
Abstract
This study was aimed to identify the potentially pathogenic gene variants that contribute to the etiology of the tuberous sclerosis complex. A Chinese pedigree with tuberous sclerosis complex was collected and the exomes of two affected individuals were sequenced using the whole exome sequencing technology. The resulting variants from whole exome sequencing were filtered by basic and advanced biological information analysis and the candidate mutation was verified as heterozygous by sanger sequencing. After basic and advanced biological information analysis, a total of 9 single nucleotide variants were identified, which were all follow the dominant inheritance pattern. Among which, the intron heterozygous mutation c.600-145 C > T transition in TSC2 was identified and validated in the two affected individuals. In silico analysis with human splicing finder (HSF) predicted the effect of the c.600-145 C > T mutations on TSC2 mRNA splicing, and detected the creation of a new exonic cryptic donor site, which would result in a frame-shift, and finally premature termination codon. Our results reported the novel intron heterozygous mutation c.600-145 C > T in TSC2 may contribute to TSC, expanding our understanding of the causally relevant genes for this disorder.
Similar content being viewed by others
Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant (95% penetrance) neurocutaneous and progressive disorder, commonly characterized by the occurrence of various tumors in different organs1. It is reported that two-thirds of TSC cases are sporadic, which reflects a high spontaneous mutation rate2. TSC can affect people of all age groups with multiple organ systems involved in different ways and at varying time3. The clinical presentation of TSC varies greatly even within a given family4,5,6.
Multisystem hamartomatous lesions in the brain, skin, kidney, lung, retina and heart are very common. The central nervous system is the most severely and commonly affected organ system in TSC patients. Cortical tubers, subependymal nodules and subependymal giant cell astrocytomas are the main structural brain lesions4,5. It is pointed out that tubers growing in the brain are closely associated with high morbidity and mortality of TSC7. Skin lesions are detected in most of TSC patients and include shagreen patches, hypomelanotic macules, confetti-like lesions, facial angiofibromas, forehead fibrous plaque and periungual and ungual fibromas8. After central nervous system and skin findings, renal manifestation is the most common abnormality in TSC patients9. Pulmonary involvement, especially lymphangioleiomyomatosis, is the third most common cause of TSC-associated morbidity9. TSC is also related to both retinal and nonretinal ocular findings10. Moreover, hamartomas are the most common retinal manifestation of TSC9. In addition, various cardiac rhabdomyomas are occurred in TSC patients9. The disease severity of TSC is variable with signs and symptoms ranging from hypomelanotic macules, to epilepsy, autism, intellectual disability and multiple hamartomas in brain, kidney, lung and heart11.
The phenotypic expression of TSC is highly variable and sometimes it can be difficult to establish the definitive clinical diagnosis. Recently, mutation analysis has become an additional diagnostic tool in TSC. It has been demonstrated that TSC is caused by mutations in either the TSC1 gene on chromosome 9q34, or the TSC2 gene on chromosome 16p13.312,13. It is worth mentioning that several TSC2 variants including A1801G, F143L, S132C, A196T, Y598H, C244R, T993M, L1511H and R1772C have been identified in individuals with symptoms of TSC14,15. Considering the genetic heterogeneity, the identity of the novel candidate genes remains a challenge. In the current study, we used whole exome sequencing to identify the novel causative gene for the two affected individuals in a Chinese TSC family. Our study may improve the understanding of this disorder and provide insight into the genetic basis for inherited TSC.
Materials and Methods
Human subjects
For the purpose of this study, a four-generation Chinese tuberous sclerosis (TSC) family with five affected individuals and five unaffected individuals was recruited. Given high suspicion for the TSC family, the two affected individuals and one unaffected individual were enrolled for the exome sequencing screen. The blood samples were collected from the participants for DNA extraction. All experiments were performed in accordance with relevant guidelines and regulations. The written informed consent was obtained from study subjects or guardian before the study. The study was approved by the licensing committee of Beijing Anzhen Hospital.
Analysis of exome capture
The genomic DNA was extracted from the blood samples according to the standard procedures. The 2 μg of genomic DNA was fragmented with about 200 bp, then ligated with adapters and amplified by ligation-mediated PCR. The qualified genomic DNA was used for exome capture and high-throughput sequencing. Agilent SureSelect Human All Exon 50 Mb Exon Kit was used to perform exome target enrichment. The captured library was sequenced on the Illumina Hiseq4000 sequencer with paired-end 125-bp and mean coverage of 100×.
Analysis of basic biological information
The fastQC was used to evaluate the quality of raw sequencing data of exome sequencing. Under tools of SeqPrep and sickle, raw data was filtered by removing adapter, contaminating reads and low quality reads, and remains were the clean ones. The exome sequencing clean reads were mapped to the reference human genome sequence (hg19) (http://hgdownload.soe.ucsc.edu/goldenPath/hg19/bigZips/) using the Burrows-Wheeler Alignment (BWA) tool (http://bio-bwa.sourceforge.net/bwa.shtml), which can do short reads alignment to a reference genome and support paired-end mapping. The sequence alignment/map (SAM) file was then generated. Picard tool (http://picard.sourceforge.net/) was used to mark and exclude the duplicate reads. Variants (single nucleotide variants (SNVs), insertions and deletions) calling was performed using the Genome Analysis Toolkit (GATK)16.
Analysis of advanced biological information
In this process, we performed the analysis of dominance/recessiveness screening and mutation site screening. To find the potential important variants, the ANNOVAR tool (http://www.openbioinformatics.org/annovar/) was used to annotate the resulting SNVs17, and the information for variant frequencies and location within genes were obtained. Moreover, the SNVs were sequentially filtered and given higher priority with the following criteria: (1) Quality By Depth (QD) < 2.0, Phred-scaled p-value using Fisher’s exact test to detect strand bias (FS) > 60.0, Mapping quality (MQ) < 40.0, Z-score From Wilcoxon rank sum test of Alt vs. Ref read mapping qualities (MQRankSum) < −12.5 and Z-score from Wilcoxon rank sum test of Alt vs. Ref read position bias (ReadPosRankSum) < −8.0; (2) minor allele frequency (MAF) < 0.05 in 1000 Genomes Project; (3) damaging as predicted by 6 bioinformatics programs including PhyloP (score > 0.85), SIFT (score < 0.05), PolyPhen (score > 0.85), GERP (score > 2), Mutation Taster (score > 0.5), and LRT (score > 0.9); (4) consistent with model of dominant disease transmission.
Variant validation
To validate the variants identified through exome sequencing, candidate SNVs were selected for sanger sequencing. The blood samples were obtained from the selected individuals. Genomic DNA was extracted and SNVs were tested in the original three individuals who underwent exome sequencing and three additional unaffected individuals in the four-generation Chinese TSC family.
Splicing analysis of variant
Human Splicing Finder (HSF) (http://www.umd.be/HSF/) is a tool to predict the effects of mutations on splicing signals, which could identify splicing motifs and evaluate the strength of branch points in any human sequence. In this study, we used this tool to predict the effects of identified mutations on mRNA splicing based on the method in the previous report of HSF use18. The detailed process of our analysis is as follows:
In order to analyze for the presence and predicted strengths of splice sites, we first chose the analysis type as “Splice site analysis” along with the option of “Automatically select the longest transcript”, and then pasted base sequence (50 bp upstream and downstream of the wild-type or variant TSC2 genomic DNA sequence) into the analysis box. Lastly, we chose the mutation position as “64” and type of mutation as “substitution”. In the end, we got the result of “Sequences” and “Interpreted data”. From the “Sequences”, we got the reference sequence and mutant sequence. From the “Interpreted data”, we found the results of the predicted signal, prediction algorithm, cDNA position and interpretation.
Results
Information of proband
We studied a Chinese family affected with TSC, in which there were five affected individuals (Sample I:2, II:1, III:1, III:2 and IV:1) (Fig. 1). The proband (III:2) was a thirty-nine-year-old woman who presented with TSC. Moreover, her mother (II:1) and grandmother (I:2) were also presented with TSC with similar phenotypes. In addition, her little sister (III:1) had been dead of epilepsy. However, the grandfather (I:1), father (II′:1), two uncles (II:2 and II:3) and spouse (III′:1) of the proband were asymptomatic. The proband developed from childhood and accompanied with coronary heart disease and polycystic kidney disease. The skin of the proband showed coffee and milk stains. The head CT scan of the proband showed low density in left caudate nucleus and right frontal cortex, multiple nodular and nodular calcifications in the left caudate nucleus head, anterior border of left cerebellar hemisphere, left temporal lobe and bilateral ventricle (Fig. 2A), multiple nodules and patchy high-density shadows in the right temprral lobe and the left frontal cortex, and microchip low density on the left side of the parietal bone. The score of mini-mental state examination was 25.

Pedigree for the Chinese family with TSC. Individuals III:2, III′:1 and IV:1 underwent exome sequencing. W: exome sequencing.

The CT scan of the proband’s head, lung, and abdomen. (A) The head CT scan of the proband. Nodular calcifications in the bilateral ventricle (arrow). (B) The lung HRCT scan of the proband. Multiple bullae in right lung (arrow). (C) The lung HRCT scan of the proband. Pulmonary nodule in right lung (arrow). (D) The abdominal CT scan of the proband. Bilateral renal angiomyolipoma (arrow).
The lung HRCT scan of the proband showed multiple bullae and pulmonary nodules in right lung (Fig. 2B,C), enlarged axillary and mediastinal lymph nodes, increased heart size, pericardial effusion, and bilateral pleural effusion. The pulmonary first pass imaging indicated that no signs of pulmonary hypertension and right-left shunt were seen.
The abdominal CT scan of the proband showed that bilateral masses with multiple hypodense (angiomyolipoma) were identified in bilateral kidney area instead of normal kidneys (Fig. 2D).
Identification of candidate genes
According to the TSC pedigree, we speculated that TSC was dominant inheritance. The pathogenic gene in the proband may be from her mother and grandmother. Therefore, exome sequencing was ideally suited to screen for the causal genes of the TSC pedigree. The whole exomes of III:2, III′:1 and IV:1 were sequenced, followed by variant detection and filtering. The exome sequencing led to the detection of 47687, 48539 and 48795 SNVs for III′:1, III:2 and IV:1 (Table 1). After further analysis of dominance/recessiveness screening and mutation site screening, a total of 9 SNVs were identified, which were all follow the dominant inheritance pattern. Detailed information of 9 SNVs was showed in Table 2. Among which, TSC2 is an intron heterozygous mutation gene, which was a rare event in the TSC. Therefore, we focused on TSC2 gene in this study.
Sanger sequencing of TSC2 variants
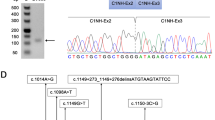
To further confirm the variant of c.600-145 C > T in TSC2 in TSC, sanger sequencing was performed in the original three individuals (III:2, III′:1 and IV:1) who underwent exome sequencing and three unaffected individuals (II′:1, II:2 and II:3) in the TSC family. The results showed that the variant was confirmed as heterozygous in the affected proband (III:2) and her daughter (IV:1) and as wild type in four unaffected individuals (III′:1, II′:1, II:2 and II:3) via Sanger sequencing (Fig. 3), which further demonstrated that the variant of c.600-145 C > T in TSC2 was closely associated with TSC.

Sanger validation results of TSC2 variants. Red arrow presented the mutation site.
In silico analysis of TSC2 variant
In silico analysis with a freely available online bioinformatics tool, human splicing finder (HSF) (http://www.umd.be/HSF3/) predicted the effect of the c.600-145 C > T mutations on TSC2 mRNA splicing. The HSF analysis detected the creation of a new exonic cryptic donor site, generating consensus values of 52.35 and 79.18 for the wild-type and mutant c.600-145 C > T nucleotides, respectively. The predicted consensus value deviation of +51.25% for the new exonic cryptic donor site indicates the loss of the wild-type splice site which would result in a frame-shift, and finally lead to a premature stop codon in the protein.
Discussion
The phenotypic expression of TSC is highly variable and sometimes it is difficult to establish a definitive clinical diagnosis. It is noted that mutation analysis has become an important diagnostic tool in familial as well as sporadic TSC. In this study, whole-exome sequencing was performed on the two affected individuals in a Chinese TSC pedigree, identifying a novel intron heterozygous mutation in TSC2 (c.600-145 C > T). Our result further demonstrated the crucial role of TSC2 in the development of TSC. The TSC2 gene comprises approximately 43 kb of genomic DNA with 41 exons encoding a 5.5 kb transcript and the 198 kDa protein of tuberin. There are various possible mechanisms for somatic inactivation of the wild-type allele of TSC2, including mutation, loss of heterozygosity and promoter methylation. It is reported that loss of function mutation in TSC2 leads to abnormal production of the end products, and finally promotes tumorigenesis of TSC9. Dabora S.L. et al. found that the disease was usually milder in patients with the TSC phenotype and no identifiable mutation in TSC22. In addition, only the p. R905Q mutation in TSC2 has been found related to milder TSC9.
TSC is an autosomal dominant neurocutaneous syndrome caused by mutations of TSC1 or TSC2 genes. Tyburczy M.E. et al. reported that 45 of 53 subjects found mutations, and TSC2 mutations and TSC1 mutations account for 82% and 18%, respectively19. TSC2 is a common intron heterozygous mutation gene in TSC. In most studies of the identified mutations in the TSC2 gene are either missense mutations or small and non-truncating insertions/deletions mutations. Heterozygous missense variant c.899 G > T, p.G300V in the TSC2 gene is found in patients with variable TSC-associated symptoms and signs20. The missense variant c.3599 G > C, p.R1200P in TSC2 gene is identified in the DNA of peripheral leukocytes of TSC patients21. It is noted that some missense changes in TSC2 are related to TSC in definite TSC patients, TSC in familial TSC patients and TSC in which patients symptoms are less severe22,23,24,25,26,27,28. In addition, the novel deletion mutant c.700–701 in the TSC2 gene was detected in patients with TSC29. In the aspect of signaling pathway, the TSC2 protein functions as a heterodimer to suppress the target of rapamycin mTOR, a serine/threonine protein kinase that play roles in the regulation of cell growth and division30,31. It is demonstrated that the small deletion mutation in TSC2 is associated with severe TSC that promotes mTOR signaling pathway29.
Herein, we identified a new intron heterozygous mutation in TSC2 (c.600-145 C > T) in a Chinese TSC pedigree, which was not reported before. The mutation type will lead to a novel variable splicing site, which might be associated with abnormal function of TSC2 protein.
Alternative splicing is a biological process of post-transcriptional RNA processing whereby the single gene can encode various distinct transcripts, which increases the diversity of mRNAs expression32,33. It is showed that alternative splicing can regulate binding between proteins, between proteins and membranes and between proteins and nucleic acids33. It is reported that the aberrant regulation of alternative splicing leads to human disease34,35,36,37,38,39,40,41,42. In addition, alternative splicing also plays roles in brain development and is involved in several neurological diseases43. Torrado et al.44 reported a novel intronic mutation (the c.2678-15 C > A variant), within intron 22 of the FBN1 gene, in a Marfan syndrome (MFS) patient with aortic dilatation. The c.2678-15 C > A variant disrupts normal splicing of intron 22 leading to frameshift, premature termination codon, and finally haploinsufciency of the FBN1 functional protein. In Lynch syndrome families45,46, c.[2635-3 T > C;2635-5 C > T] MSH2 mutation, located in intron 15, caused a significant reduction of MSH2 mRNA expression via altering the correct mRNA processing, suggesting a pathogenic role for the variant. Cariola F. et al.46 also described the variant c.2635-2 A > G in intron 15 of the MSH2 in with three members of a family manifesting the Lynch syndrome, which affects the splice site consensus sequence, and result in the absence of MSH2/MSH6 heterodimer protein. Yu et al. reported that the variant c.772 + 27 G > C in intron 6 of ACVRL1gene in a Chinese family with hereditary hemorrhagic telangiectasia (HHT) presents a decreased expression of ACVRL1 mRNA and protein in affected HHT2 patients47. Therefore, we speculated that the intron heterozygous mutation in TSC2 (c.600-145 C > T) may affect the expression of the TSC2-encoded protein tuberin through alternative splicing.
It is indicated that patients with TSC2 mutations tend to have an earlier onset, more severe cognitive deficits and higher frequency of seizures46. Northrup H. et al. found that the clearly inactivating TSC2 mutation was considered as sufficient evidence for TSC diagnosis, even in the absence of clinical signs47. Therefore, mutation analysis of the TSC2 genes in both suspected and definite TSC patients is important in genetic counselling. Our result may be helpful in the diagnosis and genetic counseling of TSC.
In summary, TSC is a complex disease with significant genetic heterogeneity. We demonstrated the presence of a novel intron heterozygous mutation c.600-145 C > T in TSC2 in the affected individuals, which may potentially contribute to TSC susceptibility. However, there is a limitation of our study. We didn’t perform the pathogenic mechanism study of identified mutation in TSC2. The animal model or cell culture experiments are needed to further investigate the potential biological function of TSC2.
References
Northrup, H., Wheless, J. W., Bertin, T. K. & Lewis, R. A. Variability of expression in tuberous sclerosis. Journal of medical genetics 30, 41–43 (1993).
Dabora, S. L. et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. American journal of human genetics 68, 64–80, https://doi.org/10.1086/316951 (2001).
Kingswood, J. C. et al. TOSCA - first international registry to address knowledge gaps in the natural history and management of tuberous sclerosis complex. Orphanet journal of rare diseases 9, 182, https://doi.org/10.1186/s13023-014-0182-9 (2014).
Curatolo, P., Bombardieri, R. & Jozwiak, S. Tuberous sclerosis. Lancet (London, England) 372, 657–668, https://doi.org/10.1016/s0140-6736(08)61279-9 (2008).
Curatolo, P. & Maria, B. L. Tuberous sclerosis. Handbook of clinical neurology 111, 323–331, https://doi.org/10.1016/b978-0-444-52891-9.00038-5 (2013).
Jozwiak, S., Schwartz, R. A., Janniger, C. K. & Bielicka-Cymerman, J. Usefulness of diagnostic criteria of tuberous sclerosis complex in pediatric patients. Journal of child neurology 15, 652–659, https://doi.org/10.1177/088307380001501003 (2000).
Avgeris, S. et al. Mutational analysis of TSC1 and TSC2 genes in Tuberous Sclerosis Complex patients from Greece. Scientific reports 7, 16697, https://doi.org/10.1038/s41598-017-16988-w (2017).
Schwartz, R. A., Fernandez, G., Kotulska, K. & Jozwiak, S. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. Journal of the American Academy of Dermatology 57, 189–202, https://doi.org/10.1016/j.jaad.2007.05.004 (2007).
Rosset, C., Netto, C. B. O. & Ashton-Prolla, P. TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: a review. Genetics and molecular biology 40, 69–79, https://doi.org/10.1590/1678-4685-gmb-2015-0321 (2017).
Rowley, S. A., O’Callaghan, F. J. & Osborne, J. P. Ophthalmic manifestations of tuberous sclerosis: a population based study. The British journal of ophthalmology 85, 420–423 (2001).
Ekong, R. et al. Variants Within TSC2 Exons 25 and 31 Are Very Unlikely to Cause Clinically Diagnosable Tuberous Sclerosis. Human mutation 37, 364–370, https://doi.org/10.1002/humu.22951 (2016).
van Slegtenhorst, M. et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science (New York, N.Y.) 277, 805–808 (1997).
Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75, 1305–1315 (1993).
Hardy, R., Shepherd, C. W., Donnelly, D. E., McKee, S. A. & Morrison, P. J. Constellation of five facial features of tuberous sclerosis in a child with a TSC2 1808A > G mutation. The oncologist 17, 925–926, https://doi.org/10.1634/theoncologist.2011-0407 (2012).
Nellist, M. et al. Functional characterisation of the TSC1–TSC2 complex to assess multiple TSC2 variants identified in single families affected by tuberous sclerosis complex. BMC medical genetics 9, 10 (2008).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Wang, K., Li, M. & Hakonarson, H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research 38, e164, https://doi.org/10.1093/nar/gkq603 (2010).
Desmet, F. O. et al. Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic acids research 37, e67, https://doi.org/10.1093/nar/gkp215 (2009).
Tyburczy, M. E. et al. Mosaic and Intronic Mutations in TSC1/TSC2 Explain the Majority of TSC Patients with No Mutation Identified by Conventional Testing. PLoS genetics 11, e1005637, https://doi.org/10.1371/journal.pgen.1005637 (2015).
Wang, F., Xiong, S. & Wu, L. A novel TSC2 missense variant associated with a variable phenotype of tuberous sclerosis complex: case report of a Chinese family. 19, 90, https://doi.org/10.1186/s12881-018-0611-z (2018).
Zivcic-Cosic, S. et al. Severe bleeding complications and multiple kidney transplants in a patient with tuberous sclerosis complex caused by a novel TSC2 missense variant. Croatian medical journal 58, 416–423 (2017).
Sancak, O. et al. Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype–phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. European journal of human genetics: EJHG 13, 731–741, https://doi.org/10.1038/sj.ejhg.5201402 (2005).
Wentink, M. et al. Functional characterization of the TSC2 c.3598C > T (p.R1200W) missense mutation that co-segregates with tuberous sclerosis complex in mildly affected kindreds. Clinical Genetics 81, 453–461 (2012).
Jansen, A. C. et al. Unusually mild tuberous sclerosis phenotype is associated with TSC2 R905Q mutation. Annals of neurology 60, 528–539, https://doi.org/10.1002/ana.21037 (2006).
Mayer, K., Goedbloed, M., van Zijl, K., Nellist, M. & Rott, H. D. Characterisation of a novel TSC2 missense mutation in the GAP related domain associated with minimal clinical manifestations of tuberous sclerosis. Journal of medical genetics 41, e64 (2004).
O’Connor, S. E., Kwiatkowski, D. J., Roberts, P. S., Wollmann, R. L. & Huttenlocher, P. R. A family with seizures and minor features of tuberous sclerosis and a novel TSC2 mutation. Neurology 61, 409–412 (2003).
Khare, L. et al. A novel missense mutation in the GTPase activating protein homology region of TSC2 in two large families with tuberous sclerosis complex. Journal of medical genetics 38, 347–349 (2001).
Hoogeveen-Westerveld, M. et al. Functional assessment of variants in the TSC1 and TSC2 genes identified in individuals with Tuberous Sclerosis Complex. Human mutation 32, 424–435, https://doi.org/10.1002/humu.21451 (2011).
Hwang, S. K. et al. Everolimus improves neuropsychiatric symptoms in a patient with tuberous sclerosis carrying a novel TSC2 mutation. Molecular brain 9, 56, https://doi.org/10.1186/s13041-016-0222-6 (2016).
Krueger, D. A. & Northrup, H. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 International Tuberous Sclerosis Complex Consensus Conference. Pediatric neurology 49, 255–265, https://doi.org/10.1016/j.pediatrneurol.2013.08.002 (2013).
Kwiatkowski, D. J. & Manning, B. D. Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Human molecular genetics 14 Spec No. 2, R251–258, https://doi.org/10.1093/hmg/ddi260 (2005).
Bush, S. J., Chen, L., Tovar-Corona, J. M. & Urrutia, A. O. Alternative splicing and the evolution of phenotypic novelty. 372 (2017).
Kelemen, O. et al. Function of alternative splicing. Gene 514, 1–30, https://doi.org/10.1016/j.gene.2012.07.083 (2013).
Ward, A. J. & Cooper, T. A. The pathobiology of splicing. The Journal of pathology 220, 152–163, https://doi.org/10.1002/path.2649 (2010).
Tazi, J. et al. Alternative splicing: regulation of HIV-1 multiplication as a target for therapeutic action. The FEBS journal 277, 867–876, https://doi.org/10.1111/j.1742-4658.2009.07522.x (2010).
Raponi, M. & Baralle, D. Alternative splicing: good and bad effects of translationally silent substitutions. The FEBS journal 277, 836–840, https://doi.org/10.1111/j.1742-4658.2009.07519.x (2010).
Hallegger, M., Llorian, M. & Smith, C. W. Alternative splicing: global insights. The FEBS journal 277, 856–866, https://doi.org/10.1111/j.1742-4658.2009.07521.x (2010).
Dhir, A. & Buratti, E. Alternative splicing: role of pseudoexons in human disease and potential therapeutic strategies. The FEBS journal 277, 841–855, https://doi.org/10.1111/j.1742-4658.2009.07520.x (2010).
Tazi, J., Bakkour, N. & Stamm, S. Alternative splicing and disease. Biochimica et biophysica acta 1792, 14–26, https://doi.org/10.1016/j.bbadis.2008.09.017 (2009).
Cooper, T. A., Wan, L. & Dreyfuss, G. RNA and disease. Cell 136, 777–793, https://doi.org/10.1016/j.cell.2009.02.011 (2009).
Kim, E., Goren, A. & Ast, G. Alternative splicing and disease. RNA biology 5, 17–19 (2008).
Wang, G. S. & Cooper, T. A. Splicing in disease: disruption of the splicing code and the decoding machinery. Nature reviews. Genetics 8, 749–761, https://doi.org/10.1038/nrg2164 (2007).
Chabot, B. & Shkreta, L. Defective control of pre-messenger RNA splicing in human disease. The Journal of cell biology 212, 13–27, https://doi.org/10.1083/jcb.201510032 (2016).
Torrado, M., Maneiro, E., Trujillo-Quintero, J. P., Evangelista, A. & Mikhailov, A. T. A Novel Heterozygous Intronic Mutation in the FBN1 Gene Contributes to FBN1 RNA Missplicing Events in the Marfan Syndrome. 2018, 3536495, https://doi.org/10.1155/2018/3536495 (2018).
Menendez, M. et al. Founder effect of a pathogenic MSH2 mutation identified in Spanish families with Lynch syndrome. Clin Genet 78, 186–190, https://doi.org/10.1111/j.1399-0004.2009.01346.x (2010).
Cariola, F. et al. Characterization of a rare variant (c.2635-2A > G) of the MSH2 gene in a family with Lynch syndrome. The International journal of biological markers, 1724600818766496, https://doi.org/10.1177/1724600818766496 (2018).
Yu, Q. et al. An intron mutation in the ACVRL1 may be associated with a transcriptional regulation defect in a Chinese family with hereditary hemorrhagic telangiectasia. PloS one 8, e58031, https://doi.org/10.1371/journal.pone.0058031 (2013).
Author information
Authors and Affiliations
Contributions
The subject was designed by Yong Zeng; the data were performed and analyzed by Yicong Ye and Yong Zeng; the manuscript was written by Yicong Ye.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ye, Y., Zeng, Y. Whole exome sequencing identifies a novel intron heterozygous mutation in TSC2 responsible for tuberous sclerosis complex. Sci Rep 9, 4456 (2019). https://doi.org/10.1038/s41598-019-38898-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-38898-9
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
