
Abstract
Genetic Creutzfeldt-Jakob disease (gCJD) with E200K mutation is one of the common subtypes of human genetic prion diseases worldwide. In this study, we systematically analyzed 30 Chinese E200K gCJD cases for their epidemiological, clinical, laboratory and genetic features. The patients came from 12 different provinces, majority in northern part of China. The onset age varied from 42 to 71 year-old (y), with the median of was 57 y. The CYP4X1 gene rs9793471 SNP was tested. Only one patient’s rs9793471 genotype was GA and the others’ were AA. The gender ratio (M: F) was 1:1.73 (11:19). The foremost symptoms and clinical progression of Chinese E200K gCJD patients were quite similar as sporadic CJD cases. Only a few cases (4/30) recalled clearly disease related family history. 74.1% (20/27), 86.7% (26/30) and 50.0% (13/26) of the cases were CSF 14-3-3 positive, sCJD associated abnormalities on MRI and special PSWC on EEG, respectively. The median clinical duration was 9 months (varying from 2 to 26 months). All 30 Chinese E200K gCJD patients were M129M and E219E homozygous. 21 members from 3 families conducted PRNP sequencing and 16 asymptomatic carriers of E200K mutation with M129M and E219E homozygous were identified. This is the largest study on E200K gCJD patients in China, which would benefit to the knowledge of E200K gCJD.
Similar content being viewed by others
Introduction
Human prion diseases are a group of rare, fatal and transmissible neurodegenerative diseases, including Kuru, Creutzfeldt-Jakob disease (CJD), Gerstmann-Sträussler-Scheinker syndrome (GSS) and fatal familial insomnia (FFI). The etiology of prion disease is known as a conformational change of the normal prion protein (PrPC) into a pathological conformation (PrPSc) that aggregates in the central nervous system (CNS)1. CJD consists of three main catalogues: sporadic, familial, variant and iatrogenic forms. Familial/genetic CJD (fCJD/gCJD) accounts for about 10–15% of CJD cases worldwide2,3, which closely links to various pathogenic mutations in the prion protein-encoding gene, PRNP. Up to now, about 50 different pathogenic mutations in PRNP have been addressed, which result in dominant hereditary prion diseases4,5.
E200K gCJD, with the substation of Glutamate for Lysine at codon 200, is the most prevalent gCJD in many European countries4,6. According to the surveillance data from Chinese CJD Surveillance Center, E200K gCJD is the third most common genetic prion disease in Chinese, after D178N FFI and T188K gCJD7,8,9. In this prospective study, the epidemiological, clinical, genetic and laboratory characteristics of 30 Chinese E200K gCJD cases were comparatively investigated based on the onset age and gender. The main examinations and laboratory data were also proposed.
Results
General information
The first Chinese E200K gCJD case was described in 200810. Up to April 2018, 30 E200K gCJD cases were identified and diagnosed by China CJD Surveillance Center, with 29 Han-Chinese and one Dong-Chinese. Analysis of the permanent addresses of the patients showed that they came from 12 provinces. 7 cases were from Tianjin (TJ), 5 from Hebei (HB), 4 from Henan (HN), 2 from Beijing (BJ), Zhejiang (ZJ), Neimenggu (NMG), Shandong (SD), and Jiangxi (JX), 1 from Guangdong (GD), Guizhou (GZ), Jilin (JL), and Heilongjiang (HLJ), respectively. The gender ratio (M:F) was 1:1.73 (11:19 cases). The onset ages varied from 42 to 71 year-old (y), with the median of 57.5 y. Most of the patients (12 cases) were in the group of 50–59 y, followed by the group of 60–69 y (10 cases). Female patients showed slightly younger onset age (median: 54 y) than male (median: 61 y), but without significant difference (Fig. 1).

Onset-age distributions of 30 Chinese E200K gCJD patients. The medians of the onset age in total and in different genders are shown in the top right. F: female; M: male.
Clinical features
The interval times of those cases from onset to the reporting to CJD surveillance center varied from 2 to 24 months, with the median of 4.5 months. 12 out 30 cases were referred to the CJD surveillance center within 3 months after onset. The main clinical and laboratory features of those patients were summarized in Table 1. Majority of the patients displayed more than one initial symptom. Progressive dementia and recognition problems were mostly described, which appeared in 73.3% (22/30) cases. Various mental problems were recorded in 16 cases, followed by pyramidal and extrapyramidal symptoms in 9 cases, cerebellum symptoms in 6 cases, and sleep disorder in 3 cases. Along with the disease progression, more sporadic CJD (sCJD) associated signs appeared. In the context of whole clinical courses, dementia was noticed in 96.7% (29/30) patients, pyramidal or extrapyramidal dysfunction in 73.3% (22/30), myoclonus in 70.0% (21/30), visual or cerebellar disturbance in 83.3% (25/30) and akinetic mutism in 53.3% (16/30). The patients were further grouped based on gender and onset age (<60 y and ≥60 y). As shown in Table 1, female patients seemed to be less frequent having cerebellum disorders (P = 0.0156) as the foremost symptoms as male. The older patients appeared higher ratio having cerebellum disorders, although without statistical difference. For the major sCJD associated neurological signs during the clinical course, there was no statistical difference between the groups of gender and onset-age. The older patients seemed to display higher ratios having more symptoms than young patients, especially mutism at later stage of diseases. Myoclonus, pyramidal and extrapyramidal symptoms were slightly frequent in male patients.
Clinical examination and laboratory tests
Lumber puncture was performed in 27 patients and the routine biochemistry items were all in the normal ranges. Western blot for CSF 14-3-3 was positive in 74.3% (20/27) cases. There was no significant difference in CSF 14-3-3 positive between genders or between young and older patients. Like the observation in sCJD patients, E200K gCJD cases with CSF 14-3-3 positive seemed to have more clinical manifestations. Half of the patients (10/20, 50.0%) of 14-3-3 positive displayed four and more than four sCJD-related symptoms, while 6 out of 7 patients who were 14-3-3 negative showed three and less three signs during their clinical courses.
EEG was recorded in the 26 cases, half (13/26) of which revealed sCJD associated periodic sharp wave complexes (PSWCs). Female patients appeared higher ratio of PSWC positive. All cases received MRI scanning, 26 showed abnormalities. Elder patients seemed to have higher rate of MRI typical abnormality, which was up to 100% in the group of 60–69 y. Additionally, 9 cases showed all positive on CSF-14–3–3, special abnormalities on MRI and PSWC on EEG. 95.0% (19/20) of the cases with CSF 14-3-3 positive displayed special abnormality on MRI. 84.6% (11/13) of the cases with PSWC on EEG showed abnormalities on MRI. Among the 11 cases with PSWC on EEG who performed CSF 14-3-3 Western blots, 9 cases (81.8%) showed CSF 14-3-3 positive. Only one case was negative in all three examinations.
Survival time
By the end of April 2018, 21 out of 30 E200K gCJD patients died, 6 lost contact and 3 are still alive. The clinical durations of the dead patients varied from 2 to 26 months, with the median of 9 months. 57.1% of the cases (12/21) died within one year after onset, and 47.6% (10/21) died within 6 months after onset. One patient lived longer than two years. The survivorship curve displayed as Fig. 2. Male patients (median: 8 m) seemed to have shorter duration than female (median: 9.7 m). There was no significant difference in the clinical duration between young and older patients, between the patients with more (≥four) and less (≤three) sCJD-related symptoms, with CSF 14-3-3 positive, PSWC in EEG and typical abnormalities in MRI.

The curves of the survival times of 30 E200K gCJD patients based on different groups. (a) male and female patients; (b) <60 or ≥60 y at onset.
PRNP, CYP4X1 rs9793471 sequencing and family history
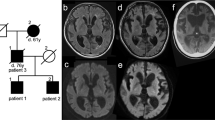
All 30 E200K gCJD patients were confirmed to have a mutation with the substation of Glutamate for Lysine at codon 200 in one of the PRNP allele. All cases were methionine homozygous genotype at codon 129 (M129M) and glutamic acid homozygous at codon 219 (E219E) of PRNP. Twenty nine cases were CYP4X1 rs9793471 AA genotype, and one case was GA. The number was too small to analyze the correlation between rs9793471 genotype and onset age. Only four cases recorded possible disease-related family history. Case 3 was 58 year-old women, who showed progressive dementia, cerebellar disturbance and akinetic mutism at the end stage. She died in 5 months afterwards. Her son, elder daughter, young daughter and four grandkids donated their blood samples for PRNP sequencing. Six of them were E200K mutation carries. Her elder daughter died of sudden cerebral hemorrhage at the age of 29 year-old three years after PRNP sequencing (Fig. 3A). Case 20 appeared neurological symptoms at 67 year-old. Her elder sister was recalled to die with the similar clinical manifestation but without referable neurological diagnosis. One of her elder brothers died at 62 year-old without definite diagnosis. The family member recalled that he had visual impairment and died a couple of months after onset. 7 members of this family were tested with PRNP sequencing. Her two daughters, her young sister and one niece were E200K mutation carries. The other three tested members, a nephew, a niece and a grandnephew, were wild-type PRNP (Fig. 3B). Case 23 was male who displayed clinical symptoms at 52 year-old. His father was recalled to die with the similar symptoms. PRNP sequencing revealed his son and one of his brothers were E200K mutation carries (Fig. 3C). Case 24 appeared neurological symptoms at 62 year-old. His mother was dead with similar clinical manifestation (Fig. 3D). The family members also recalled that one of his younger female cousins was dead of neurological problems. None of his family member donated the blood for PRNP sequencing. He is still alive, with the symptoms of dementia, pyramidal and extrapyramidal dysfunctions, and is now bedridden. In addition, all asymptomatic E200K carriers in this study remained healthy by the end of April, 2018.

Disease related family histories and E200K mutations within PRNP in four families. (A) Case 3; (B) Case 20; (C). Case 23; (D) Case 24. The significances of various shapes, lines and colors are summarized in the panel below.
Discussion
In this study, we systematically analyzed 30 Chinese E200K gCJD cases for their epidemiological, clinical, laboratory and genetic features. From our surveillance data of human prion diseases since 2006, about 150 cases of genetic prion diseases have been identified, which contain 15 different subtypes of PRNP mutations, which accounts about 10% of all referring human prion diseases7. E200K gCJD is the third most commonly observed genetic prion disease in China after D178N FFI and T188K gCJD7,8,11. E200K mutation is the largest cluster of gCJD existing in the North African Jewish community of Libyan and Tunisian ancestry12. In many European countries E200K is the most prevalent mutation in Caucasian4,13,14,15,16. Among the 30 E200K gCJD cases in this study, the number of female patients is slightly more than that of male ones. All E200K gCJD cases in this study appear sporadic onset, without any relationship of consanguinity. Although the majority of those E200K gCJD cases are come from northern provinces of China, the relatively small numbers of the cases seems not to be suitable for further analysis of geography association.
The onset and clinical characteristics of Chinese E200K gCJD are more like as sCJD, although the onset age of E200K gCJD is younger than that of sCJD8,17. CYP4X1 SNP rs9793471 has an influence on age at onset of disease in patients with E200K genetic and sporadic CJD18. In this study, twenty nine cases were CYP4X1 rs9793471 AA genotype, and only one case was GA. So the number was too small to analyze the correlation between rs9793471 genotype and onset age. Progressive dementia and mental symptoms are the most reported foremost ones. Along with the disease progression, dementia and other main sCJD-associated neurological signs are frequently observable. Like the other studies19,20,21, three patients in this study complain sleeping problems as the foremost symptoms. The first Chinese E200K patient who was reported in 2010 also had sleeping disturbance as the initial symptom10. The positive rates of CSF 14-3-3, PSWC in EEG and special abnormalities in MRI of those E200K gCJD cases are similar as those of Chinese sCJD8. Therefore, without PRNP sequencing, it is almost impossible to distinguish E200K gCJD and sCJD clinically. The median survival time of E200K gCJD cases is longer than that of 257 Chinese sCJD patients (4.5 moths)8. However, similar as the data of sCJD, the exact clinical duration each case varies largely, while no special associations of the survival time with gender, onset age and other clinical features are addressed.
Clinically, the disease associated family history among 30 Chinese E200K gCJD patients is less frequent, that only four cases are recalled to have disease-associated family history during our follow-up survey. Several E200K mutation carries are identified in the three families having performed PRNP sequencing. It is reported that carriers of the E200K mutation are born with this dominant mutation in the PRNP gene, but most remain asymptomatic until middle age and some only develop the disease after the age of 80 years22,23. Although the clinical durations of three dead patients with positive family history seem to be slightly shorter, however, without showing significance by statistical analysis. Additionally, all 30 Chinese E200K gCJD patients are M129M and E219E homozygous, which reflects a typical profile of PRNP genotype of Northeastern Asian. Actually, all Chinese patients with various genetic prion diseases are M129M so far, whose rate is obviously higher than that of normal Chinese population (about 93%) and sCJD patients (98.5%)8.
The features of neuropathology and PrPSc deposit of Chinese E200K gCJD patients remain unknown, as none of those patients undergoes postmortem or biopsy assay. Typical spongiform changes and synaptic-type PrPSc deposition have been described in the brains of E200K gCJD patients in Japan, which resembles MM1-type sCJD24. A more detailed neuropathological study has proposed that the immunoreactivity of PrP of E200K gCJD cases, defined as multiple globular structures, is distinct from sCJD with intraneuronal small dot like profile25. On the other hand, higher positive rate of E200K gCJD patients in CSF RT-QuIC has been reported repeatedly26,27,28. Moreover, the CSF samples of E200K gCJD cases show higher maximal intensity of ThT fluorescent signal and shorter lag phase of positive conversion than those of sCJD ones. It possibly reflects more prion agent releasing from brain tissues to CSF. In line with those observations, higher ThT values and shorter lag phases in CSF RT-QuIC assays are also observed in Chinese E200K gCJD cases compared with the data of sCJD, as well as T188K gCJD and D178N FFI (unpublished data).
Nowadays, therapeutic intervention in human prion diseases is still unsuccessful. Some protocols have showed the abilities to reduce the speed of deterioration of prion disease in short periods of time, but none shows the effect to reverse the severe neurological deficits apparent already at diagnosis29,30,31. Several mouse models for human gCJD have been constructed, such as mouse models for E200K gCJD32, P102L GSS33, A117V GSS34 and D178N FFI35. Recently we have constructed a transgenic mice expressing human PRNP with T188K mutation for T188K gCJD (unpublished data). Using those mouse models, the pathogenesis of human genetic prion diseases and potential therapeutic and prophylactic tools will be further exploded.
Materials and Methods
Ethics statement
This study was approved by the Ethical Committee of the National Institute for Viral Disease Control and Prevention (Beijing, China) under the protocol 2011ZX10004-101. The written informed content of each patient was got either by a family member or a relative of the patient prior to inclusion in the study, according to the requirement of CJD surveillance. And all methods were performed in accordance with the relevant guidelines and regulations.
Study population and data collection
30 genetic-confirmed E200K gCJD cases from Chinese CJD Surveillance Center from 2011 to 2018 were enrolled in this study. The diagnosis of CJD was based on the diagnostic criteria issued by China National Health Commission, requiring a progressive dementia with at least two out of four following manifestations clinically: myoclonus, visual or cerebellar symptoms, pyramidal or extrapyramidal dysfunction or akinetic mutism. Diagnosis of probable CJD was supported by typical changes in magnetic resonance imaging (MRI) with the presences of high signal in caudate/putamen and/or symmetrical or dissymmetrical cortical ribbon syndrome on diffusion-weighted imaging (DWI), or electroencephalography (EEG) with the presences of periodic sharp wave complexes (PSWC), or CSF 14-3-3 positive. Diagnosis of E200K gCJD needs the sequencing of PRNP. All patients were followed up till death by the specialist in Chinese CJD Surveillance Center. The survival time was evaluated from disease onset to death.
Laboratory tests
The whole blood and CSF samples of the referred patients were collected by the clinicians from local hospitals according to the CJD surveillance protocol7,17,36. The whole DNAs of the blood samples were extracted by a commercial kit (Qiagen 51104). The PRNP gene was amplified by a PCR protocol with the specific primers (forward: 5-GGCAAACCTTGGATGCTGG-3, reverse: 5-CCCACTATCAGGAAGATGAGG-3) and subjected into sequencing analysis of PRNP and polymorphisms of codon 129 and 21917,36. The CYP4X1 rs9793471 SNP was amplified and sequenced with specifically designed primers(forward: TGCGTTAACTTCACAAGGTGACA, reverse: GGTCAGGGCCTAATGATCCTCC) to test rs9793471 genotype. Then analyze the relation between CYP4X1 and the age at onset. The CSF samples were employed into Western blot for 14-3-3 protein based the protocol described previously8,17,36.
Statistical analysis
The statistical analyses were performed using the SAS 9.4 statistical software program. Significance was determined using fisher’s precise test. A difference was considered statistically significant if the p value was <0.05.
References
Prusiner, S. B. Prions. Proceedings of the National Academy of Sciences of the United States of America 95, 13363–13383 (1998).
Chen, C. & Dong, X. P. Epidemiological characteristics of human prion diseases. Infectious diseases of poverty 5, 47, https://doi.org/10.1186/s40249-016-0143-8 (2016).
Collinge, J. Prion diseases of humans and animals: their causes and molecular basis. Annual review of neuroscience 24, 519–550, https://doi.org/10.1146/annurev.neuro.24.1.519 (2001).
Kovacs, G. G. et al. Genetic prion disease: the EUROCJD experience. Human genetics 118, 166–174, https://doi.org/10.1007/s00439-005-0020-1 (2005).
Schmitz, M. et al. Hereditary Human Prion Diseases: an Update. Molecular neurobiology 54, 4138–4149, https://doi.org/10.1007/s12035-016-9918-y (2017).
Kovacs, G. et al. Genetic Creutzfeldt-Jakob disease associated with the E200K mutation: characterization of a complex proteinopathy. Acta Neuropathol. 121, 39–57 (2011).
Shi, Q. et al. The Features of Genetic Prion Diseases Based on Chinese Surveillance Program. PloS one 10, e0139552, https://doi.org/10.1371/journal.pone.0139552 (2015).
Shi, Q. et al. The Characteristics of Chinese Prion Diseases Based On 10 Years Surveillance Data from 2006 To 2015. Neuropsychiatry (London) 8, 739–750, https://doi.org/10.4172/Neuropsychiatry.1000550 (2018).
Shi, Q. et al. Fatal Familial Insomnia: Insight of the Most Common Genetic Prion Disease in China Based On the Analysis of 40 Patients. Neuropsychiatry (London) 8, 739–747, https://doi.org/10.4172/Neuropsychiatry.1000524 (2018).
Gao, C. et al. The first Chinese case of Creutzfeldt-Jakob disease with mutation of E200K in PRNP. Biomedical and environmental sciences: BES 23, 158–160 (2010).
Shi, Q. et al. Rare genetic Creutzfeldt-Jakob disease with T188K mutation: analysis of clinical, genetic and laboratory features of30 Chinese patients. Journal of neurology, neurosurgery, and psychiatry 88, 889–890, https://doi.org/10.1136/jnnp-2016-314868 (2017).
Goldfarb, L. G., Mitrova, E., Brown, P., Toh, B. K. & Gajdusek, D. C. Mutation in codon 200 of scrapie amyloid protein gene in two clusters of Creutzfeldt-Jakob disease in Slovakia. Lancet 336, 514–515 (1990).
Mitrova, E., Kosorinova, D., Gajdos, M., Sebekova, K. & Tomeckova, I. A pilot study of a genetic CJD risk factor (E200K) in the general Slovak population. European journal of epidemiology 29, 595–597, https://doi.org/10.1007/s10654-014-9937-9 (2014).
Mancuso, M. et al. Creutzfeldt-Jakob disease with E200K PRNP mutation: a case report and revision of the literature. Neurological sciences: official journal of the Italian Neurological Society and of the Italian Society of Clinical Neurophysiology 30, 417–420, https://doi.org/10.1007/s10072-009-0118-7 (2009).
Heinemann, U. et al. Creutzfeldt-Jakob disease in Germany: a prospective 12-year surveillance. Brain: a journal of neurology 130, 1350–1359, https://doi.org/10.1093/brain/awm063 (2007).
Ladogana, A. et al. High incidence of genetic human transmissible spongiform encephalopathies in Italy. Neurology 64, 1592–1597, https://doi.org/10.1212/01.WNL.0000160118.26865.11 (2005).
Gao, C. et al. The epidemiological, clinical, and laboratory features of sporadic Creutzfeldt-Jakob disease patients in China: surveillance data from 2006 to 2010. PloS one 6, e24231, https://doi.org/10.1371/journal.pone.0024231 (2011).
Poleggi, A. et al. Age at onset of genetic (E200K) and sporadic Creutzfeldt-Jakob diseases is modulated by the CYP4X1 gene. Journal of neurology, neurosurgery, and psychiatry. https://doi.org/10.1136/jnnp-2018-318756 (2018).
Taratuto, A. L. et al. Insomnia associated with thalamic involvement in E200K Creutzfeldt-Jakob disease. Neurology 58, 362–367 (2002).
Cohen, O. S. et al. Characterization of sleep disorders in patients with E200K familial Creutzfeldt-Jakob disease. Journal of neurology 262, 443–450, https://doi.org/10.1007/s00415-014-7593-3 (2015).
Givaty, G., Maggio, N., Cohen, O. S., Blatt, I. & Chapman, J. Early pathology in sleep studies of patients with familial Creutzfeldt-Jakob disease. Journal of sleep research 25, 571–575, https://doi.org/10.1111/jsr.12405 (2016).
Chapman, J., Ben-Israel, J., Goldhammer, Y. & Korczyn, A. D. The risk of developing Creutzfeldt-Jakob disease in subjects with the PRNP gene codon 200 point mutation. Neurology 44, 1683–1686 (1994).
Cohen, O. S. et al. Familial Creutzfeldt-Jakob disease with the E200K mutation: longitudinal neuroimaging from asymptomatic to symptomatic CJD. Journal of neurology 262, 604–613, https://doi.org/10.1007/s00415-014-7615-1 (2015).
Higuma, M. et al. Relationships between clinicopathological features and cerebrospinal fluid biomarkers in Japanese patients with genetic prion diseases. PloS one 8, e60003, https://doi.org/10.1371/journal.pone.0060003 (2013).
Kovacs, G. G. et al. Intraneuronal immunoreactivity for the prion protein distinguishes a subset of E200K genetic from sporadic Creutzfeldt-Jakob Disease. Journal of neuropathology and experimental neurology 71, 223–232, https://doi.org/10.1097/NEN.0b013e318248aa70 (2012).
Sano, K. et al. Early detection of abnormal prion protein in genetic human prion diseases now possible using real-time QUIC assay. PloS one 8, e54915, https://doi.org/10.1371/journal.pone.0054915 (2013).
Cramm, M. et al. Characteristic CSF prion seeding efficiency in humans with prion diseases. Molecular neurobiology 51, 396–405, https://doi.org/10.1007/s12035-014-8709-6 (2015).
Franceschini, A. et al. High diagnostic value of second generation CSF RT-QuIC across the wide spectrum of CJD prions. Scientific reports 7, 10655, https://doi.org/10.1038/s41598-017-10922-w (2017).
Geschwind, M. D. Clinical trials for prion disease: difficult challenges, but hope for the future. The Lancet. Neurology 8, 304–306, https://doi.org/10.1016/S1474-4422(09)70050-X (2009).
Gandini, A. & Bolognesi, M. L. Therapeutic Approaches to PrionDiseases. Progress in molecular biology and translational science 150, 433–453, https://doi.org/10.1016/bs.pmbts.2017.06.013 (2017).
Haik, S. et al. Doxycycline in Creutzfeldt-Jakob disease: a phase 2, randomised, double-blind, placebo-controlled trial. The Lancet. Neurology 13, 150–158, https://doi.org/10.1016/S1474-4422(13)70307-7 (2014).
Canello, T. et al. Copper is toxic to PrP-ablated mice and exacerbates disease in a mouse model of E200K genetic prion disease. Neurobiology of disease 45, 1010–1017, https://doi.org/10.1016/j.nbd.2011.12.020 (2012).
Asante, E. A. et al. Transmission Properties of Human PrP 102L Prions Challenge the Relevance of Mouse Models of GSS. PLoS pathogens 11, e1004953, https://doi.org/10.1371/journal.ppat.1004953 (2015).
Yang, W. et al. A New Transgenic Mouse Model of Gerstmann-Straussler-Scheinker Syndrome Caused by the A117V Mutation of PRNP. The Journal of neuroscience: the official journal of the Society for Neuroscience 29, 10072–10080, https://doi.org/10.1523/JNEUROSCI.2542-09.2009 (2009).
Bouybayoune, I. et al. Transgenic fatal familial insomnia mice indicate prion infectivity-independent mechanisms of pathogenesis and phenotypic expression of disease. PLoS pathogens 11, e1004796, https://doi.org/10.1371/journal.ppat.1004796 (2015).
Shi, Q. et al. Quality evaluation for the surveillance system of human prion diseases in China based on the data from 2010 to 2016. Prion 10, 484–491, https://doi.org/10.1080/19336896.2016.1229731 (2016).
Acknowledgements
We thank the staffs in Chinese National CJD Surveillance Network. This work was supported by National Science and Technology Major Project (2018ZX10711001), Chinese National Natural Science Foundation Grants (81630062, 81572048), Research Project (2016YFC1202700, 2017YFC1200500) and SKLID Development Grant (2015SKLID503 and 2016SKLID603).
Author information
Authors and Affiliations
Contributions
X.P.D. and Q.S. designed the experiments. L.P.G., K.X. and W.Z. performed the experiments. L.P.G., Q.S. and X.P.D. prepared the manuscript, while J.W., C.C. provided suggestions for revision.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gao, LP., Shi, Q., Xiao, K. et al. The genetic Creutzfeldt-Jakob disease with E200K mutation: analysis of clinical, genetic and laboratory features of 30 Chinese patients. Sci Rep 9, 1836 (2019). https://doi.org/10.1038/s41598-019-38520-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-38520-y
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
