVS) and hippocampus (bottom, HIP-PVS) on T1-weighted axial magnetic resonance images. b, Circular Manhattan plot. The inner circle corresponds to the cross-ancestry GWAS meta-analyses results, the middle circle to the results of the MTAG analysis and the outer circle to gene-based test results. Results for WM-PVS are in blue, for BG-PVS in purple and for HIP-PVS in green. The gray line corresponds to the genome-wide significance threshold (P = 5 × 10−8, two-sided, correcting for multiple testing at the genome-wide level).
We performed conditional logistic regression using Genome-wide Complex Trait Analysis (GCTA)-COJO (Methods) to seek independent association signals within genome-wide significant loci. Consistent with linkage disequilibrium (LD)-based clumping, this identified two independent signals at chr3p25.1 (WNT7A) and six at chr20q13.12 (SLC13A3; Supplementary Fig. 1 and Supplementary Table 5a), four of which with low-frequency variants (Table 1). The six polymorphisms at chr20q13.12 generated eight haplotypes with haplotypic R2 (percentage of haplotypic variability explained by observed genotypes) > 0.7 in the Three-City Dijon Study (3C-Dijon) cohort, of European ancestry (N = 1,500; Supplementary Table 5b). The two common rs2425881-A and rs2425884-C alleles, in very low LD with each other (r2 (a measure of correlation of alleles for two genetic variants) ~ 0.05, D′ (a pairwise r2 standardized for allele frequencies) ~ 0.50), generated a common haplotype that was more frequent in individuals with extensive WM-PVS than in those without (0.50 versus 0.47, odds ratio (OR) = 1.19 (95% confidence interval (95% CI), 0.99–1.43)). The effect of this haplotype was amplified by 1.7 in the presence of the rs112407396-T allele (MAF = 0.03), which has a high probability of being a regulatory variant (HaploReg, GTex, RegulomeDB). Next, to account for allelic heterogeneity between ancestries, we conducted cross-ancestry meta-analyses with MR-MEGA (Methods). There were no loci showing high heterogeneity in allelic effects across ancestries (PHet < 0.01) and reaching genome-wide significance (Supplementary Table 6).
Using MAGMA and VEGAS, we performed gene-based association analyses in participants of European ancestry, testing the combined association of variants within a gene with PVS (Methods). MAGMA identified 28 gene-wide significant associations (P < 2.63 × 10−6), of which 12 in 8 loci did not reach genome-wide significance in the GWAS (WM-PVS: 3 (INS-IGF2/IGF2, PRKAG2, LRP4/CKAP5); BG-PVS: 4 (SH3PXD2A, WNT3, ZMYND15, KCNRG/TRIM13/SPRYD7); and HIP-PVS: 1 (PDZRN4); Fig. 1 and Supplementary Table 7). VEGAS identified one additional gene (NSF) for BG-PVS (same locus as WNT3; Supplementary Table 7). All were in suggestive GWAS loci (P < 5 × 10−6; Supplementary Table 8).
Using LD-score regression, we estimated heritability at 11% for WM-PVS, 5% for BG-PVS and 8% for HIP-PVS (Methods and Supplementary Table 9). We found moderate genetic correlation between BG-PVS and HIP-PVS (rg (SE) = 0.63 (0.14), P = 7.23 × 10−6), and modest genetic correlation of WM-PVS with BG-PVS (rg (SE) = 0.24 (0.12), P = 0.055) and HIP-PVS (rg (SE) = 0.27 (0.09), P = 0.003). The genetic correlation of PVS in CHARGE and UKB was moderate to high for WM-PVS and HIP-PVS and weaker for BG-PVS (Supplementary Table 10). Associations with genome-wide significant PVS loci were highly consistent between the UKB and CHARGE contributions and between the two dichotomous and the continuous PVS measures in UKB (Methods and Supplementary Tables 11 and 12). In sensitivity analyses in two representative cohorts (UKB and 3C-Dijon), continuous and dichotomous PVS measures were strongly correlated (Spearman’s ρ, 0.61–0.80; Supplementary Table 13).
To increase statistical power, we conducted secondary multivariate association analyses using Multi-Trait Analysis of GWAS (MTAG) (Methods), including summary statistics from GWAS of other cSVD markers (WMH volume, lacunes; Supplementary Table 14). We observed the highest gain in power for BG-PVS: ten additional loci reached genome-wide significance, of which two also for HIP-PVS (STN1, DEGS2/EVL). Two MTAG BG-PVS loci (CACNB2, NSF/WNT3) and one MTAG WM-PVS locus (VWA2) were not described before with any MRI marker of cSVD. Six loci showed greater significance in MTAG than with PVS, WMH volume or lacunes alone: at VWA2 (WM-PVS); SH3PXD2A/STN1, COL4A2, CACNB2 and NSF/WNT3 (BG-PVS); and DEGS2/EVL (BG-PVS, HIP-PVS).
Using WES data and exome content of WGS data in 19,010 participants from UKB and the Brain Imaging, cognition, dementia, and next-generation genomics (BRIDGET) consortium (Methods and Supplementary Table 1), of whom 4,531, 4,424 and 4,497 had extensive PVS in WM, BG and hippocampus, we identified 19 variants in the chr1q25.3 locus associated with HIP-PVS, including two missense variants (rs20563 and rs20558) and one splice donor insertion (rs34133998) in LAMC1 at P < 5 × 10−8, in strong LD with the GWAS sentinel variant (Supplementary Table 15a). Gene-based burden tests exploring protein-modifying rare variants (MAF < 0.01) did not identify any gene-wide significant association (Supplementary Table 15b).
Follow-up of findings across the lifespan and ancestries
We explored associations of WM-PVS and BG-PVS risk variants with these phenotypes in young adults (Internet-based Students’ HeAlth Research Enterprise (i-Share) study, N = 1,748, 22.1 ± 2.3 yr) and in older Japanese community-dwelling people (Nagahama study, N = 2,862, 68.3 ± 5.3 yr; Methods). We used an artificial intelligence-based method to derive quantitative WM-PVS and BG-PVS burden (HIP-PVS not available) and dichotomized it (top quartile versus the rest; Supplementary Table 2). In total, 67% of WM-PVS loci reached nominally significant associations in at least one of the two follow-up cohorts (P < 0.05 in i-Share and/or Nagahama), 43% of which at P < 1.09 × 10−3 (correcting for the number of loci tested), with consistent directionality of effect (a binomial test showed significant concordance of risk alleles; Supplementary Table 12b). In i-Share, 52% of WM-PVS risk variants were associated with WM-PVS (P < 0.05, of which 4 at P < 1.09 × 10−3; Table 2 and Supplementary Table 12a). A WM-PVS rescaled weighted genetic risk score (rwGRS) derived from European GWAS loci was associated with WM-PVS in i-Share (OR = 1.16 (95% CI, 1.08–1.24), P = 5.89 × 10−6 and β (SE) = 0.064 (0.007), P = 2.06 × 10−19 for dichotomous and continuous measures; Supplementary Fig. 2). Although meta-regression suggested larger effect sizes at younger ages for lead variants at OPA1 and SLC13A3, differences were not significant after removing the much younger i-Share cohort (Supplementary Fig. 3). In Nagahama, out of 15 available WM-PVS risk loci (six were rare or monomorphic), eight loci (53%) were associated with continuous PVS burden at P < 0.05, of which four at P < 1.09 × 10−3 and one at genome-wide significance (at SLC13A3; Table 2 and Supplementary Table 12a). A European WM-PVS weighted genetic risk score (wGRS) combining 14 independent loci (1000 Genomes project (1000G) Japanese reference panel) was associated with WM-PVS in Nagahama (OR = 1.18 (95% CI, 1.13–1.24), P = 5.68 × 10−13 and β (SE) = 0.01 (0.001), P = 7.18 × 10−18 for dichotomous and continuous measures). Although HIP-PVS data were not available in the follow-up cohorts, two of the three HIP-PVS loci were shared with WM-PVS and replicated with that phenotype.
Clinical correlates of identified PVS loci
We examined whether PVS risk loci (lead and proxy variants with r2 > 0.9) were associated with MRI markers of brain aging, putative risk factors (vascular risk factors and sleep patterns) and common neurological diseases (stroke, Alzheimer’s disease, Parkinson’s disease), using the largest published GWAS (Methods). Of 24 independent PVS risk loci, five (21%) were significantly (P < 3.3 × 10−5) associated with WMH volume and five (21%) with blood pressure traits (in the same and opposite directions; Fig. 2). Colocalization analyses suggested a shared causal variant for two-thirds of these associations (posterior probability for a shared causal variant, PP4 > 0.75; Supplementary Table 16). Sixteen PVS loci (67%) did not show any association with vascular or neurological traits, thus pointing to pathways that do not seem mediated by established risk factors (Methods and Supplementary Tables 16 and 17).

a, Venn diagram displaying significant association of genome-wide significant risk loci for PVS burden with vascular risk factors and other MRI markers of cSVD: in italics for BG-PVS; underlined for HIP-PVS; underlined and in bold for HIP- and WM-PVS; all others for WM-PVS only (P < 3.3 × 10−5, two-sided, correcting for multiple testing (21 independent phenotypes, 3 PVS locations and 24 independent loci)); *6 independent loci; **2 independent loci; †genome-wide significant in Europeans only; ‡in colocalization analyses the posterior probability PP4 was higher than 75% for these loci (only with WMH at NBEAL1-ICA1L). Exact P values are provided in Supplementary Table 16. b, Direction of association and level of significance of pleiotropic SNPs displayed in a: in red when the risk allele for extensive PVS burden is positively associated with the trait, in blue when the PVS risk allele is negatively associated with the trait (unexpected direction), in dark red and dark blue for genome-wide significant associations and in light red and light blue for significant association after multiple-testing correction (P < 3.3 × 10−5, two-sided, correcting for multiple testing (21 independent phenotypes, 3 PVS locations and 24 independent loci)). PP, pulse pressure; BMI, body mass index; LDL, LDL cholesterol.
Second, we explored genetic correlations of PVS burden with the same traits using LD-score regression (Methods, Fig. 3 and Supplementary Table 9). We observed significant (P < 7.9 × 10−4) genetic correlation of BG-PVS with larger WMH and caudate nucleus volumes, and of HIP-PVS with larger hippocampal volume. BG-PVS and HIP-PVS showed significant genetic correlation with higher systolic blood pressure (SBP), diastolic blood pressure (DBP), any stroke and ischemic stroke, and nominally significant genetic correlation with (deep) intracerebral hemorrhage (ICH). Genetic correlations were consistent in secondary analyses conducted separately in CHARGE and UKB (Supplementary Table 9).

a–c, Genetic correlation using LD-score regression of extensive PVS burden with putative risk factors (a), neurological diseases (b) and other MRI markers of brain aging (c); two-sided exact P values are provided for nominally significant results (*P < 0.05) and significant results after multiple-testing correction (**P < 7.9 × 10−4, correcting for 21 independent phenotypes and the three PVS locations); full results are provided in Supplementary Table 9. Larger colored squares correspond to more significant P values and the colors represent the direction of the genetic correlation (positive in red, negative in blue). HDL, high-density lipoprotein; amygdala, accumbens (nucleus), caudate (nucleus), pallidum, and putamen correspond to the volumes of these subcortical structures.
Third, we used two-sample Mendelian randomization (MR) to seek evidence for a causal association of putative risk factors with PVS burden and of PVS burden with neurological diseases, using generalized summary-data-based MR (GSMR), and confirming significant associations (P < 1.19 × 10−3) with RadialMR, TwoSampleMR and MR-CAUSE (Methods). Genetically determined higher SBP and DBP were consistently associated with BG-PVS, HIP-PVS and WM-PVS, although for WM-PVS the association with SBP was only nominally significant in RadialMR (Supplementary Table 18 and Extended Data Fig. 2). There was no evidence for reverse causation using MR-Steiger, but some evidence of residual pleiotropy after removal of outlier variants for SBP and DBP (RadialMR), with significant evidence for a causal model in MR-CAUSE for BG-PVS. Genetic liability to BG-PVS and HIP-PVS derived from a multi-trait analysis accounting for other MRI markers of cSVD (MTAG) was associated with an increased risk of any stroke, ischemic stroke and small vessel stroke (SVS) for BG-PVS, and SVS for HIP-PVS, suggesting that shared pathways between PVS, WMH and lacunes may be causally associated with stroke (Supplementary Table 18 and Extended Data Fig. 3). In multivariable MR analyses accounting for SBP and DBP, genetic liability to BG-PVS and HIP-PVS was significantly associated with an increased risk of any stroke, ischemic stroke and SVS (Supplementary Table 19).
Functional exploration of identified PVS loci
Using MAGMA and VEGAS2Pathway (Methods), we identified significant enrichment of PVS loci in pathways involved in extracellular matrix (ECM) structure and function, lymphatic endothelial cell differentiation, cell motility and thyroid hormone transport (Supplementary Tables 20 and 21).
Genes closest to PVS lead risk variants were significantly enriched in genes mutated in Online Mendelian Inheritance in Man (OMIM) syndromes associated with leukodystrophy, leukoencephalopathy or WMH, with a 20-fold enrichment in genes containing an intragenic lead variant. This enrichment was 30-fold when focusing on WM-PVS loci only, comprising several genes involved in early-onset leukodystrophies: GFAP (chr17q21.31), mutations of which cause Alexander disease, a rare neurodegenerative disorder of astrocytes leading to psychomotor regression and death; SLC13A3 (chr20q13.12), causing acute reversible leukoencephalopathy with increased urinary alpha-ketoglutarate; and PNPT1 (chr2p16.1), causing Aicardi–Goutières syndrome and cystic leukoencephalopathy (Methods, Extended Data Fig. 4 and Supplementary Table 22). Although several genes near PVS lead risk variants were described to be involved in glioma, we found no significant enrichment for glioma genes (Methods).
To seek evidence for a causal implication of specific genes and variants, we performed transcriptome-wide association studies (TWAS) using TWAS-Fusion (Methods), with European PVS GWAS summary statistics and the GTEx v7 multi-tissue (RNA sequencing) database, focusing on brain, vascular and blood tissues. We found 36 transcriptome-wide significant expression–trait associations for WM-PVS, 25 for BG-PVS and seven for HIP-PVS that were significant in colocalization analyses (TWAS-COLOC), providing evidence of a shared causal variant between the corresponding gene expression and PVS (Supplementary Table 23). Most genes with significant expression–trait associations (12) were in genome-wide significant PVS risk loci: eight genes in five WM-PVS GWAS loci (C6orf195, ITGB5, LPAR1, LRRC25, RP11-71H17.9, SLC20A2, SMIM19, UMPS), two genes in one BG-PVS GWAS locus (ICA1L, NBEAL1) and two genes in an HIP-PVS GWAS locus (LAMC1 and RP11-181K3.4), while nine were outside GWAS loci, requiring confirmation (Fig. 4). TWAS-COLOC signals were mostly observed in brain tissues (17 genes), but also in vascular tissues (ten genes) and blood (two genes).

We used precomputed functional weights from 22 publicly available gene expression reference panels from brain (GTEx v7, CommonMind Consortium (CMC)), peripheral nerve tissues (GTEx v7), heart and arteries (GTEx v7), and blood (Netherlands Twin Registry (NTR) and Young Finns Study (YFS)). Transcriptome-wide significant genes (eGenes) and the corresponding eQTLs were determined using Bonferroni correction, based on the average number of features (4,235 genes) tested across all tissues and correcting for the three independent PVS locations (P < 3.93 × 10−6). *Significant result in the TWAS and conditional analyses; **significant result in the TWAS and conditional analyses, and with a COLOC PP4 > 0.75; eGenes for loci identified in the GWAS (†), gene-based test (‡) or both GWAS and gene-based test (§).
To identify enrichment in specific brain cell types, we used a recently developed pipeline combining three cell type enrichment methods, stratified LD-score, MAGMA and H-MAGMA (Supplementary Table 24). We observed significant enrichment in brain vascular endothelial cells for all PVS locations, based on a human single-cell atlas of fetal gene expression, and in pericytes and astrocytes for WM-PVS (Supplementary Tables 24 and 25).
We explored brain expression patterns from development to adulthood of genes nearest to PVS loci, prioritizing TWAS-COLOC genes (Methods). Several genes showed important variations in expression levels throughout the life course, some peaking in the prenatal period (for example, LAMC1, UMPS), suggestive of developmental mechanisms (Extended Data Fig. 5 and Supplementary Fig. 4).
Finally, we conducted an exploratory search for enrichment of PVS genes in targets of drugs validated in other indications (Methods). We found significant enrichment of BG-PVS genes in targets for anti-infectives, driven by CRHR1 (chr17q21.31, target for telavancin), and for diseases of the nervous system, driven by MAPT (chr17q21.31, target for davunetide); and of HIP-PVS genes in targets for ear disease drugs, driven by SERPIND1 (chr22q11.21, target for sulodexide, also used for venous thrombosis prevention; Extended Data Figs. 6 and 7). We also observed significant enrichment of TWAS-significant HIP-PVS genes in vascular disease drugs, including simvastatin, vincamine and macitentan (Extended Data Fig. 8).
Discussion
In up to 40,095 participants from older population-based cohorts, we identified 24 genome-wide significant risk loci for extensive PVS burden, predominantly for WM-PVS, and six additional loci after accounting for other MRI markers of cSVD. Consistent with distinct risk factor profiles2,10, the genetic architecture of PVS differed across PVS locations, with WM-PVS showing the highest heritability and low genetic correlation with BG-PVS and HIP-PVS1,2,16. In line with the hypothesis that PVS is a marker of cSVD, moderate to high genetic correlation was observed with other MRI markers of cSVD, primarily for BG- and HIP-PVS. Pathway analyses highlight ECM structure and function, known to play an important role in cSVD5,20,21, and several loci include genes involved in the matrisome (ECM and associated proteins), perturbations of which were proposed as a convergent pathologic pathway in cSVD (LAMC1, EFEMP1, COL4A2, SH3PXD2A, VWA2)5,21. Several PVS risk loci (at FOXF2, EFEMP1, KCNK2 and NBEAL1-ICA1L) are known risk loci for other cSVD features (WMH, SVS)5,22,23, and mutations in two MTAG genes cause monogenic SVD (at COL4A1-COL4A2 and STN1)24,25.
PVS have been described early in life7,26, but their clinical significance at young ages is unknown. Our results suggest shared molecular mechanisms underlying PVS in young and older age. This corroborates recently described associations of WMH risk variants with changes in MRI-detected WM microstructure at age 20 yr (ref. 5). The significant enrichment of PVS risk loci in genes involved in early-onset leukodystrophies and expressed in fetal brain vascular endothelial cells supports involvement of developmental processes. In spontaneously hypertensive stroke-prone rats, closely modeling cSVD, intrinsic endothelial cell dysfunction was observed at birth, including reduced tight junctions, as well as altered oligodendrocyte maturation and myelination27. At the most significant WM-PVS locus in young adults, OPA1 harbors mutations causing autosomal-dominant optical atrophy, sometimes associated with multiple sclerosis-like illness, parkinsonism and dementia28, and endothelial OPA1 plays an important role in developmental angiogenesis29. These observations corroborate epidemiological associations of early-life factors with cSVD severity in older age30.
The present effort has the largest East-Asian contribution compared with other large GWAS of MRI-defined phenotypes31,32, with over half of available WM-PVS loci reaching nominally significant, directionally consistent associations in the Japanese follow-up study. The prevalence of cSVD is higher in East-Asian than European populations33. Our results are an important initial step to establish the generalizability of cSVD genetic associations across ancestries. Efforts to further enhance the non-European contribution to MRI cSVD genomic studies, including in populations of African-ancestry in whom cSVD is also more frequent34, are of paramount importance.
The combination of PVS GWAS findings with TWAS and WES/WGS strongly supports putative causal genes. WM-PVS associates with lower LPAR1 expression in vascular tissues. LPAR1 (chr9q31.3), expressed in oligodendrocytes, encodes a receptor for lysophosphatidic acid, an extracellular signaling small lipid, and is involved in postnatal myelination and functional connectivity across brain regions35. An LPAR1 antagonist was found to attenuate brain damage after transient arterial occlusion, by decreasing inflammation36, and LPAR1 modulation may also impact neural regeneration37. Several drugs targeting LPAR1 are available (for example, the antidepressant mirtazapine38) or in development39. WNT7A (chr3p25.1) encodes a secreted signaling protein that targets the vascular endothelium, and was implicated in brain angiogenesis and blood brain barrier regulation40. Loss of Wnt7a/b function in mice results in severe WM damage41.
WM-PVS was associated with lower ITGB5 (chr3q21.2) expression in whole blood. ITGB5 encodes a beta subunit of integrin, and plays a central role in monogenic SVD42. Higher ITGB5 plasma levels were associated with decreased odds of cognitive impairment or dementia, lower brain amyloid burden and slower brain atrophy rates43. HIP-PVS was associated with lower expression of LAMC1 (chr1q25.3, encoding Laminin gamma-1) in brain and higher expression in vascular tissues, while WES/WGS identified a splice donor variant at LAMC1. Laminins are ECM glycoproteins, and the major noncollagenous constituent of basement membranes. Genes encoding other basement membrane proteins (NID2, COL4A1/2) are implicated in cSVD5,22. Laminin regulates blood vessel diameter44 and blood brain barrier integrity and function45, and astrocytic laminin loss decreases expression of tight junction proteins and aquaporin-4 (AQP4)45, a key modulator of glymphatic flow in experimental models7.
Some genes point to complex pleiotropic mechanisms. At chr2q33.2, also associated with WMH, SVS, Alzheimer’s disease and caudate volume5,23,46,47, BG-PVS was associated with higher expression of ICA1L in brain tissues and of NBEAL1 in vascular tissues, similar to TWAS of WMH and SVS5,22. ICA1L (encoding islet cell autoantigen-1-like and predominantly expressed in endothelial cells) harbors mutations causing juvenile amyotrophic lateral sclerosis48, while NBEAL1 (encoding neurobeachin-like 1 protein) modulates low-density lipoprotein (LDL)-receptor expression49.
Our study points to an important involvement of solute carriers (SLCs), the largest family of transporters and candidates for drug target development50, in PVS pathophysiology. The most significant PVS risk variants involve an intronic haplotype of SCL13A3, encoding a plasma membrane Na+/dicarboxylate cotransporter expressed in kidney, astrocytes and choroid plexus51. Mutations in SLC13A3 cause acute reversible leukoencephalopathy with increased urinary alpha-ketoglutarate51, where SLC13A3 loss-of-function may affect elimination of organic anions and xenobiotics from the cerebrospinal fluid (CSF)51. At the same locus (Supplementary Fig. 1), other genome-wide significant variants are located near SLC2A10, harboring mutations causing arterial tortuosity syndrome52, described to be associated with PVS burden and cSVD53. WM-PVS was associated with lower SLC20A2 expression in brain tissue. SLC20A2, involved in phosphate transport, harbors loss-of-function mutations causing idiopathic familial BG calcification, a neurodegenerative disorder with inorganic phosphate accumulation in the ECM54. Given their role in CSF secretion and substance transport at the blood–CSF barrier55, SLCs could be involved in interstitial fluid accumulation adjacent to the PVS56.
Consistent with other SVD phenotypes, we observed evidence for a causal association of blood pressure with PVS. Experimental work suggests that the perivascular pump becomes less efficient with increasing blood pressure, reducing net forward flow in the PVS. These effects were found to be larger at more distal locations, where arteries have thinner and less muscular walls57. Such hemodynamic and anatomic differences1,2,18 could, perhaps, at least partly explain the more significant association of blood pressure with BG-PVS and HIP-PVS compared with WM-PVS. In contrast, WM-PVS were previously found to be associated with CAA11 and with higher brain amyloid deposition on positron emission tomography, across the clinical spectrum of CAA12. The updated Boston Criteria (v.2.0) for CAA include severe WM-PVS as a new diagnostic criterion13.
The clinical relevance of PVS is strongly supported by the significant genetic correlation of BG-PVS and HIP-PVS with any stroke and ischemic stroke and robust evidence for a possible causal association of BG-PVS and HIP-PVS with any stroke, ischemic stroke and SVS, accounting for blood pressure. The nominally significant genetic correlation of BG-PVS and HIP-PVS with (deep) ICH, based on smaller GWAS and thus less statistical power, is also consistent with epidemiological findings10. Considering the association of HIP-PVS with lower LAMC1 expression in brain, it is striking to note that conditional knock-out of laminin in astrocytes leads to deep ICH in mice58. This is reminiscent of known associations of variants in COL4A1/A2, encoding another basement membrane protein, with monogenic and multifactorial deep ICH46,59.
Significant enrichment of PVS genes in targets of drugs validated or under investigation for vascular and cognitive disorders (for example, telavancin and davunetide) highlights the potential of PVS genetics for cSVD drug discovery.
To our knowledge, this is the first study exploring the genetic determinants of PVS, using a comprehensive gene-mapping strategy and extensive bioinformatics follow-up. We acknowledge limitations. To account for heterogeneity in PVS quantification methods, we pragmatically dichotomized PVS variables based on the top quartile of the distribution, which may be less powerful than continuous measures. This may have been most prominent for BG-PVS, for which the genetic correlation pattern between CHARGE and UKB was low, in contrast with WM-PVS and HIP-PVS. Reassuringly, loci identified using dichotomous PVS phenotypes were also associated with continuous PVS burden in studies where computational methods were available (UKB, i-Share, Nagahama), mostly with more significant P values. A conservative approach will also have helped minimize the effect of accidentally including WMH in the PVS measures, a problem which some computational PVS methods have not yet overcome. Strikingly, 67% of WM-PVS loci were associated at least nominally with WM-PVS in one or both follow-up cohorts, despite considerably smaller samples and distinct age and ancestry, with consistent directionality. This suggests that our genomic discovery approach, although likely conservative, led to robust findings. With increasing development of artificial intelligence-based computational methods for PVS quantification, future genomic studies will likely have even greater power to detect genetic associations, to enable studying the genomics of total PVS volume, accounting for differences in individual PVS volume, width, length, shape60, density, location and anatomical predominance, and to run sex-specific analyses.
In conclusion, in this gene-mapping study of PVS, one of the earliest MRI markers of cSVD, we describe 24 genome-wide significant risk loci, with six additional loci in secondary multivariate analyses accounting for other cSVD markers. Our findings provide insight into the biology of PVS across the adult lifespan and its contribution to cSVD pathophysiology, with potential for genetically informed prioritization of drug targets for prevention trials of cSVD, a major cause of stroke and dementia worldwide.
Methods
Study design
This study complies with all relevant ethical regulations, and all participants gave written, informed consent. Analyses were performed on stroke-free participants from 22 population-based cohorts (18 for the GWAS meta-analysis), taking part in UKB, the CHARGE consortium and the BRIDGET initiative. Institutional review boards approved individual studies: UKB (National Research Ethics Service Committee North West–Haydock), 3C-Dijon (Ethical Committee of the University Hospital of Kremlin-Bicêtre), Austrian Stroke Prevention Study and Austrian Stroke Prevention Family Study (ASPS/ASPS-Fam) (Ethics Committee of the Medical University of Graz), Epidemiology of Dementia in Singapore Study (EDIS) (the Singapore Chinese Eye Study/Singapore Malay Eye Study-2, Singapore Eye Research Institute and the National Healthcare Group Domain-Specific Review Board), Framingham Heart Study (FHS) (Institutional Review Board of Boston University Medical Center), Investigating Silent Strokes in Hypertensives Study (ISSYS) (Comité de ética de investigacion con medicamentos, Hospital Universitari Vall d’Hebron), Lothian Birth Cohort 1936 (LBC1936) (Lothian and Scottish Multicentre Research Ethics Committees), Northern Manhattan Study (NOMAS) (Columbia University Medical Center Institutional Review Board and the University of Miami Institutional Review Board), Rotterdam Study I, II and III (RS-I, RS-II and RS-III) (Ministry of Health, Welfare, and Sport of the Netherlands), Study of Health in Pomerania (SHIP) (SHIP-2, SHIP-Trend Batch 1 and 2, Ethics Commission of the University of Greifswald), i-Share study (Comités de Protection des Personnes (CPP) Sud-Ouest Outre-Mer III, Sydney Memory and Ageing Study (MAS) (Ethics Committees of the University of New South Wales, South-Eastern Sydney, and the Illawarra Area Health Service), Older Australian Twins Study (OATS) (Ethical Committees of the Australian Twin Registry, the University of New South Wales, the University of Melbourne, the Queensland Institute of Medical Research, and the South-Eastern Sydney and Illawarra Area Health Service) and the Nagahama Study (Ethics Committee of Kyoto University Graduate School of Medicine and the Nagahama Municipal Review Board) (Supplementary Table 1). Characteristics of study participants are provided in Supplementary Tables 1–3 and 26 and Supplementary Fig. 5.
PVS burden definition
PVS were defined as fluid-filled spaces with a signal identical to that of CSF, of round, ovoid or linear shape depending on the slice direction, with usually a maximum diameter smaller than 3 mm, no hyperintense rim on T2-weighted or FLAIR sequences, and located in areas supplied by perforating arteries3. In most CHARGE cohorts, visual semiquantitative rating scales were used to quantify PVS burden. As different scales were used across studies, we dichotomized PVS burden into ‘extensive PVS burden’ versus the rest, defined by a cut-off closest to the top quartile of the semiquantitative scale distribution within each cohort (Supplementary Tables 2, 27 and 28). This cohort-specific threshold definition was chosen because (1) small PVS counts are very sensitive to MRI field strength and less prominently associated with age and vascular risk factors61; (2) extreme burden of other MRI markers of cSVD (for example, extensive WMH burden within the top quartile of the distribution) was previously shown to facilitate the identification of genetic variants underlying cSVD62; and (3) PVS burden is highly dependent on participant characteristics, especially age, PVS quantification methods and image acquisition parameters. In RS-III and in UKB, a recently developed automated method was used to quantify the number of PVS (Supplementary Table 27), dichotomized according to the same cut-off (top quartile). For sensitivity analyses, we also compared results obtained in UKB with the dichotomized and continuous (log-transformed) PVS variables.
Covariates and descriptive variables
Intracranial volume (sum of gray matter, WM and CSF volumes) was available in all studies except ASPS, where brain parenchymal fraction was used (ratio of brain parenchymal tissue volume to total volume within the surface contour of the whole brain). Other covariates are described in Supplementary Table 1.
Genotyping and imputation
Genome-wide genotypes were imputed to the 1000G project (1000G pIv3) or the Haplotype Reference Consortium reference panels (Supplementary Table 3).
PVS genome-wide association analyses in individual cohorts
Ancestry-specific logistic regression analyses with an additive genetic model were performed, adjusting for age, sex (genetically determined) and intracranial volume (or brain parenchymal fraction for ASPS), principal components of population stratification, and study site.
As sensitivity analyses, we ran linear mixed models in UKB, (1) using the log-transformed (log(variable + 1)) continuous PVS measurements, adjusting for the same covariates as above; (2) generating residuals adjusting for the same covariates and then dichotomizing the residuals (instead of adjusting for covariates after dichotomization).
PVS genome-wide association meta-analyses
We performed quality control in each study following the recommendations of Winkler et al.63. Analyses were done on autosomal biallelic markers. Duplicate markers were removed, marker names and alleles were harmonized across studies, and PZ-plots, quantile–quantile plots and allele frequency plots were constructed63. In each study, rare variants (MAF < 0.01) and variants with low imputation accuracy (R², oevar_imp or info score < 0.5) or extensive effect size values (β > 5 or β < −5) were removed. We reported the number of SNPs passing quality control for each study (Supplementary Table 4). GWAS were run within each cohort using logistic regression (or linear regression for sensitivity analyses), using software described in Supplementary Table 3. We then conducted GWAS meta-analyses across participating cohorts in METAL, using sample size-weighted meta-analysis as PVS were measured on different scales. Meta-analyses were conducted within each ancestry (European (EUR), Asian (ASN), African-American (AA), Hispanic (HISP)) using METAL (https://github.com/statgen/METAL), followed by meta-analyses across ancestries. Ancestry was genetically inferred using principal components of population stratification (Supplementary Tables 1 and 3). Genomic control was applied to each study-specific GWAS with a genomic inflation factor greater than 1.00. Variants with an effective allele count (twice the product of MAF, imputation accuracy and number of participants with extensive PVS) < 10 and significant heterogeneity (PHet < 5.0 × 10−8) were excluded from the meta-analysis. We performed LD-clumping, sorting the genome-wide significant SNPs by P value, keeping the most significant SNP and removing SNPs with an r² > 0.1 within 1 megabase (Mb). Only variants present in at least half of participants of the final meta-analysis were used to construct quantile–quantile and Manhattan plots. In secondary analyses, we ran inverse variance-weighted meta-analyses to obtain effect estimates and standard errors for follow-up bioinformatics analyses.
Conditional and joint multiple-SNP analysis
We used GCTA-COJO64 to perform conditional and joint multiple-SNP analysis of PVS GWAS summary statistics, to identify secondary association signals at each of the genome-wide significant loci within 1 Mb of the lead SNP. We used European GWAS summary statistics as recommended to avoid population stratification. This method relied on a stepwise selection procedure to select SNPs based on the conditional P values, and the joint effects of all selected SNPs after optimization of the model were estimated64. We used genotypes of 6,489 unrelated participants of European ancestry from the 1000G-imputed 3C-Dijon study data for LD correction. We performed haplotype association analyses on the six independent lead variants at chr20q13.12 (Supplementary Table 5b).
Cross-ancestry meta-regression of GWAS
We conducted cross-ancestry meta-analyses using MR-MEGA65, which uses meta-regression to model allelic effects, including axes of genetic variation as covariates in the model.
Gene-based analyses
We performed gene-based analyses on European PVS GWAS meta-analyses. We included variants within 10 kilobase (kb) of the 3′ and 5′ untranslated regions (UTRs) of a gene to capture regulatory variants. We used the MAGMA software implemented in FUMA66 to perform a gene-based association study, including 19,037 protein-coding genes. This method is based on a multiple linear principal components regression model. Gene-wide significance was defined at P < 2.63 × 10−6. We also performed gene-based tests using VEGAS2 (ref. 67), including 18,371 autosomal genes, leading to a gene-wide significance at P < 2.72 × 10−6. Genes were considered in the same locus if they were within 200 kb of each other.
PVS heritability estimates
We used LD-score regression (ldsc package https://github.com/bulik/ldsc/) to estimate the heritability of extensive PVS burden in each location, overall and, in secondary analyses, separately, in CHARGE and UKB.
Multi-trait GWAS with PVS and other MRI markers of cSVD
We conducted a joint analysis of summary statistics from GWAS of PVS, WMH and lacunes using MTAG68, with the expectation to gain in power because of the genetic correlation between these MRI markers of cSVD. MTAG is a generalization of inverse variance-weighted GWAS meta-analysis of two or more traits, which accounts for sample overlap between GWAS results for different traits by employing LD-score regression. MTAG is based on the assumption that all SNPs share the same variance–covariance matrix of effect sizes across traits. We prioritized variants with a P < 5 × 10−8 in the PVS MTAG analysis and P < 0.05 in the univariate PVS GWAS, which showed greater significance for association with PVS in MTAG than in univariate analyses for PVS, WMH and lacunes.
PVS next-generation sequencing association analyses
Using WES data and exome content of WGS data in 19,010 participants from UKB and BRIDGET, of whom 4,531, 4,424 and 4,497 had extensive PVS in WM, BG and HIP, respectively, we performed a whole-exome association study to identify (rare) exonic variants associated with extensive PVS (Supplementary Tables 1 and 29).
Follow-up of findings across lifespan and ancestries
We explored associations of WM-PVS and BG-PVS risk variants identified in the GWAS meta-analysis with these phenotypes in young adults (i-Share study, N = 1,748, mean age 22.1 ± 2.3 yr) and in older Japanese population-based cohort participants (Nagahama study, N = 2,862, 68.3 ± 5.3 yr; Supplementary Tables 1 and 3). In each study, we used both quantitative PVS measurements derived from a computational artificial intelligence-based method (Supplementary Tables 27 and 28) and dichotomized PVS burden (top quartile of PVS distribution; Supplementary Table 2). HIP-PVS data were not available. Continuous PVS measurements were log-transformed (log(variable + 1)) to obtain a normal distribution.
In i-Share participants of European ancestry, we also explored the association of WM-PVS with a wGRS of WM-PVS burden derived from the 21 independent genome-wide significant SNPs identified in the European GWAS meta-analysis (r² < 0.10 based on the 1000G European reference panel). SNPs were weighted by the SNP effect sizes in the European GWAS meta-analysis (for the allele associated with larger PVS burden); the wGRS was rescaled (rwGRS) so that one unit of the wGRS corresponds to one additional WM-PVS risk allele. We tested for significant modifying effects of age on associations with WM-PVS for the three genome-wide significant WM-PVS loci in young adults (at chr2p16.1, chr3q29 and chr20q13.12). We collected effect estimates and standard errors for the lead SNPs at these three loci in each individual cohort, and fitted a meta-regression of the lead SNPs’ effect sizes onto an intercept and age. Meta-regression analysis was performed using Metafor69, and any statistical evidence of linear association was corrected for multiple testing (P < 0.05/3 = 1.7 × 10−2).
In Nagahama we explored the association of WM-PVS with a rwGRS of WM-PVS burden, including the 14 available independent SNPs identified in the European GWAS meta-analysis (r² < 0.10 based on 1000G Japanese reference panel); SNPs were weighted by the SNP effect sizes in the European GWAS meta-analysis.
Shared genetic variation with other phenotypes
In the European ancestry meta-analysis, we explored shared genetic variation with vascular and neurological phenotypes: (1) putative risk factors (SBP, DBP, pulse pressure, body mass index, high-density lipoprotein cholesterol, LDL cholesterol, triglycerides, type 2 diabetes and sleep patterns); (2) other MRI markers of brain aging (WMH burden, covert MRI-defined brain infarcts and lacunes, and hippocampal, nucleus accumbens, amygdala, caudate nucleus, pallidum and putamen volumes); and (3) the most common neurological conditions previously reported to be associated with PVS, namely stroke (any stroke, any ischemic stroke, large artery stroke, cardio-embolic stroke, SVS, ICH), Alzheimer’s disease and Parkinson’s disease (Supplementary Table 30).
We explored whether genome-wide significant PVS risk loci (lead variants or in LD with r² > 0.9, based on the 1000G European reference panel) were associated with these traits. A P value threshold <3.3 × 10−5, correcting for 21 independent phenotypes, three PVS locations and 24 independent loci tested, was used (Supplementary Table 30). We performed a colocalization analysis using COLOC to search for evidence for a single causal variant between PVS and the other phenotypes, a posterior probability (PP4) > 75% supporting a single causal variant for both traits70.
Second, we used LD-score regression (ldsc package: https://github.com/bulik/ldsc/) to estimate the genetic correlation of extensive PVS burden with these phenotypes (P < 7.9 × 10−4 was used as a significance threshold, correcting for 21 phenotypes and three PVS locations). To decrease potential bias due to poor imputation quality, the summary statistics were filtered to the subset of HapMap3 SNPs for each trait. In secondary analyses, we estimated genetic correlation of PVS burden with the same traits separately in CHARGE and UKB.
We used FUMA to obtain extensive functional annotation for genome-wide significant SNPs and to identify SNPs associated with other traits at genome-wide significance from the GWAS catalog66.
MR
We used an MR approach to explore the possible causal relation of putative risk factors (vascular risk factors and sleep patterns) with extensive PVS burden, and of extensive PVS burden with neurological traits (stroke, Alzheimer’s disease and Parkinson’s disease).
We used the GSMR method implemented in GCTA71. Summary statistics were clumped using 1000G-imputed 3C-Dijon study data (r² < 0.05 and P < 5 × 10−8) using only SNPs with MAF > 0.01. The heterogeneity in independent instrument (HEIDI)-outlier method was used to remove genetic instruments that showed pleiotropic effects on both the exposure and the outcome.
For (at least) nominally significant GSMR associations, we conducted secondary MR analyses using both TwoSampleMR and RadialMR72,73. Only independent SNPs (r2 < 0.01 based on 1000G European, window size = 1 Mb) reaching P < 5 × 10−8 in the primary meta-analysis were included as recommended. Effect estimates (β values) and SE values were derived from the inverse variance-weighted GWAS meta-analyses. With TwoSampleMR, we estimated the effect of each exposure on each outcome using weighted median, random-effect inverse variance weighting (IVW) and MR-Egger. In addition, we confirmed the directionality of the observed associations with the Steiger test74. With RadialMR (https://github.com/WSpiller/RadialMR), the putative causal effect of each exposure on each outcome was estimated using the fixed-effect IVW method using the modified second-order inverse variance weight73. Cochran’s Q statistic was used to test for the heterogeneity (P < 0.05) due to horizontal pleiotropy73. We excluded outlier SNPs, identified by regressing the predicted causal estimate against the inverse variance weights73, and re-ran IVW tests, as well as MR-Egger regression, assessing heterogeneity with Rücker’s Q′ statistic73. When the ratio of Q′ (Egger) on Q (IVW) (QR) was close to 1, indicating that both IVW and MR-Egger models fit the data equally, we selected the IVW model. We formally ruled out horizontal pleiotropy when the MR-Egger intercept after exclusion of outliers was nonsignificant (P ≥ 0.05). To account for potential residual correlated pleiotropy, we used MR-CAUSE75. Finally, we explored the association between genetic liability to PVS and stroke, conditioning on blood pressure (SBP and DBP separately), by running multivariable MR analyses using TwoSampleMR72. A P < 1.19 × 10−3, correcting for 14 independent phenotypes and the three PVS locations, was considered significant.
Pathway analyses
We used MAGMA gene set analyses (in FUMA66) to identify pathways overrepresented in the associations. We identified genes associated with extensive PVS burden and estimated the correlation between genes. The P values and gene correlation matrix were used in a generalized least squares model. A P < 3.2 × 10−6 correction for 15,496 gene sets was considered significant. As a sensitivity analysis, we used VEGAS2Pathway76, which aggregates association strengths of individual markers into prespecified biological pathways using VEGAS-derived gene association P values for extensive PVS burden, with an empirical significance threshold of P < 1 × 10−5 (accounting for 6,213 correlated pathways).
Enrichment analyses in OMIM and COSMIC genes
Using hypergeometric tests, we performed enrichment analyses of genes within 1 Mb, 100 kb or 10 kb of the lead variants, but also of genes within 10 kb of the lead variants with intragenic variants, and genes within 10 kb of the genetic loci with intragenic lead variants. We used the rest of the protein-coding genome as a reference. We performed the analysis first combining loci of all PVS locations, and second including only WM-PVS loci. We searched for an enrichment in different gene groups from the OMIM database77, including PVS (‘perivascular space’ OR ‘virchow-robin space’), WMH (‘leukoaraiosis’ OR ‘white matter lesion’ OR ‘white matter hyperintensities’) and leukodystrophy (‘leukodystrophy’ OR ‘leukoencephalopathy’) genes. We also searched for an enrichment of genes involved in glioma and glioblastoma, identified in the Catalog Of Somatic Mutations In Cancer (COSMIC) (https://cancer.sanger.ac.uk).
TWAS
We performed TWAS using TWAS-Fusion78, to identify genes whose expression is significantly associated with PVS burden without directly measuring expression levels. We restricted the analysis to tissues considered relevant for cerebrovascular disease, and used precomputed functional weights from 22 publicly available gene expression reference panels from blood, arterial, brain and peripheral nerve tissues (Fig. 4). TWAS-Fusion was then used to estimate the TWAS association statistics between predicted gene expression and PVS burden by integrating information from expression reference panels (SNP expression weights), GWAS summary statistics (SNP PVS effect estimates) and LD reference panels (SNP correlation matrix). Transcriptome-wide significant genes (eGenes) and the corresponding expression quantitative trait loci (eQTLs) were determined using Bonferroni correction (P < 3.93 × 10−6, correcting for 4,235 genes tested and three PVS locations). eGenes were then tested in conditional analyses as implemented in TWAS-Fusion. Next, we performed a genetic colocalization analysis of gene expression and PVS burden for each conditionally significant gene (P < 0.05) using COLOC70, to estimate the posterior probability of a shared causal variant between the gene expression and the trait (PP4 ≥ 0.75). Gene regions with eQTLs not reaching genome-wide significance in association with PVS, and not in LD (r2 < 0.01) with the lead SNP for genome-wide significant PVS risk loci, were considered as novel.
Cell type enrichment analysis
We conducted a cell type enrichment analysis using Single cell Type Enrichment Analysis for Phenotypes (https://github.com/erwinerdem/STEAP/). This is an extension to CELLECT and uses S-LDSC, MAGMA and H-MAGMA for enrichment analysis. PVS GWAS summary statistics were munged. Then, expression specificity profiles were calculated using human and mouse single-cell RNA sequencing databases (PsychENCODE DER-22, GSE67835, GSE101601, DroNc Human Hippocampus, Allen Brain Atlas MTG and LNG, Mousebrain, Tabula Muris, Descartes Human Cerebrum and Cerebellum; Supplementary Table 24). Cell type enrichment was calculated with MAGMA, H-MAGMA (incorporating chromatin interaction profiles from human brain tissues in MAGMA) and stratified LD-score regression. P values were corrected for the number of independent cell types in each database.
Lifetime brain gene expression profile
We studied the lifetime expression of genes identified in the TWAS-COLOC analysis, and the three genes associated with WM-PVS burden in both the old and young populations, to search for developmental processes. We used a public database (https://hbatlas.org/) comprising genome-wide exon-level transcriptome data from 1,340 tissue samples from 16 brain regions (cerebellar cortex, mediodorsal nucleus of the thalamus, striatum, amygdala, hippocampus and 11 neocortex areas) of 57 postmortem human brains, from embryonic development to older adults of different ancestries.
Enrichment in drug target genes
We used the GREP (Genome for Repositioning)79 software tool, which quantifies an enrichment of gene sets from GWAS summary statistics in drugs of certain Anatomical Therapeutic Chemical Classification (ATC) classes, or indicated for some ICD10 (10th revision of the International Statistical Classification of Diseases and Related Health Problems) disease categories, and captures potentially repositionable drugs targeting the gene set. Genes with false discovery rate FDR q < 0.1 in MAGMA were used for enrichment analyses (in GREP) of target genes for approved or investigated drugs curated in DrugBank and the Therapeutic Target Database.
We used the Trans-Phar (integration of TWAS and Pharmacological database) software to identify drug target candidates in a specific tissue or cell type80, using first FOCUS to identify up- and downregulated genes in participants with extensive PVS burden, followed by a negative Spearman’s rank correlation analysis between the gene expression (Z-score) of the top 10% genes with the highest expression variation and the LINCS CMap L1000 library database (Extended Data Fig. 8).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
Genome-wide summary statistics for the European and cross-ancestry meta-analysis generated and analyzed during the current study are deposited on the GWAS Catalog (study code GCST90244151-GCST90244156). As for other meta-analyses of GWAS or sequencing data, individual cohort data are subject to controlled access, for privacy and legal issues (national and European regulations, including GDPR). This applies to all participating cohorts (cohorts included in the meta-analyses and follow-up cohorts). UKB data (GWAS and sequencing) are accessible by submitting an application to the UKB portal (this research has been conducted under Application Number 23509). We used publicly available data for analyses described in this manuscript, including data from GTEx (https://gtexportal.org/home/), the Gusev laboratory (http://gusevlab.org/projects/fusion/), the CommonMind Consortium (https://www.nimhgenetics.org/resources/commonmind), the Netherlands Twin Registry (https://tweelingenregister.vu.nl/), the Young Finns Study (https://youngfinnsstudy.utu.fi/), OMIM (https://www.omim.org/), OMIM genes description are publicly available: GFAP (https://www.omim.org/entry/137780); SLC13A3 (https://www.omim.org/entry/618384); PNPT1 (https://www.omim.org/entry/610316), COSMIC (https://cancer.sanger.ac.uk), RNA sequencing datasets: PsychENCODE DER-22 (www.ncbi.nlm.nih.gov/geo/, accession code GSE97942), GSE67835 (www.ncbi.nlm.nih.gov/geo/, accession code GSE67835), GSE101601, DroNc_Human Hippocampus (https://www.gtexportal.org/home/datasets), Allen Brain Atlas (http://portal.brain-map.org/), Descartes_Human (https://descartes.brotmanbaty.org/), Mousebrain (http://mousebrain.org/), Tabula Muris (https://tabula-muris.ds.czbiohub.org/). All other data supporting the findings of this study are available within the article, the supplementary information or the supplementary data files.
Code availability
The PVS quantification method used in the Nagahama Study is available using this link: https://github.com/pboutinaud/SHIVA_PVS. We used publicly available data from METAL (https://github.com/statgen/METAL), GCTA-cojo (https://yanglab.westlake.edu.cn/software/gcta/#COJO), FUMA (https://fuma.ctglab.nl/), MAGMA (https://ctg.cncr.nl/software/magma), LD-Score Regression (https://github.com/bulik/ldsc/), MTAG (https://github.com/JonJala/mtag), coloc (https://chr1swallace.github.io/coloc/) R package, RadialMR (https://github.com/WSpiller/RadialMR), TwoSampleMR (https://mrcieu.github.io/TwoSampleMR/), STEAP (https://github.com/erwinerdem/STEAP/), CELLECT (https://github.com/perslab/CELLECT), S-LSC (https://github.com/bulik/ldsc), H-MAGMA (https://github.com/thewonlab/H-MAGMA), HBT (https://hbatlas.org/). Drug discovery analysis was conducted using the following publicly available tools: GREP (https://github.com/saorisakaue/GREP), Trans-Phar (https://github.com/konumat/Trans-Phar).
References
Pollock, H., Hutchings, M., Weller, R. O. & Zhang, E. T. Perivascular spaces in the basal ganglia of the human brain: their relationship to lacunes. J. Anat. 191, 337–346 (1997).
Wardlaw, J. M. et al. Perivascular spaces in the brain: anatomy, physiology and pathology. Nat. Rev. Neurol. 16, 137–153 (2020).
Wardlaw, J. M. et al. Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 12, 822–838 (2013).
Jessen, N. A., Munk, A. S. F., Lundgaard, I. & Nedergaard, M. The glymphatic system: a beginner’s guide. Neurochem. Res. 40, 2583–2599 (2015).
Sargurupremraj, M. et al. Cerebral small vessel disease genomics and its implications across the lifespan. Nat. Commun. 11, 6285 (2020).
Debette, S., Schilling, S., Duperron, M. G., Larsson, S. C. & Markus, H. S. Clinical significance of magnetic resonance imaging markers of vascular brain injury: a systematic review and meta-analysis. JAMA Neurol. 76, 81–94 (2019).
Mestre, H., Kostrikov, S., Mehta, R. I. & Nedergaard, M. Perivascular spaces, glymphatic dysfunction, and small vessel disease. Clin. Sci. (Lond.) 131, 2257–2274 (2017).
Deramecourt, V. et al. Staging and natural history of cerebrovascular pathology in dementia. Neurology 78, 1043–1050 (2012).
Bacyinski, A., Xu, M., Wang, W. & Hu, J. The paravascular pathway for brain waste clearance: current understanding, significance and controversy. Front. Neuroanat. 11, 101 (2017).
Duperron, M. G. et al. High dilated perivascular space burden: a new MRI marker for risk of intracerebral hemorrhage. Neurobiol. Aging 84, 158–165 (2019).
Charidimou, A. et al. MRI-visible perivascular spaces in cerebral amyloid angiopathy and hypertensive arteriopathy. Neurology 88, 1157–1164 (2017).
Tsai, H. H. et al. Centrum semiovale perivascular space and amyloid deposition in spontaneous intracerebral hemorrhage. Stroke 52, 2356–2362 (2021).
Charidimou, A. et al. The Boston criteria version 2.0 for cerebral amyloid angiopathy: a multicentre, retrospective, MRI-neuropathology diagnostic accuracy study. Lancet Neurol. 21, 714–725 (2022).
Mestre, H. et al. Cerebrospinal fluid influx drives acute ischemic tissue swelling. Science 367, eaax7171 (2020).
Månberg, A. et al. Altered perivascular fibroblast activity precedes ALS disease onset. Nat. Med 27, 640–646 (2021).
Duperron, M. G. et al. Burden of dilated perivascular spaces, an emerging marker of cerebral small vessel disease, is highly heritable. Stroke 49, 282–287 (2018).
Yao, M. et al. Hippocampal perivascular spaces are related to aging and blood pressure but not to cognition. Neurobiol. Aging 35, 2118–2125 (2014).
Bouvy, W. H. et al. Visualization of perivascular spaces and perforating arteries with 7 T magnetic resonance imaging. Invest. Radiol. 49, 307–313 (2014).
Psaty, B. M. et al. Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) Consortium: design of prospective meta-analyses of genome-wide association studies from 5 cohorts. Circ. Cardiovasc. Genet. 2, 73–80 (2009).
Bordes, C., Sargurupremraj, M., Mishra, A. & Debette, S. Genetics of common cerebral small vessel disease. Nat. Rev. Neurol. 18, 84–101 (2022).
Joutel, A., Haddad, I., Ratelade, J. & Nelson, M. T. Perturbations of the cerebrovascular matrisome: a convergent mechanism in small vessel disease of the brain? J. Cereb. Blood Flow. Metab. 36, 143–157 (2016).
Traylor, M. et al. Genetic basis of lacunar stroke: a pooled analysis of individual patient data and genome-wide association studies. 20(5):351-361 Lancet Neurol. 20, 351–361 (2021).
Persyn, E. et al. Genome-wide association study of MRI markers of cerebral small vessel disease in 42,310 participants. Nat. Commun. 11, 2175 (2020).
Simon, A. J. et al. Mutations in STN1 cause Coats plus syndrome and are associated with genomic and telomere defects. J. Exp. Med. 213, 1429–1440 (2016).
Whittaker, E. et al. Systematic review of cerebral phenotypes associated with monogenic cerebral small-vessel disease. J. Am. Heart Assoc. 11, e025629 (2022).
Piantino, J. et al. Characterization of MR imaging-visible perivascular spaces in the white matter of healthy adolescents at 3T. AJNR Am. J. Neuroradiol. 41, 2139–2145 (2020).
Rajani, R. M. et al. Reversal of endothelial dysfunction reduces white matter vulnerability in cerebral small vessel disease in rats. Sci. Transl. Med. 10, eaam9507 (2018).
Carelli, V. et al. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol. 78, 21–38 (2015).
Herkenne, S. et al. Developmental and tumor angiogenesis requires the mitochondria-shaping protein Opa1. Cell Metab. 31, 987–1003.e1008 (2020).
Backhouse, E. V. et al. Early life predictors of late life cerebral small vessel disease in four prospective cohort studies. Brain 144, 3769–3778 (2021).
Zhao, B. et al. Common genetic variation influencing human white matter microstructure. Science 372, eabf3736 (2021).
Grasby, K. L. et al. The genetic architecture of the human cerebral cortex. Science 367, eaay6690 (2020).
Mok, V. et al. Race-ethnicity and cerebral small vessel disease – comparison between Chinese and White populations. Int. J. Stroke 9, 36–42 (2014).
Akinyemi, R. O. et al. Stroke in Africa: profile, progress, prospects and priorities. Nat. Rev. Neurol. 17, 634–656 (2021).
Mollink, J. et al. The spatial correspondence and genetic influence of interhemispheric connectivity with white matter microstructure. Nat. Neurosci. 22, 809–819 (2019).
Gaire, B. P., Sapkota, A., Song, M. R. & Choi, J. W. Lysophosphatidic acid receptor 1 (LPA1) plays critical roles in microglial activation and brain damage after transient focal cerebral ischemia. J. Neuroinflammation 16, 170 (2019).
Gross, I. & Brauer, A. U. Modulation of lysophosphatidic acid (LPA) receptor activity: the key to successful neural regeneration? Neural Regen. Res. 15, 53–54 (2020).
Hisaoka-Nakashima, K. et al. Mirtazapine increases glial cell line-derived neurotrophic factor production through lysophosphatidic acid 1 receptor-mediated extracellular signal-regulated kinase signaling in astrocytes. Eur. J. Pharm. 860, 172539 (2019).
Allanore, Y. et al. Lysophosphatidic acid receptor 1 antagonist SAR100842 for patients with diffuse cutaneous systemic sclerosis: a double-blind, randomized, eight-week placebo-controlled study followed by a sixteen-week open-label extension study. Arthritis Rheumatol. 70, 1634–1643 (2018).
Stenman, J. M. et al. Canonical Wnt signaling regulates organ-specific assembly and differentiation of CNS vasculature. Science 322, 1247–1250 (2008).
Chavali, M. et al. Wnt-dependent oligodendroglial-endothelial interactions regulate white matter vascularization and attenuate injury. Neuron 108, 1130–1145.e1135 (2020).
Capone, C. et al. Reducing Timp3 or vitronectin ameliorates disease manifestations in CADASIL mice. Ann. Neurol. 79, 387–403 (2016).
Tanaka, T. et al. Plasma proteomic signatures predict dementia and cognitive impairment. Alzheimers Dement. (N. Y.) 6, e12018 (2020).
Jakobsson, L., Domogatskaya, A., Tryggvason, K., Edgar, D. & Claesson-Welsh, L. Laminin deposition is dispensable for vasculogenesis but regulates blood vessel diameter independent of flow. FASEB J. 22, 1530–1539 (2008).
Yao, Y., Chen, Z. L., Norris, E. H. & Strickland, S. Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nat. Commun. 5, 3413 (2014).
Chung, J. et al. Genome-wide association study of cerebral small vessel disease reveals established and novel loci. Brain 142, 3176–3189 (2019).
Armstrong, N. J. et al. Common genetic variation indicates separate causes for periventricular and deep white matter hyperintensities. Stroke 51, 2111–2121 (2020).
Hadano, S. et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 29, 166–173 (2001).
Bindesbøll, C. et al. NBEAL1 controls SREBP2 processing and cholesterol metabolism and is a susceptibility locus for coronary artery disease. Sci. Rep. 10, 4528 (2020).
Wang, W. W., Gallo, L., Jadhav, A., Hawkins, R. & Parker, C. G. The druggability of solute carriers. J. Med. Chem. 63, 3834–3867 (2020).
Dewulf, J. P. et al. SLC13A3 variants cause acute reversible leukoencephalopathy and α-ketoglutarate accumulation. Ann. Neurol. 85, 385–395 (2019).
Beyens, A. et al. Arterial tortuosity syndrome: 40 new families and literature review. Genet. Med. 20, 1236–1245 (2018).
Chen, Y. C. et al. Correlation between internal carotid artery tortuosity and imaging of cerebral small vessel disease. Front. Neurol. 11, 567232 (2020).
Wang, C. et al. Mutations in SLC20A2 link familial idiopathic basal ganglia calcification with phosphate homeostasis. Nat. Genet. 44, 254–256 (2012).
Ho, H. T., Dahlin, A. & Wang, J. Expression profiling of solute carrier gene families at the blood-CSF barrier. Front. Pharm. 3, 154 (2012).
Wang, H. et al. Structure, function, and genomic organization of human Na+-dependent high-affinity dicarboxylate transporter. Am. J. Physiol. Cell Physiol. 278, C1019–1030 (2000).
Mestre, H. et al. Flow of cerebrospinal fluid is driven by arterial pulsations and is reduced in hypertension. Nat. Commun. 9, 4878 (2018).
Chen, Z. L. et al. Ablation of astrocytic laminin impairs vascular smooth muscle cell function and leads to hemorrhagic stroke. J. Cell Biol. 202, 381–395 (2013).
Rannikmäe, K. et al. COL4A2 is associated with lacunar ischemic stroke and deep ICH: meta-analyses among 21,500 cases and 40,600 controls. Neurology 89, 1829–1839 (2017).
Ballerini, L. et al. Computational quantification of brain perivascular space morphologies: associations with vascular risk factors and white matter hyperintensities. A study in the Lothian Birth Cohort 1936. Neuroimage Clin. 25, 102120 (2020).
Bouvy, W. H. et al. Perivascular spaces on 7 Tesla brain MRI are related to markers of small vessel disease but not to age or cardiovascular risk factors. J. Cereb. Blood Flow. Metab. 36, 1708–1717 (2016).
Mishra, A. et al. Gene-mapping study of extremes of cerebral small vessel disease reveals TRIM47 as a strong candidate. Brain 30, 1992–2007 (2022).
Winkler, T. W. et al. Quality control and conduct of genome-wide association meta-analyses. Nat. Protoc. 9, 1192–1212 (2014).
Yang, J. et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet. 44, S361–363 (2012).
Magi, R. et al. Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum. Mol. Genet. 26, 3639–3650 (2017).
Watanabe, K., Taskesen, E., van Bochoven, A. & Posthuma, D. Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017).
Mishra, A. & Macgregor, S. VEGAS2: software for more flexible gene-based testing. Twin Res. Hum. Genet. 18, 86–91 (2015).
Turley, P. et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 50, 229–237 (2018).
Viechtenbauer, W. Conducting meta-analysis in R with the metafor package. J. Stat. Softw. 36(3), 1–48 (2010).
Giambartolomei, C. et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet. 10, e1004383 (2014).
Zhu, Z. et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 9, 224 (2018).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. eLife 7, e34408 (2018).
Bowden, J. et al. Improving the visualization, interpretation and analysis of two-sample summary data Mendelian randomization via the Radial plot and Radial regression. Int. J. Epidemiol. 47, 1264–1278 (2018).
Hemani, G., Tilling, K. & Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 13, e1007081 (2017).
Morrison, J., Knoblauch, N., Marcus, J. H., Stephens, M. & He, X. Mendelian randomization accounting for correlated and uncorrelated pleiotropic effects using genome-wide summary statistics. Nat. Genet. 52, 740–747 (2020).
Mishra, A. & MacGregor, S. A novel approach for pathway analysis of GWAS data highlights role of BMP signaling and muscle cell differentiation in colorectal cancer susceptibility. Twin Res. Hum. Genet. 20, 1–9 (2017).
Amberger, J. S., Bocchini, C. A., Schiettecatte, F., Scott, A. F. & Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Res. 43, D789–798 (2015).
Gusev, A. et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat. Genet. 48, 245–252 (2016).
Sakaue, S. & Okada, Y. GREP: Genome for REPositioning drugs. Bioinformatics 35, 3821–3823 (2019).
Konuma, T., Ogawa, K. & Okada, Y. Integration of genetically regulated gene expression and pharmacological library provides therapeutic drug candidates. Hum. Mol. Genet. 30, 294–304 (2021).
Acknowledgements
Austrian Stroke Prevention Study (ASPS)/Austrian Stroke Prevention Family Study (ASPS-Fam) (E.H., P.G.G., H.S. and R.S.): We thank the staff and the participants for their valuable contributions. We thank B. Reinhart for her long-term administrative commitment, E. Hofer for the technical assistance in creating the DNA bank, J. Semmler and A. Harb for DNA sequencing and DNA analyses by TaqMan assays, and I. Poelzl for supervising the quality management processes after ISO9001 in the biobanking and DNA analyses. The Medical University of Graz and the Steiermärkische Krankenanstaltengesellschaft support the databank of the ASPS/ASPS-Fam. The research reported in this article was funded by the Austrian Science Fund (FWF) (grant nos. PI904, P20545-P05 and P13180) and supported by the Austrian National Bank Anniversary Fund (grant no. P15435) and the Austrian Ministry of Science under the aegis of the EU Joint Programme–Neurodegenerative Disease Research (JPND): www.jpnd.eu. Epidemiology of Dementia in Singapore (EDIS) (S.H., C.Chen, C.-Y.C., T.Y.W. and W.Z.): The EDIS study is supported by the National Medical Research Council (NMRC), Singapore (NMRC/CG/NUHS/2010 (grant no. R-184-006-184-511), NMRC/CSA/038/2013) and a Ministry of Education Tier 1 grant (no. A-0006106-00-00). Framingham Heart Study (FHS) (J.R.R., A.B., J.J.H., S.L., P.P., C.L.S., Q.Y. and S.Seshadri): This work was supported by the National Heart, Lung and Blood Institute’s FHS Contract (no. N01-HC-25195, no. HHSN268201500001I and no. 75N92019D00031). This study was also supported by grants from the National Institute of Aging (R01 grant nos. AG031287, AG054076, AG049607, AG059421, AG059725; U01 grant nos. AG049505, AG052409) and the National Institute of Neurological Disorders and Stroke (R01 grant no. NS017950). Funding for SHARe Affymetrix genotyping was provided by NHLBI Contract no. N02-HL64278. The computational work reported in this paper was performed on the Shared Computing Cluster which is administered by Boston University’s Research Computing Services. We also thank all the FHS study participants. Internet-based Students’ Health Research Enterprise (i-Share) study (C.B., J.Z., M.M., Q.LG., S. Schilling, Y.-C.Z., A.Tsuchida, M.-G.D., B.M., S.D. and C.T.): The i-Share study is conducted by the Universities of Bordeaux and Versailles Saint-Quentin-en-Yvelines (France). The i-Share study has received funding by the French National Agency (Agence Nationale de la Recherche, ANR), via the Investment for the Future program (grant nos. ANR-10-COHO-05 and ANR-18-RHUS-0002) and from the University of Bordeaux Initiative of Exellence (IdEX). This project has also received funding from the European Research Council under the European Union’s Horizon 2020 research and innovation program under grant agreement no. 640643 and from the Fondation pour la Recherche Médicale (grant no. DIC202161236446). Q.L.G. was supported by the Digital Public Health Graduate Program (DPH), a PhD program supported by the French Investment for the Future Program (grant no. 17-EURE-0019). Investigating Silent Strokes in Hypertensives: a Magnetic Resonance Imaging Study (ISSYS) (P.D., C.C. and I.F.-C.): This research was funded by the Instituto de Salud Carlos III (grant nos. PI10/0705, PI14/01535, PI17/02222), cofinanced by the European Regional Development Fund. Lothian Birth Cohort 1936 (LBC1936) (M.L., M.E.B., I.J.D., Z.M., S.M.M., M.C.V.H. and J.M.W.): We thank the LBC1936 cohort members and research staff involved in data collection, processing and preparation. The LBC1936 is supported by Age UK (Disconnected Mind program grant). The work was undertaken by The University of Edinburgh Centre for Cognitive Ageing and Cognitive Epidemiology, part of the cross-council Lifelong Health and Wellbeing Initiative (grant no. MR/K026992/1). The brain imaging was performed in the Brain Research Imaging Centre (www.bric.ed.ac.uk), a center in the SINAPSE Collaboration (www.sinapse.ac.uk) supported by the Scottish Funding Council and Chief Scientist Office. Funding from the UK Biotechnology and Biological Sciences Research Council (BBSRC), the UK Medical Research Council (MRC), the Row Fogo Charitable Trust (M.C.V.H.) and the UK Dementia Research Institute, which receives its funding from the UK Medical Research Council, Alzheimer’s Society and Alzheimer’s Research UK (J.M.W.), is gratefully acknowledged. Genotyping was supported by a grant from the BBSRC (no. BB/F019394/1). The Nagahama Study (T.K., S.M., M.O., K.S., Y.T., K.Y., A.Tsuchida, P.B., B.M., M.J., M.-G.D. and F.M.): We are grateful to the Nagahama City Office and nonprofit organization Zeroji Club for their help in conducting the study. This project is supported by operational funds of Kyoto University and the Top Global University Project of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) in Japan. We also received a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science, research grants from the Japan Agency for Medical Research and Development for the Practical Research Project for Rare/Intractable Diseases, and the Comprehensive Research on Aging and Health Science for Dementia R&D. We thank C. Galmiche for rating PVS in the validation dataset for the artificial intelligence-based method. The Northern Manhattan Study (NOMAS) (N.D.D., T.J. and R.L.S.): We gratefully acknowledge and thank the NOMAS participants. Funding was awarded through grants from the National Institute of Neurological Disorders and Stroke (R01 grant no. NS 29993) and the Evelyn F. McKnight Brain Institute. Rotterdam Study (M.J.K., F.D., M.W.V., M.A.I. and H.H.H.A.): The Rotterdam Study is funded by Erasmus Medical Center and Erasmus University, Rotterdam, the Netherlands Organization for Health Research and Development (ZonMw), the Research Institute for Diseases in the Elderly (RIDE), the Ministry of Education, Culture and Science, the Ministry for Health, Welfare and Sports, the European Commission (DG XII), and the Municipality of Rotterdam. The authors are grateful to the study participants, the staff from the Rotterdam Study and the participating general practitioners and pharmacists. The generation and management of GWAS genotype data for the Rotterdam Study (RS I, RS II, RS III) were executed by the Human Genotyping Facility of the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, Rotterdam, the Netherlands. The GWAS datasets are supported by the Netherlands Organisation for Scientific Research (NWO) Investments (no. 175.010.2005.011, 911-03-012), the Genetic Laboratory of the Department of Internal Medicine, Erasmus MC, the Research Institute for Diseases in the Elderly (grant no. 014-93-015; RIDE2), the Netherlands Genomics Initiative/NWO, the Netherlands Consortium for Healthy Aging, project no. 050-060-810. We thank P. Arp, M. Jhamai, M. Verkerk, L. Herrera, M. Peters and C. Medina-Gomez for their help in creating the GWAS database; and K. Estrada, Y. Aulchenko and C. Medina-Gomez for the creation and analysis of imputed data. H.H.H.A. is supported by ZonMW grant no. 916.19.151. Study of Health in Pomerania (SHIP) (S.F., R.B., A.T., K.W., H.J.G. and U.V.): SHIP is part of the Community Medicine Research net (CMR) (http://www.medizin.uni-greifswald.de/icm) of the University Medicine Greifswald, which is funded by the Federal Ministry of Education and Research (grant nos. 01ZZ9603, 01ZZ0103 and 01ZZ0403), the Ministry of Cultural Affairs as well as the Social Ministry of the Federal State of Mecklenburg-West Pomerania, and the network ‘Greifswald Approach to Individualized Medicine (GANI_MED)’ funded by the Federal Ministry of Education and Research (grant no. 03IS2061A). Genome-wide data have been supported by the Federal Ministry of Education and Research (grant no. 03ZIK012) and a joint grant from Siemens Healthineers, Erlangen, Germany and the Federal State of Mecklenburg-West Pomerania. The University of Greifswald is a member of the Caché Campus program of the InterSystems GmbH. This study was further supported by the EU-JPND Funding for BRIDGET (grant no. FKZ:01ED1615). H.J.G. has received travel grants and speakers’ honoraria from Fresenius Medical Care, Servier, Neuraxpharm and Janssen Cilag, as well as research funding from Fresenius Medical Care. Sydney Memory and Ageing Study (MAS) & Older Australian Twins Study (OATS) (R.M.T., N.J.A., H.B., J.J., M.P., A.T., J.N.T., P.S.S., W.W., K.A.M. and M.J.W.): Sydney MAS: The Sydney MAS has been funded by three National Health & Medical Research Council (NHMRC) Program Grants (grant nos. ID350833, ID568969 and APP1093083). Collection of WGS data was supported by the NHMRC National Institute for Dementia Research Grants no. APP1115575 and no. APP1115462. MRI scans were processed with the support of NHMRC Project Grants (grant nos. 510175 and 1025243) and an Australian Research Council (ARC) Discovery Project Grant (no. DP0774213) and the John Holden Family Foundation. We also thank the MRI Facility at NeuRA. We thank the participants and their informants for their time and generosity in contributing to this research. We also acknowledge the MAS research team: https://cheba.unsw.edu.au/research-projects/sydney-memory-and-ageing-study. OATS: The OATS study has been funded by an NHMRC and ARC Strategic Award Grant of the Ageing Well, Ageing Productively Program (grant no. 401162); NHMRC Project (seed) Grants (grant nos. 1024224 and 1025243); NHMRC Project Grants (grant nos. 1045325 and 1085606); and NHMRC Program Grants (grant nos. 568969 and 1093083). Collection of WGS data was supported by the NHMRC National Institute for Dementia Research Grants no. APP1115575 and no. APP1115462. This research was facilitated through access to Twins Research Australia, a national resource supported by a Centre of Research Excellence Grant (no. 1079102) from the National Health and Medical Research Council. We thank the participants for their time and generosity in contributing to this research. We acknowledge the contribution of the OATS research team (https://cheba.unsw.edu.au/project/older-australian-twins-study) to this study. Three-City Dijon Study (3C-Dijon) (S.D., M.-G.D., S. Schilling, C.T., B.M. and A.M.): This project is supported by a grant overseen by the French National Research Agency (ANR) as part of the ‘Investment for the Future Program’ no. ANR-18-RHUS-0002. It is also supported by a JPND project through the following funding organizations under the aegis of JPND: www.jpnd.eu: Australia, National Health and Medical Research Council; Austria, Federal Ministry of Science, Research and Economy; Canada, Canadian Institutes of Health Research; France, French National Research Agency; Germany, Federal Ministry of Education and Research; the Netherlands, the Netherlands Organisation for Health Research and Development; United Kingdom, Medical Research Council. This project has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement nos. 643417, 640643, 667375 and 754517. The project also received funding from the French National Research Agency (ANR) through the VASCOGENE and SHIVA projects, and from the Initiative of Excellence of the University of Bordeaux (C-SMART project). Computations were performed on the Bordeaux Bioinformatics Center (CBiB) computer resources, University of Bordeaux. Funding support for additional computer resources at the CREDIM (Centre de Recherche et Développement en Informatique Médicale, University of Bordeaux) has been provided to S.D. by the Fondation Claude Pompidou. The Three-City (3C) Study: The 3C Study is conducted under a partnership agreement among the Institut National de la Santé et de la Recherche Médicale (INSERM), the University of Bordeaux and Sanofi-Aventis. The Fondation pour la Recherche Médicale funded the preparation and initiation of the study. The 3C Study is also supported by the Caisse Nationale Maladie des Travailleurs Salariés, Direction Générale de la Santé, Mutuelle Générale de l’Education Nationale (MGEN), Institut de la Longévité, Conseils Régionaux of Aquitaine and Bourgogne, Fondation de France and Ministry of Research–INSERM program ‘Cohortes et collections de données biologiques.’ C.T. and S.D. have received investigator-initiated research funding from the French National Research Agency (ANR) and from the Fondation Leducq. M.-G.D. received a grant from the ‘Fondation Bettencourt Schueller’. We thank P. Amouyel, U1167 Institut Pasteur de Lille - University of Lille - Inserm, for supporting funding of genome-wide genotyping of the 3C Study. This work was supported by the National Foundation for Alzheimer’s disease and related disorders, the Institut Pasteur de Lille, the labex DISTALZ and the Centre National de Génotypage. We thank A. Boland (CNG) for her technical help in preparing the DNA samples for analyses. UK Biobank (UKB) (M.J.K., F.D., M.W.V., M.A.I., H.H.H.A., A.M. and T.E.): This research has been conducted using the UK Resource under application no. 23509. McGill Genome Center (M.B., P.M., G.B. and M.Lathrop): Work done at the Canadian Center for Computational Genomics was supported by Genome Canada. Data analyses were enabled by computing and storage resources provided by Compute Canada and Calcul Québec. G.B. is supported by the Fonds de Recherche Santé Québec and the Canada Research Chair program. We thank all the participating cohorts for contributing to this study. We thank H. Jacqmin-Gadda, Bordeaux Population Health research center, University of Bordeaux/Inserm U1219 for statistical advice. We thank J. Thomas-Crusells, Bordeaux Population Health Research Center, University of Bordeaux/Inserm U1219, for editorial assistance. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
Author information
Authors and Affiliations
Consortia
Contributions
F.M., C.T., J.M.W., S.Seshadri, H.H.H.A. and S.D. jointly supervised research. M.-G.D., M.J.K., Q.LG., T.E.E., A.M. and A.Tsuchida contributed equally. M.-G.D., M.J.K., H.H.H.A. and S.D. designed and conceived the study. J.R.R., S.F., M.L., E.H., M.B., N.D.D., P.D., S.H., R.M.T., F.D., J.S., Y.S., N.J.A., C.B., M.E.B., A.B., H.B., R.B., C. Carrera, C.Chen, C.-Y.C., I.J.D., P.G.G., J.J.H., J.J., T.K., S.L., M.M., P.M., Z.M., S.M.M., S.M., M.O., M.P., P.P., T.R., M.S., S. Schilling, K.S., O.S., Y.T., A.T., A.Thalamuthu, J.N.T., M.C.V.H., M.W.V., U.V., K.W., T.Y.W., M.J.W., J.Z., W.Z., Y.-C.Z., H.S., P.S.S., W.W., K.Y., C.L.S., R.L.S., G.B., M.Lathrop, I.F.-C., Q.Y., B.M., P.B., H.J.G., K.A.M., R.S., M.J. and M.A.I. generated the PVS phenotype and genomic data and conducted cohort-wise GWAS analyses. G.R., T.K., D.-A.T., A.J., T.P. and Y.O. contributed to bioinformatics analyses. M.-G.D., S.D., M.J.K., Q.L.G., T.E.E., A.M., A.Tsuchida, F.M., C.T., J.M.W., S.Seshadri and H.H.H.A. wrote and edited the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Medicine thanks Andreas Charidimou, Timothy Frayling and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 Manhattan and QQ plots of extensive PVS burden in the cross-ancestry and European meta-analyses.
Manhattan and Quantile-Quantile (QQ) plots of the p-values (observed versus expected) in the cross-ancestry (A) and European (B) GWAS meta-analyses are presented along with the genomic inflation factor (λ). For QQ plots the observed -log10(p) is represented in the y-axis and the expected -log10(p) in x-axis. The dotted line corresponds to the genome-wide significance threshold (p = 5×10−8, two sided). PVS indicates perivascular spaces; WM, white matter; BG, basal ganglia; HIP, hippocampus.
Extended Data Fig. 2 Two-sample Mendelian randomization analysis between putative risk factors and extensive PVS burden.
Two-sample Mendelian randomization was conducted using European PVS GWAS summary statistics (N = 38,598 (WM-PVS), N = 38,903 (BG-PVS) and N = 38,871 (HIP-PVS)), combined with summary statistics for lacunes (N = 1,715 cases / N = 15,096 controls) and WMH volume (N = 48,454) in MTAG, for the outcomes and European GWAS summary statistics for blood pressure traits (N = 757,601) to generate instruments for exposures (Supplementary Tables 1 and 29). Only significant associations after multiple testing correction (p < 1.19×10−3) in GSMR are shown. Each dot (or triangle if p < 1.19×10−3) represents the beta estimate from Mendelian randomization with lines representing the 95% confidence interval. Two-sided p-values of GSMR are reported. PVS indicates perivascular spaces; WM, white matter; BG, basal ganglia; HIP, hippocampus; MTAG, multi-trait analysis of genome-wide association summary statistics; DBP, diastolic blood pressure; PP, pulse pressure; SBP, systolic blood pressure; NS, not significant; GSMR, Generalised Summary-data-based Mendelian Randomisation; IVW, Inverse variance weighted; 2SMR, Two-SampleMR.
Extended Data Fig. 3 Two-sample Mendelian randomization analysis between extensive PVS burden and neurological traits.
Two-sample Mendelian randomization was conducted using European GWAS summary statistics for any stroke (N = 40,585 cases / N = 406,111 controls), ischemic stroke (N = 34,217 cases / N = 400,201 controls) and small vessel stroke (N = 5,386 cases / N = 254,558 controls) for outcomes and European PVS GWAS summary statistics (N = 38,598 (WM-PVS), N = 38,903 (BG-PVS) and N = 38,871 (HIP-PVS)), combined with summary statistics for lacunes (N = 1,715 cases / N = 15,096 controls) and WMH volume (N = 48,454) in MTAG, to generate instruments for exposures (Supplementary Tables 1 and 29). Only significant associations after multiple testing correction (p < 1.19×10−3) in GSMR are shown. Each dot (or triangle if p < 1.19×10−3) represents the beta estimate from Mendelian randomization with lines representing the 95% confidence interval. Two-sided p-values of GSMR are reported. PVS indicates perivascular spaces; WM, white matter; BG, basal ganglia; HIP, hippocampus; MTAG, multi-trait analysis of genome-wide association summary statistics; NS, not significant; GSMR, Generalised Summary-data-based Mendelian Randomisation; IVW, Inverse variance weighted; 2SMR, Two-SampleMR.
Extended Data Fig. 4 Enrichment of PVS risk loci in genes mutated in OMIM syndromes.
Enrichment of all perivascular spaces (PVS) loci (left) and WM-PVS loci only (right) in genes mutated in OMIM syndromes associated with white matter hyperintensities, leukodystrophy, leukoencephalopathy, according to distance from the lead variant; * p < 0.05; ** p < (0.05/5); *** p < (0.05/5/2). Exact p-values for all PVS loci: p = 0.041 (genes with intragenic SNPs), p = 9,27×10−06 (genes with intragenic lead SNPs); exact p-values for WM-PVS loci only: p = 0,018 (genes within 10 kb distance), p = 0.008 (genes with intragenic SNPs), p = 8,61×10−08 (genes with intragenic lead SNPs), full results are provided in Supplementary Table 22.
Extended Data Fig. 5 Brain expression pattern across the lifespan of genes near genome-wide significant PVS loci that are also identified in the TWAS and peaking in the pre-natal period (UMPS and LAMC1).
The figure displays brain expression patterns across the lifespan of the 2 significant PVS TWAS-COLOC genes from genome-wide significant PVS GWAS loci peaking in the pre-natal period (UMPS for WM-PVS and LAMC1 for HIP-PVS) (see Supplementary Table 4 for brain expression pattern across the lifespan of other genes). The spatio-temporal gene expression level is plotted as log2-transformed exon array signal intensity (y-axis) against the post conception days (x-axis) as provided by the Human Brain Transcriptome project database. Periods of human development and adulthood are indicated by vertical dashed lines: 4-8 post conception weeks [PCW] (period 1), 8-10 PCW (period 2), 10-13 PCW (period 3), 13-16 PCW (period 4), 16-19 PCW (period 5), 19-24 PCW (period 6), 24-38 PCW (period 7), birth-6 postnatal months (period 8), 6-12 postnatal months (period 9), 1-6 years (period 10), 6-12 years (period 11), 12-20 years (period 12), 20-40 years (period 13), 40-60 years (period 14), and 60 years + (period 15). The boundary between pre- and postnatal periods is indicated by the solid vertical line. Each colored point represents the expression level of each gene across 16 anatomical brain regions and ages. Brain structure includes 11 neocortical areas (NCX, blue), and 5 subcortical regions: hippocampus (HIP, cyan), amygdala (AMY, orange), striatum (STR, black), mediodorsal nucleus of thalamus (MD, dark green), and cerebellar cortex (CBC, red). Neocortical areas include orbital prefrontal cortex (OFC), dorsolateral prefrontal cortex (DFC), ventrolateral prefrontal cortex (VFC), medial prefrontal cortex (MFC), primary motor cortex (M1C), primary somatosensory cortex (S1C), posterior inferior parietal cortex (IPC), primary auditory cortex (A1C), posterior superior temporal cortex (STC), inferior temporal cortex (ITC), and primary visual cortex (V1C).
Extended Data Fig. 6 Enrichment of PVS genes in targets of drugs validated in other indications (Genome for REPositioning drugs, using ICD10 codes).
Using the Genome for REPositioning drugs (GREP) software (A), we found a significant enrichment of BG-PVS genes in targets of drugs for diseases of the nervous system (ICD10 codes G50-G59, G70-G73, and G80-G83, OR = 51.0, 49.7, and 60.4, p = 3.90×10−2, 4.00×10−2, and 3.32×10−2) and for symptoms and signs involving cognition, perception, emotional state and behaviour (ICD10 codes R40-R46, OR = 48.4, p = 4.10×10−2), driven by MAPT (chr17q21.31), a target for davunetide (B). We also found a significant enrichment of HIP-PVS genes in targets of drugs for diseases of the ear (ICD10 codes H90-H95, OR = 57.6, p = 2.34×10−2), driven by SERPIND1 (chr22q11.21) a target for sulodexide, also used for the prophylaxis and treatment of vascular diseases with increased risk of thrombosis (B). PVS indicates perivascular spaces; ATC, Anatomical Therapeutic Chemical classification; ICD10, the 10th revision of the International Statistical Classification of Diseases and Related Health Problems; TTD, Therapeutic Target Database.
Extended Data Fig. 7 Enrichment of PVS genes in targets of drugs validated in other indications (Genome for REPositioning drugs, using ATC codes).
We found a significant enrichment of BG-PVS genes in targets for antiinfectives for systemic use (ATC J01, OR = 252.4, P = 8.85×10−3), driven by CRHR1 (chr17q21.31), a target for telavancin (A, B); of note, CRHR1 antagonists were shown to attenuate blood brain barrier permeability, cortical vascular hyperpermeability and tight junction disruption. ATC indicates Anatomical Therapeutic Chemical classification.
Extended Data Fig. 8 Enrichment of PVS genes in targets of drugs validated in other indications (Trans-Phar).
We used the Integration of Transcriptome-wide Association Study and Pharmacological Database (Trans-Phar) software, leveraging TWAS on all GTEx v7 tissues and a database of compound-regulated gene expression (C-map) (Methods). A TWAS using FOCUS, which demonstrates fine-mapping of causal gene sets from TWAS results, and 27 tissues in the GTEx v7 database (corresponding to defined 13 tissue-cell-type categories assigned by the 27 tissues in GTEx v7 database and 77 LINCS CMap L1000 library cell types) was performed to identify up- and down-regulated genes in participants with extensive PVS burden, and select the top 10% genes with the highest expression variation. We observed significant enrichment of HIP-PVS in drugs for vascular diseases, including simvastatin (lipid-lowering drug, p = 1.64×10−4), vincamine (vasodilator increasing cerebral blood flow, p = 2.87×10−4), and macitentan (used for pulmonary arterial hypertension, p = 7.46×10−4). PVS indicates perivascular spaces.
Supplementary information
Supplementary Information
Supplementary figures.
Supplementary Tables
Supplementary tables.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Duperron, MG., Knol, M.J., Le Grand, Q. et al. Genomics of perivascular space burden unravels early mechanisms of cerebral small vessel disease. Nat Med 29, 950–962 (2023). https://doi.org/10.1038/s41591-023-02268-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41591-023-02268-w
