
Abstract
Juvenile idiopathic arthritis (JIA) is a complex rheumatic disease with both autoimmune and autoinflammatory components. Recently, familial cases of systemic-onset JIA have been attributed to mutations in LACC1/FAMIN. We describe three affected siblings from a Moroccan consanguineous family with an early-onset chronic, symmetric and erosive arthritis previously diagnosed as rheumatoid factor (RF)-negative polyarticular JIA. Autozygosity mapping identified four homozygous regions shared by all patients, located in chromosomes 3, 6 (n:2) and 13, containing over 330 genes. Subsequent whole exome sequencing identified two potential candidate variants within these regions (in FARS2 and LACC1/FAMIN). Genotyping of a cohort of healthy Moroccan individuals (n: 352) and bioinformatics analyses finally supported the frameshift c.128_129delGT mutation in the LACC1/FAMIN gene, leading to a truncated protein (p.Cys43Tyrfs*6), as the most probable causative gene defect. Additional targeted sequencing studies performed in patients with systemic-onset JIA (n:23) and RF-negative polyarticular JIA (n: 44) revealed no pathogenic LACC1/FAMIN mutations. Our findings support the homozygous genotype in the LACC1/FAMIN gene as the defect underlying the family here described with a recessively inherited severe inflammatory joint disease. Our evidences provide further support to the involvement of LACC1/FAMIN deficiency in different types of JIA in addition to the initially described systemic-onset JIA.
Similar content being viewed by others

Introduction
Juvenile idiopathic arthritis (JIA) refers to an arthritis of unknown origin, starting before the 16th birthday and lasting for at least 6 weeks1. It represents the most common pediatric rheumatic condition2. Its diagnosis relies on the criteria of the International League of Associations of Rheumatology, defining seven different subtypes: Systemic-onset JIA (SoJIA), oligoarticular, rheumatoid factor (RF)-positive polyarticular, RF-negative polyarticular, enthesitis-related, psoriatic and undifferentiated arthritis1. All JIA subtypes are genetically complex disorders that lack a well-defined mode of inheritance, however, as with other complex disorders, there are a small number of cases following Mendelian inheritance. Genome-wide association studies (GWAS) have identified different loci as susceptibility factors, including the MHC and PTPN22 loci3,4,5. Recently, recessively inherited LACC1/FAMIN mutations have been identified in families with monogenic forms of arthritis including SoJIA6,7, severe debilitating arthropathy and Crohn’s disease8, oligoarticular JIA7,9, polyarticular JIA9 and enthesitis-related JIA9.
We herein describe three siblings born from a consanguineous couple and presenting with RF-negative polyarticular JIA, in which we identified a novel homozygous LACC1/FAMIN mutation as the causative defect. These results add novel evidences of the role of this gene in the pathogenesis of a severe form of early-onset inflammatory joint disease and support the clinical diversity of this rare disease. In addition, we present the results of the genetic screening of LACC1/FAMIN in 67 JIA patients with different JIA subtypes, performed to assess the role of LACC1/FAMIN in sporadic cases of JIA.
Results
Clinical description
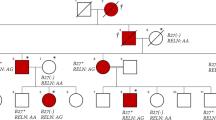
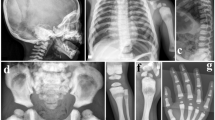
We describe three siblings born from a consanguineous Moroccan couple (pedigree in Fig. 1A) with no familial history of autoimmune disease, primary immunodeficiency, metabolic disease or rheumatological disease. All three siblings were afflicted by an early-onset, severe, chronic and symmetric polyarthritis affecting both large and small joints. Fever was detected at disease onset in only one patient, whereas none of the patients presented with skin manifestations at disease onset. In patient II-4, recurrent, self-limited painful erysipelas-like plaques on the legs have appeared in the past two years. Laboratory analyses revealed leukocytosis, thrombocytosis, anemia, marked increases of inflammatory markers, and negative results for RF, anti-nuclear antibodies (ANAs) and HLA-B*27 (See Table 1 and Supplementary Fig. 1 for a detailed description of each patient). These features lead to a proposed diagnosis of RF-negative polyarticular JIA. All patients received both local and systemic treatments, including DMARDs, steroids, anti-TNF, anti-CD20 and anti-IL-6 drugs. All administered treatments were non-effective or provoked only partial responses, with the only exception of the anti-IL-6 tocilizumab that resulted in complete response in Patient II-4 (Table 1).

Panel (A). Family’s pedigree. Black filled symbols represent affected individuals, open symbols, unaffected individuals, squares, male individuals, and circles, female individuals. The asterisks indicate individuals evaluated by whole-exome sequencing. Panel (B). (top) Genomic organization of the LACC1/FAMIN gene and (bottom) Sanger sense chromatograms from a homozygous wild-type healthy control (left box), heterozygous individuals (middle box), and homozygous mutated patients (right box). The asterisks indicate the two deleted nucleotide positions detected in the patients. Panel (C). (top) Normal structure of laccase protein, and (bottom) predicted structure of the truncated protein encoded by the mutated p.Cys43Tyrfs*6 LACC1/FAMIN allele.
Molecular genetics
We postulated an autosomal recessive mode of inheritance for the disease based on the rare phenotype, its presence in individuals of both genders in the same generation, and the presence of familial consanguinity. To identify the underlying gene defect, a combination of genome wide SNP genotyping and WES was performed. SNP genotyping in three affected and one unaffected individuals revealed four different homozygosity regions, located at chromosomes 3, 6 (n: 2) and 13, exclusively shared by all patients. These regions covered approximately 50.61 Mb and contained over 330 genes (Supplementary Table 1). No rare CNVs overlapping with genes were identified in the selected regions. WES was performed in two affected (II-2, II-4) and one unaffected (II-3) siblings. After filtering for novel or rare (minor allele frequency (MAF) <0.005) homozygous variants, two candidate variants within the homozygosity regions were identified: c.506A > T; p.Asp169Val in the FARS2 gene and c.128_129delGT; p.Cys43Tyrfs*6 in the LACC1/FAMIN gene (Table 2). Sanger sequencing in all family members confirmed intrafamilial co-segregation of both variants with the phenotype, following a recessive mode of inheritance. These variants were subsequently genotyped in a control group of healthy Moroccan people. Of the two variants, only the FARS2 variant was present in the control group (MAF: 0.0014%). Therefore, based on the ACMG classification10 we classified the FARS2 variant as a variant of unknown significance (VUS), as it has not previously been related to disease, it is classified in ClinVar database as a VUS, it is located on an amino acid residue that is not fully conserved through evolution (Supplementary Fig. 2), and the previously FARS2-associated phenotypes (spastic paraplegia and combined oxidative phosphorylation deficiency) are not related to arthritis. By contrast, the LACC1/FAMIN variant was classified as pathogenic, as it is a null variant (a frameshift 2-bp deletion) that leads to a much shorter open reading frame (p.Cys43Tyrfs*6) (Fig. 1C), it is absent in public databases and in the group of healthy Moroccan controls, and the LACC1/FAMIN gene has been linked to various arthritis related phenotypes. Thus, the LACC1/FAMIN variant is the most likely causative gene defect for the disorder detected in this family.
LACC1/FAMIN sequencing in JIA patients
Lately, four articles have described pathogenic or likely pathogenic variants in the LACC1/FAMIN gene in patients with different types of JIA6,7,8,9. To investigate the potential role of this gene in the pathogenesis of sporadic JIA, we sequenced LACC1/FAMIN in two different groups of unrelated, sporadic patients of Spanish ancestry with (1) SoJIA (n: 23) and (2) RF-negative polyarticular JIA (n: 44). The sequencing revealed two missense variants, which were not predicted to be pathogenic by functional prediction algorithms. Variant p.Lys38Glu was detected in heterozygosis in one soJIA case, while variant p.Ile254Val was present in heterozygosis in 9 SoJIA and 13 RF-negative polyarticular JIA cases and in homozygosis in two RF-negative polyarticular JIA cases (Table 3). This variant is also present in the Spanish general population. Based on data from the Geuvadis exome variant server (geevs.crg.eu) or the collaborative Spanish variant server (www.csvs.babelomics.org), the populational frequency of the variant is similar to that observed in our dataset (observed MAF in JIA cases 0.194; MAF for Spanish samples 0.2197/0.202; Table 3). This suggests that LACC1/FAMIN mutations are not major factors to cause these JIA subtypes.
Discussion
We describe a consanguineous Moroccan family with a severe joint inflammatory disease that shows a recessive mode of inheritance. Genetic analyses identified the homozygous p.Cys43Tyrfs*6 LACC1/FAMIN mutation as the most probable causative gene defect. The LACC1/FAMIN gene, previously known as C13orf31, encodes for laccase domain containing 1, a member of the blue multicopper oxidases. These enzymes were first identified in plants and fungi, where they catalyze the oxidation of aromatic substrates concomitantly to the reduction of molecular oxygen to water11. Previous articles have associated LACC1/FAMIN variants with inflammatory diseases, suggesting a potential relationship of human laccase with inflammatory processes12,13. Functional studies of wild-type and variant LACC1/FAMIN provided a mechanism for its involvement in the inflammatory response as a controller of energy homeostasis in macrophages and show that a reduced or complete loss of function may promote sterile inflammation14.
Recent reports have described LACC1/FAMIN gene variants in patients with different monogenic JIA subtypes. The first described pathogenic variant in LACC1, p.Cys284Arg, cosegregated with disease in a consanguineous family from Saudi Arabia with a complex phenotype including Crohn’s disease and a severe arthropathy8. This same variant was also identified in five additional consanguineous families from Saudi Arabia diagnosed with SoJIA6. Subsequently, different homozygous LACC1/FAMIN variants have been detected in consanguineous families from various ancestries with different types of JIA7,9, including start codon mutations (p.Met1Ile), in-frame deletions (p.Ile330del) and truncating variants (p.Thr276fs*2, p.Arg414Ter). We have identified a novel frameshift truncating LACC1/FAMIN mutation in homozygosity in three JIA cases. This variant, p.Cys43Tyrfs*6, generates a shorter open reading frame of only 49 amino acids with a new stop codon in exon 2, which likely leads to nonsense mediated mRNA decay and absence of human laccase in the affected siblings.
All described families with LACC1/FAMIN mutations seem to share some clinical features such as the age at disease onset, the hematological and biochemical profiles, and the pattern of joint involvement, including its chronic course, symmetry, and number and type of affected joints. Nevertheless, marked differences in their phenotypes may be also observed. Thus, most of the patients of the families reported by Wakil et al., Patel et al., and Kallinich et al. had fever, cutaneous lesions, serositis, organomegaly or lymphadenopathy, supporting their diagnosis as SoJIA6,7,8. By contrast, most of the patients reported by Karacan et al. and the family here described did not present with fever or cutaneous lesions at disease onset. In these patients the joint involvement was the most prominent manifestation at disease onset and showed a chronic and polyarticular course, suggesting the diagnosis of polyarticular JIA9. Despite LACC1/FAMIN mutations have been repeatedly identified in Mendelian forms of JIA, we failed to detect homozygous or compound heterozygous carriers of rare pathogenic LACC1 variants in the two groups of Spanish JIA patients analyzed, suggesting that mutations in LACC1 are not a major cause of JIA, and reinforcing the genetic complexity of this disorder. In fact, although common SNPs in LACC1/FAMIN (including variant p.Ile254Val) have been associated to non-systemic JIA in a Swedish cohort15, we do not replicate this association in our small dataset, which has a similar carrier frequency to that of the Spanish population obtained from exome sequencing databases. Consequently, all available evidences strongly suggest that biallelic LACC1/FAMIN mutations may provoke a severe form of early-onset inflammatory arthritis, and its screening could only benefit those patients with very early-onset JIA.
In summary, our findings describe a novel LACC1/FAMIN mutation as the causative defect of a recessively inherited, severe inflammatory joint disease. The clinical features of these patients add novel evidences that the phenotype of this rare genetic disease includes forms of JIA other than SoJIA. The collected data support a relevant role of the LACC1/FAMIN gene in inflammatory processes, indicating that further research into its function and its role as a therapeutic target may improve diagnosis and treatment of patients with JIA and other common inflammatory diseases.
Methods
Patients
The patients’ data were collected by direct interviews and by the review of their clinical charts. Blood samples were collected for genetic and molecular studies after obtaining written-informed consent from patients or patients’ parents (<18 years), and approval by the ethics committee of Hospital Clinic. All protocols were approved by the Ethics Committee of Hospital Clinic and all methods were performed in accordance with the relevant guidelines and regulations. The control groups included a group of healthy Moroccan individuals (n: 352), and two groups of sporadic Spanish patients with either SoJIA (n: 23) or RF-negative polyarticular JIA (n: 44), which were diagnosed according the criteria of the International League of Associations of Rheumatology. Genetic studies were performed in accordance with the Declaration of Helsinki.
Homozygosity mapping
DNA was isolated from whole blood using QIAmp DNA Blood Mini Kit (QIAgen, Germany). SNPs were genotyped with HumanCNV370-Duo Beadchip (Illumina Inc, USA), and analyzed for homozygosity mapping using AutoSNPa16. Copy number variants (CNVs) detection from SNP genotyping was performed using PennCNV17.
Whole-exome sequencing (WES)
Libraries were prepared with the SureSelect Human All exon V2 kit (Agilent Technologies Inc, USA). Paired-end sequencing was performed on the Illumina Genome Analyzer II platform (Illumina Inc, USA). The sequence reads were aligned to the Human Reference Genome Build hg19 using the BWA software18, followed by GATK base quality score recalibration, duplicate marking and local realignment. SNPs and indels were simultaneously called in all samples using the GATK HaplotypeCaller algorithm, applying hard-filtering parameters according to GATK best practices recommendations19. Variants were annotated and prioritized using ediva, our in-house pipeline (www.ediva.crg.eu), which provides information on minor allele frequencies from various databases, including the Exome Aggregation Consortium, and Exome Variant Server-NHBLI, variant functional effect prediction scores by SIFT, Polyphen2, MutationAssessor and CADD, and variant conservation scores PhyloP and Gerp++. Fastq files can be accessed through EGA (EGAS00001003510).
LACC1/FAMIN Sanger sequencing
Coding exons of LACC1/FAMIN were amplified by PCR, purified with Illustra ExoStar 1-Step kit (GE Healthcare, USA), fluorescence sequenced using ABI BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA) and run on an automated ABI 3730XL DNA analyzer.
References
Petty, R. E. et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol 31, 390–392 (2004).
Ravelli, A. & Martini, A. Juvenile idiopathic arthritis. Lancet 369, 767–778, https://doi.org/10.1016/S0140-6736(07)60363-8 (2007).
Hinks, A. et al. Identification of a novel susceptibility locus for juvenile idiopathic arthritis by genome-wide association analysis. Arthritis Rheum 60, 258–263, https://doi.org/10.1002/art.24179 (2009).
Hinks, A. et al. Dense genotyping of immune-related disease regions identifies 14 new susceptibility loci for juvenile idiopathic arthritis. Nat Genet 45, 664–669, https://doi.org/10.1038/ng.2614 (2013).
Ombrello, M. J. et al. Genetic architecture distinguishes systemic juvenile idiopathic arthritis from other forms of juvenile idiopathic arthritis: clinical and therapeutic implications. Ann Rheum Dis 76, 906–913, https://doi.org/10.1136/annrheumdis-2016-210324 (2017).
Wakil, S. M. et al. Association of a mutation in LACC1 with a monogenic form of systemic juvenile idiopathic arthritis. Arthritis Rheumatol 67, 288–295, https://doi.org/10.1002/art.38877 (2015).
Kallinich, T. et al. Juvenile arthritis caused by a novel FAMIN (LACC1) mutation in two children with systemic and extended oligoarticular course. Pediatr Rheumatol Online J 14, 63, https://doi.org/10.1186/s12969-016-0124-2 (2016).
Patel, N. et al. Study of Mendelian forms of Crohn’s disease in Saudi Arabia reveals novel risk loci and alleles. Gut 63, 1831–1832, https://doi.org/10.1136/gutjnl-2014-307859 (2014).
Karacan, I. et al. LACC1 Gene Defects in Familial Form of Juvenile Arthritis. J Rheumatol 45, 726–728, https://doi.org/10.3899/jrheum.170834 (2018).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17, 405–424, https://doi.org/10.1038/gim.2015.30 (2015).
Giardina, P. et al. Laccases: a never-ending story. Cell Mol Life Sci 67, 369–385, https://doi.org/10.1007/s00018-009-0169-1 (2010).
Zhang, F. R. et al. Genomewide association study of leprosy. N Engl J Med 361, 2609–2618, https://doi.org/10.1056/NEJMoa0903753 (2009).
Umeno, J. et al. Meta-analysis of published studies identified eight additional common susceptibility loci for Crohn’s disease and ulcerative colitis. Inflamm Bowel Dis 17, 2407–2415, https://doi.org/10.1002/ibd.21651 (2011).
Cader, M. Z. et al. C13orf31 (FAMIN) is a central regulator of immunometabolic function. Nat Immunol 17, 1046–1056, https://doi.org/10.1038/ni.3532 (2016).
Assadi, G. et al. LACC1 polymorphisms in inflammatory bowel disease and juvenile idiopathic arthritis. Genes Immun 17, 261–264, https://doi.org/10.1038/gene.2016.17 (2016).
Carr, I. M., Flintoff, K. J., Taylor, G. R., Markham, A. F. & Bonthron, D. T. Interactive visual analysis of SNP data for rapid autozygosity mapping in consanguineous families. Hum Mutat 27, 1041–1046, https://doi.org/10.1002/humu.20383 (2006).
Wang, K. et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17, 1665–1674, https://doi.org/10.1101/gr.6861907 (2007).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
Van der Auwera, G. A. et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43, 11 10 11–33, https://doi.org/10.1002/0471250953.bi1110s43 (2013).
Acknowledgements
The authors would like to thank the CRG Genomics Unit for assistance with whole exome sequencing. This work has been partially funded by: CERCA Programme/Generalitat de Catalunya (JIA, XE, SO), the PERIS program of the Generalitat de Catalunya grant SLT002/16/00310 (RR), the Spanish Ministry of Economy and Competitiveness co-financed by European Regional Development Fund (ERDF) grant SAF2015-68472-C2-1-R (JIA), the Instituto de Salud Carlos III/Transnational Research Projects on Rare Diseases (JIA) grant AC15/00027, the Spanish Society of Pediatric Rheumatology (JIA), the Secretaria d’Universitats i Recerca del Departament d’Economia grant 2009-SGR-1502 (XE) and the European Union Seventh Framework Programme (FP7/2007-2013) grant agreement no. 262055 (XE). We also acknowledge support of the Spanish Ministry of Economy and Competitiveness (MEIC) to the EMBL partnership, ‘Centro de Excelencia Severo Ochoa’.
Author information
Authors and Affiliations
Contributions
R.R., J.Y., R.M., X.E. and J.I.A. designed research and discussed data. R.R. and J.I.A. wrote the manuscript. R.R., A.M.-V., E.G.-R., O.D., E.R.-O., A.P., D.C., S.O., X.E. and J.I.A. performed genetic investigations, discussed data and reviewed the manuscript. A.R., S.M., R.A., R.M., J.A., E.I. and C.M. provided clinical data and blood samples, discussed data and reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Rabionet, R., Remesal, A., Mensa-Vilaró, A. et al. Biallelic loss-of-function LACC1/FAMIN Mutations Presenting as Rheumatoid Factor-Negative Polyarticular Juvenile Idiopathic Arthritis. Sci Rep 9, 4579 (2019). https://doi.org/10.1038/s41598-019-40874-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-40874-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
