
Abstract
RT-qPCR is a widely used technique for the analysis of gene expression. Accurate estimation of transcript abundance relies strongly on a normalization that requires the use of reference genes that are stably expressed in the conditions analyzed. Initially, they were adopted from those used in Northern blot experiments, but an increasing number of publications highlight the need to find and validate alternative reference genes for the particular system under study. The development of high-throughput sequencing techniques has facilitated the identification of such stably expressed genes. Nicotiana benthamiana has been extensively used as a model in the plant research field. In spite of this, there is scarce information regarding suitable RT-qPCR reference genes for this species. Employing RNA-seq data previously generated from tomato plants, combined with newly generated data from N. benthamiana leaves infiltrated with Pseudomonas fluorescens, we identified and tested a set of 9 candidate reference genes. Using three different algorithms, we found that NbUbe35, NbNQO and NbErpA exhibit less variable gene expression in our pathosystem than previously used genes. Furthermore, the combined use of the first two is sufficient for robust gene expression analysis. We encourage employing these novel reference genes in future RT-qPCR experiments involving N. benthamiana and Pseudomonas spp.
Similar content being viewed by others

Introduction
Plants are in constant interaction with beneficial and pathogenic microorganisms. For detection of these microbes, plants use pattern-recognition receptors (PRRs) that perceive conserved features named microbe- (or pathogen-) associated molecular patterns (MAMPs or PAMPs), activating pattern-triggered immunity (PTI), the first layer of inducible plant defense1,2,3. PTI is associated with the production of reactive oxygen species, activation of mitogen-activated protein kinases (MAPKs), changes in intracellular calcium concentrations and changes in gene expression that prevent the infection of many potentially pathogenic microbes4,5,6,7,8. Some bacterial pathogens such as Pseudomonas syringae use a type III secretion system to introduce virulence proteins (effectors) into the plant cell cytoplasm to counteract PTI5,9 and to manipulate host metabolic processes in order to facilitate growth and proliferation in the apoplast10,11. The second layer of plant immune response, referred as effector-triggered immunity (ETI), consists of the intracellular detection of pathogen effectors by resistance proteins (R proteins)12,13. This immune response is often associated with a hypersensitive response (HR) that leads to localized cell death, which restricts pathogen spread5,14. Some effectors are involved in the suppression of this plant-immunity associated cell death15.
Pathogens cause up to 30% of crop loss, which has a detrimental economical impact16. Therefore, scientific progress aimed at understanding how plants respond to infections is an important step for the design of new technologies to increase food production and quality. Pseudomonas syringae pv. tomato (Pst) has been used as a model bacteria in the molecular studies of plant-pathogen interactions17. One of the reasons for this is that Pst can be manipulated to infect tomato, Arabidopsis and also Nicotiana benthamiana plants17.
The Australian endemic plant N. benthamiana, is an important model organism in plant biology18. This species belongs to the Solanaceae family along with several economically important crops such as tomato, eggplant, potato, tobacco and petunia. It was adopted for virology studies because of its susceptibility to different virus strains19. Nowadays, several reasons make this species a model for plant research, including amenability to genetic transformation, high efficiency using virus-induced gene silencing (VIGS) and efficient transient protein expression and the availability of a draft genome sequence18,19,20.
Several experimental methods are available for gene expression quantification. Reverse transcription-quantitative PCR (RT-qPCR) is a widely used technique for the determination of mRNA level changes in different biological systems21. Although it is considered an important and frequently used method in laboratories, RT-qPCR results can be misinterpreted if several critical steps are not carefully followed22,23. One of them is the selection of optimal reference genes for accurate normalization of transcript abundance. A reference gene should have a minimal expression variation in the analyzed conditions23,24. Most of the RT-qPCR reference genes traditionally used such as glyceraldehyde-3-phosphate dehydrogenase (GADPH), 18 S ribosomal RNA (18 S RNA), beta-tubulin-4 (TUB4), elongation factor 1 alpha (EF1α), polyubiquitin (UBQ), actin (ACT), have been adopted from Northern blot and semi-quantitative RT-PCR experiments24. However, recent studies indicate that traditional reference genes are not always stably expressed and suggest the necessity of a systematic selection and validation of reference genes for each particular experimental condition. Despite the importance of N. benthamiana as a research model plant, to our knowledge only two studies have evaluated reference genes for this species. These reports analyzed the expression stability of traditional reference genes in plants infected with virus25 or plants used for VIGS experiments26.
RNA-seq is a powerful high-throughput technology used for transcriptome analysis in different organisms under diverse conditions and treatments27,28,29,30. Previously, RNA-seq was used to study transcriptional changes during the activation of PTI in tomato and the subsequent inhibition of this response by Pst AvrPto and AvrPtoB effectors7. Following a similar approach, genes specifically induced or repressed during PTI or ETI activation were identified8. Analyzing this large set of data, combined with newly generated data, novel RT-qPCR reference genes in the tomato-Pseudomonas pathosystem were recently identified and validated31. Taking advantage of these data previously generated for tomato, we aimed here at transferring the information to N. benthamiana by identifying the most closely related genes, with the hypothesis that these genes would also have stable expression in N. benthamiana. We generated new RNA-seq data for N. benthamiana challenged with Pseudomonas fluorescens 55 (PTI activation) and used this information to establish a set of candidate genes for validation. These novel reference genes were then tested using three different algorithms (geNorm, NormFinder and Bestkeeper) and their performance compared with two traditional reference genes (NbEF1α and NbGADPH) and NbPP2a, a previously validated reference gene for virus-infected N. benthamiana25. Our analysis allowed the identification of three novel RT-qPCR reference genes (NbUbe35, NbNQO and NbErpA) that can be used in the N. benthamiana-Pseudomonas pathosystem or related systems.
Results
Selection of stably expressed genes based on tomato and Nicotiana benthamiana RNA-seq data
In order to identify genes with low expression variation to be used as reference in RT-qPCR experiments, we took advantage of a previous analysis of gene stability based on RNA-seq data from tomato leaves with different treatments (37 treatments/time points with an average of 3 biological replicates generated in independent experiments)31. This large set of tomato data was narrowed down to 50 genes with the most stable expression across all treatments. Performing BlastX analysis using these tomato genes as input and N. benthamiana proteins as database, we identified the closest putative orthologs and hypothesized they would also be stably expressed genes. To assist in the selection of reference genes, we performed RNA-seq analysis with N. benthamiana leaves vacuum-infiltrated with a suspension of Pseudomonas fluorescens 55 and MgCl2 as a mock. This bacterial treatment, that results in a strong PTI induction and large transcriptomic changes at 6 h after infiltration (hai)7, allowed the identification of 10,300 differentially expressed genes from the 57,139 predicted genes in N. benthamiana (Supplementary Table S1). Using this information generated from N. benthamiana tissue, we calculated the coefficient of variation (CV) of the 50 selected genes, using the expression values (RPKMs, reads per kilobase of transcript per million mapped reads) of each biological replicate individually. The lower the CV is, the more stable the expression of the gene is across the conditions. In this way we were able to select 9 genes with the lowest CV (ranging from 5.14% to 10.27%) for analysis (Supplementary Table S2). We also selected a gene named NbPP2a (Niben101Scf09716g01002.1) previously validated as a stable reference gene in virus-infected N. benthamiana plants25 and two traditional plant reference genes NbEF1α and NbGADPH.
Analysis of candidate reference genes expression profiles indicated high efficiencies and unique transcript amplifications
We evaluated the amplification efficiencies of each selected gene performing RT-qPCR using cDNA dilutions (1:5, 1:10, 1:100, 1:1000). Amplification efficiency E was measured as 10−1/slope and expressed in percentage (Supplementary Table S3). All the primers showed high E values ranging from 92% to 100%. We also analyzed the specificity of the amplification through melting curves for all pairs of primers used and in all cases observed a single peak accounting for a single PCR product (Supplementary Fig. S1). We checked the presence of contamination and primer dimers with the analysis of the non-template control melting curves. Only in one of the three technical replicates corresponding to NbTspan gene, we observed a small peak. Analysis in detail of its corresponding Cq value (35.25) showed that was nearly 10 Cq-values lower than those obtained when using template (<25.80). Considering the mentioned difference in Cq values, this amplification was ignored22.
Cycle amplification values (Cq) allowed narrowing down the number of selected genes
We designed an experiment for the evaluation of our set of genes under different immune responses using the model plant N. benthamiana. A summary of the infiltrations performed is shown in Table 1. In order to induce PTI activation we infiltrated N. benthamiana leaves with Pseudomonas fluorescens 55 (Pf)32 and used 10 mM MgCl2 as a mock treatment. Additionally, we infiltrated N. benthamiana leaves with Pst DC300033 and Pst DC3000 ΔhopQ1-134. The comparison of these last two treatments allows dissecting ETI response. We collected leaf tissue from 3 biological replicates at 6 and 12 hai and later on we visually monitored the development of symptoms on the plants to confirm activation of the expected defense responses.
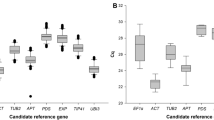
Average Cq values (Fig. 1) for most of the genes were within the recommended values for a RT-qPCR reference gene (higher than 15 and lower than 30)35. Two genes were the exception, with mean Cq values of 34.4 (NbLip) and 30.6 (NbP5βR), and were consequently excluded from further analysis. Among the 10 remaining genes, NbEF1α and NbTspan had the largest (5.2) and smallest (1.4) difference between maximum and minimum Cq values, respectively.

Cycle quantification (Cq) values of selected genes. Box and whisker plot graph showing Cq values of each selected gene in all treatments (Table 1) for all the samples analyzed (three biological replicates per treatment, three technical replicates per sample, n = 24). Black lines and boxes represent the Cq medians and 25/75 percentiles, respectively. Whisker caps represent the minimum and maximum Cq values. ○, indicate outliers.
Based on different algorithms, three newly identified reference genes are the most stably expressed
To estimate gene expression stability, we analyzed RT-qPCR data with three different software tools. We first used geNorm software36 to establish the average expression stability value M. This program determines the pairwise variation of each gene with all other analyzed genes under the same experimental conditions. The lower the M value, the more stable the gene is. Three genes presented the highest variability, with M values over the usually proposed cutoff value of M ≤0.537. These genes were NbKLC (M = 0.659), NbEF1α (M = 0.618) and NbGADPH (M = 0.556). This software also selects an optimal pair of reference genes and in this experiment the most stable ones were NbCENPO and NbUbe35 with M value of 0.306 (Fig. 2A).

geNorm analysis of selected reference genes in N. benthamiana leaves infiltrated with different Pseudomonas strains. (A) N. benthamiana reference genes were ranked based on expression stability calculated by geNorm. M values represent the average pairwise variation of the gene compared with all other control genes. (B) Pairwise variation (Vn/Vn + 1) for determination of the optimal number of reference genes. The pairwise variation was calculated considering all the samples treatments and time-points together (Total), mock and P. fluorescens (PTI), Pst DC3000 and Pst DC3000 ΔhopQ1-1 (ETI), samples taken at 6 hai (6 h) or samples taken at 12 hai (12 h).
We then estimated the minimal number of reference genes to be used. To achieve this, we determined the pairwise variation (V) of a normalization factor (NF) calculated by introducing reference genes one by one, starting from the two least variable and adding the rest in a decreasing stability order until the whole set was included36. We decided to analyze our data as a whole, only including PTI activation (Pf 55 and mock), only including ETI activation (Pst DC3000 and Pst DC3000 ΔhopQ1-1), only including samples taken at 6 hai (6 h) and only including samples taken at 12 hai (12 h) (Fig. 2B). Regardless of using the complete dataset, the defense response or time-point subsets, the results were very similar. In all of the cases, the V2/3 value obtained was smaller than the proposed cut-off of 0.1536, suggesting that only the two most stable reference genes (NbCENPO and NbUbe35) identified by geNorm software are sufficient for a good normalization of RT-qPCR data, regardless of the type of response evaluated (PTI or ETI) or time-point (6 or 12 h).
Another algorithm that also calculates an M index is NormFinder38. To calculate this index, NormFinder estimates the intragroup (within each sample/treatment) and then the intergroup (within different groups of samples/treatments) variation. Similarly to geNorm analysis, NormFinder selected NbEF1α, NbKLC and NbGADPH as the most variable genes, with M values of 0.402, 0.387 and 0.352, respectively. However, the most suitable reference genes derived from NormFinder analysis were NbPP2a (M = 0.167), NbErpA (M = 0.216) and NbNQO (M = 0.242) (Fig. 3).

NormFinder expression stability of selected reference genes in N. benthamiana-Pseudomonas pathosystem. N. benthamiana reference genes were ranked based on expression stability calculated by NormFinder. The analysis was performed using expression data from all biological replicates and treatments (n = 24).
To further analyze the candidate gene stability, we used BestKeeper39. This tool allows the analysis of 10 genes in two steps. First, it calculates different statistical parameters and then, a coefficient of correlation (r) is obtained by comparing a BestKeeper index with each particular gene (Table 2). According to standard deviation (SD) values all the genes under study were suitable to be considered as reference genes (SD [±Cq] <1 and SD [±x-fold] <2)39. We analyzed their variation parameters (SD [±Cq] and CV [% Cq]), and observed that NbTspan, NbUbe35 and NbNQO were the most stable genes and NbEF1α, NbGADPH and NbKLC, the most variable ones. The calculated r values for the comparison of each gene with the BestKeeper index were inconsistent with the SD-based analysis described above. For example, according with SD [±Cq] value, NbEF1α is the most variable gene but its coefficient of correlation was high (0.81). We therefore used SD [±Cq] and CV [% Cq] parameters to rank the genes based on their stability (Table 2). This approach is employed in the RefFinder tool40 as a ranking method for the BestKeeper output.
When we compared the outputs of the statistical programs used, we observed a certain degree of discrepancy mainly in the selection of the genes with lower variability across the experiment. This degree of divergence among the stability ranking generated by geNorm, NormFinder and BestKeeper has been previously reported and could attributed to the fact that these tools are based on different algorithms31,41,42,43. In order to analyze the results globally, we calculated the arithmetical mean of the ranking value obtained for each gene using all three algorithms31,41,42,44. As a result, NbUbe35 was rated as the most stable with a mean ranking value of 2.33 (Table 3). With this overall stability ranking we reanalyzed the pairwise variation in order to establish the minimum number of reference genes for normalization, considering the proposed threshold of 0.1536. Based on this analysis, the combination of NbUbe35 and NbNQO is sufficient for accurate normalization (Supplementary Fig. S2) and we consequently used this combination for further experiments.
Validation of the selected genes confirmed their suitability as reference genes
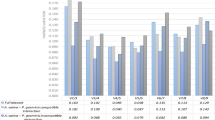
Using BlastP we identified two putative N. benthamiana orthologs of a previously described tomato gene (Solyc02g069960) that is induced by PTI8: Niben101Scf04323g01009.1 and Niben101Ctg15860g00004.1 (NbNAC042). According to our RNA-seq data the first one had very low expression (0-0.46 RPKM), while NbNAC042 was induced by PTI with RKPM values ranging between 0 and 20.53 (Supplementary Table S4). For this reason we chose NbNAC042 to put to test the two most stable genes described here (NbUbe35 and NbNQO) and compare their performance to a middle-ranked gene (NbTspan) and a traditionally used reference gene (NbEF1α). Regardless of the reference genes used the trend of transcript abundance increase of NbNAC042 upon PTI induction (6 and 12 hai), was similar (Fig. 4A). Nevertheless, the combined use of NbUbe35 and NbNQO resulted in lower standard deviation values allowing establishing statistically significant differences at both time-points and at a lower significance level. To our surprise NbNAC042 gene expression increased with ETI activation (Pst DC3000 vs. Pst DC3000 ΔhopQ1-1), given Solyc02g069960 in tomato is not affected by ETI at 6 hai8. Again, the use of the combination of NbUbe35 and NbNQO lead to lower deviations and lower significance levels (Fig. 4B). These results highlight the relevance of the selection of accurate reference genes for the experimental system under study.

Relative expression of NbNAC042 analyzed using different reference genes. Relative expression analysis by RT-qPCR at two time points (6 and 12 hai) using plants infiltrated with: (A), mock (10 mM MgCl2) or 108 cfu/ml Pseudomonas fluorescens 55 (Pf); (B), 5 × 106 cfu/ml of Pseudomonas syringae pv. tomato DC3000 (Pst DC3000) or Pst DC3000 ΔhopQ1-1 (ΔhopQ1-1) strains. In both cases, the geometric mean of the two best (NbUbe35/NbNQO), the intermediate (NbTspan) or the worst (NbEF1α) reference genes was used for normalization of the data. The relative expression was expressed as E−ΔΔCq, where E corresponds to the primer efficiency value. Calibration samples are 10 mM MgCl2 6 h in A and Pst DC3000 6 h in B (biological replicate 1 in both cases). Bars represent the mean of three biological replicates and three technical replicates with their corresponding standard deviation. ** or * indicate significant differences using Student t-test with p-values < 0.01 or <0.05, respectively.
Discussion
Due to its amenability to genetic transformation, virus induced gene silencing and transient protein expression, Nicotiana benthamiana has become very popular in the plant biology field18. Particularly, its susceptibility to a wide variety of pathogens made this model plant one of the most widely used in molecular studies of plant-pathogen interactions19. RT-qPCR is a frequently used technology for detection and quantification of gene expression, but accurate data interpretation highly depends on the use of appropriate reference genes whose expression should have minimal variations in the tissue, treatment or condition to be analyzed35. In this sense, several reports have contributed to the development of reference genes for the analysis of plant gene expression in the interaction with different pathogens. Some examples are tomato-virus41, tomato-bacteria31, wheat-fungus45, soybean-nematode and insect46 and rice-virus47 interactions. In spite of the extensive use of N. benthamiana, to our knowledge there are only two reports that analyzed the expression stability of traditional reference genes for plants infected with virus25 and for VIGS experiments26.
RNA-seq has become a powerful technology used for transcriptomic analysis in different organisms and treatments27,28,48. The information generated using this technique was used in the plant research field for the selection of new and more robust RT-qPCR reference genes in grape, soybean, potato, Lycoris and tomato31,49,50,51,52. In this work we used a new approach for the selection of novel stably expressed genes that can be used in N. benthamiana-bacterial interaction studies. We have taken advantage of previously generated tomato RNA-seq information7,8,31. These studies include transcriptional changes of tomato leaves for studying PTI and ETI activation and the influence of bacterial effectors on plant defenses, through the infiltration of MAMPs and bacterial strains and mutants along with untreated tomato plants. Together these experiments constitute a robust and large set of data that allowed the identification of novel reference genes in the tomato-Pseudomonas pathosystem31. Due to the closeness between the two species, we were able to find the N. benthamiana gene orthologs of the stably expressed tomato genes previously reported. This strategy was earlier useful to find reference genes in pepper using microarray information generated from tomato53. In this work we produced new RNA-seq data from N. benthamiana leaves infiltrated with Pseudomonas fluorescens 55, whose comparison with a mock treatment, accounts for a strong transcriptional PTI induction7. In agreement with this, we were able to identify 10,300 genes differentially expressed in N. benthamiana when challenged with this strain (Supplementary Table S1), This new dataset assisted in the selection of 9 novel genes with low coefficient of variation in the N. benthamiana-Pseudomonas pathosystem. This strategy led to the identification of a set of stably expressed genes in N. benthamiana that mostly differs from those previously found in tomato by an RNA-seq approach31, further confirming that it is necessary to evaluate reference genes for every system.
Using different algorithms such as geNorm36, NormFinder38 and BestKeeper39 we identified 3 reference genes (NbUBE35, NbNQO, NbErpA) that are more stably expressed compared to the commonly used ones that we selected for comparison (NbEF1α and NbGAPDH). We strongly recommend the use of NbUbe35 jointly with NbNQO as reference genes for expression studies that involve N. benthamiana leaf tissue infiltrated with Pseudomonas spp. or related experiments. According to our pairwise variation analysis, the use of two of these genes is sufficient to obtain accurate results (Supplementary Fig. S2). We also included in our analysis the NbPP2a gene, previously found to be the most stably expressed gene in N. benthamiana-virus interactions25. Although this gene performed fairly well in our study system, we found 3 genes whose overall expression was more stable (Table 3). When we put to test the combination of NbUbe35/NbNQO against a middle-ranked gene (NbTspan) or NbEF1α as reference, we found discordant results in terms of the capability of identifying statistically significant differences. Although the expression trend was similar, the high standard deviation obtained using either NbTspan or NbEF1α as reference prevented the detection of significant differences between the samples (Fig. 4). These results highlight the importance of the selection and validation of reliable reference genes for an accurate interpretation of the results. We therefore encourage the use of the information generated in this work in future RT-qPCR experiments involving N. benthamiana-Pseudomonas spp. pathosystems.
Material and Methods
Bacterial strains and growth conditions
Bacterial strains used were: Pseudomonas fluorescens 55 (P. fluorescens)32, Pseudomonas syringae pv. tomato (Pst) DC300033, Pst DC3000 ΔhopQ1-134. All of them were grown on King’s B medium at 30 °C. Antibiotics used were: ampicillin (100 μg/ml) for P. fluorescens and rifampicin (10 μg/ml) for Pst DC3000 and mutants.
Plant material and treatments
RNA-seq analysis was performed using 6-week old Nicotiana benthamiana plants vacuum-infiltrated with 108 cfu/ml P. fluorescens suspension or 10 mM MgCl2 as a mock treatment. Leaves were sampled at 6 h after infiltration (hai), frozen in liquid N2 and stored at −80 °C until processed.
For RT-qPCR studies, 6-week old N. benthamiana leaves were syringe-infiltrated with a suspension of 108 cfu/ml P. fluorescens, 5 × 106 cfu/ml Pst DC3000, 5 × 106 cfu/ml Pst DC3000 ΔhopQ1-1 or 10 mM MgCl2. Leaf samples were collected at 6 and 12 hai, frozen in liquid N2 and stored at −80 °C until processed.
In all the experiments, three biological replicates per infiltration were used. Details of the experiments are shown in Table 1.
RNA-seq library preparation and analysis
Total RNA was isolated using TRIzol reagent (Life Technologies, NY, USA) and libraries prepared as described previously8. Barcoded libraries were multiplexed by 12 in each lane and sequenced on an Illumina HiSeq 2000 equipment with 101 bp pair-end read mode. Sequence reads generated in this work have been deposited in the NCBI sequence read archive (SRA) under accession number SRP118889. Analysis of the RNA-seq data was performed as described previously8.
Selection of the candidate genes and primer design
Taking advantage of tomato gene expression stability ranking previously generated based on RNA-seq data31, using BlastX analysis from Sol Genomics Network54, we identified 50 N. benthamiana orthologs which we hypothesized had stable gene expression. Using the newly generated RNA-seq data (N. benthamiana leaves challenged with P. fluorescens 55) we discarded those with low expression (RPKM <3) and ranked them based on their coefficient of variation (CV) across treatments and biological replicates. We then selected the 9 most stably expressed genes for validation (Supplementary Table S2). Additionally, two traditional reference genes used in N. benthamiana RT-qPCR experiments (NbGADPH and NbEF1α) and NbPP2a, the most stably expressed gene identified in a previous report using virus-infected N. benthamiana plants25, were included for the analysis.
We used BlastP to identify two putative N. benthamiana orthologs of a previously reported tomato gene (Solyc02g069960) that is induced by PTI8. From the two closest found, Niben101Scf04323g01009.1 and Niben101Ctg15860g00004.1 (NbNAC042), we selected NbNAC042 based on its gene expression level and PTI induction (Supplementary Table S4), to test the performance of the most stable reference genes described in this work.
The nucleotide sequence of each gene was downloaded from the Sol Genomics Network webpage54 and primers were designed using PrimerQuest tool (Integrated DNA Technologies). Primer efficiencies were checked by RT-qPCR using different cDNA dilutions. This information, along with the accession number of all N. benthamiana genes used in this work, is show in Supplementary Table S3. Dissociation curves were performed to confirm amplification specificity (Supplementary Fig. S1).
RNA isolation and cDNA preparation
Total RNA was isolated using the Tri-Reagent (Sigma Aldrich) following the manufacturer’s instructions. RNA integrity was assayed by 1% agarose gel electrophoresis. Total RNA (8 μg) was processed with RQ1 RNase-free DNase (Promega) for 60 minutes at 37 °C to eliminate potential DNA contamination and then purified using a chloroform:octanol mix (24:1). RNA concentration and purity was determined using a CLARIOstar microplate reader (BMG Labtech). Purified RNA (2.4 μg) was used to prepare cDNA using M-MLV reverse transcriptase (Promega) with random primers according to the manufacturer’s instructions.
RT-qPCR reactions
RT-qPCR was performed as described previously55 in 96-well plates (Thermo Fisher Scientific) on a StepOnePlus system (Applied Biosystems). Primer sequences and characteristics are shown in Supplementary Table S3. The reaction mix was performed using: 5 μl of FastStart Universal SYBR Green Master (Rox) (Roche Life Sciences), 2 μl of 2 μM primer mix, 2 μl of a diluted 1:10 cDNA and water to complete a final volume of 10 μl. Cycling conditions were 95 °C for 10 min, and 40 cycles of 95 °C for 15 s, 60 °C for 1 min. All RT-qPCR experiments were performed using three biological and three technical replicates.
Evaluation and validation of reference gene expression stability
Data obtained from the RT-qPCR experiments were analyzed using three statistical programs: geNorm36, NormFinder38 and BestKeeper39. The relative expression of NbNAC042 gene was expressed as E−ΔΔCq, where E corresponds to the primer efficiency value. When a pair of reference genes were used (NbUbe35/NbNQO) the geometric Cq mean and efficiency average were employed.
References
Boller, T. & He, S. Y. Innate immunity in plants: an arms race between pattern recognition receptors in plants and effectors in microbial pathogens. Science 324, 742–744 (2009).
Schwessinger, B. & Ronald, P. C. Plant innate immunity: perception of conserved microbial signatures. Annu Rev Plant Biol 63, 451–482 (2012).
Couto, D. & Zipfel, C. Regulation of pattern recognition receptor signalling in plants. Nat Rev Immunol 16, 537–552 (2016).
Segonzac, C. & Zipfel, C. Activation of plant pattern-recognition receptors by bacteria. Curr Opin Microbiol 14, 54–61 (2011).
Dodds, P. N. & Rathjen, J. P. Plant immunity: towards an integrated view of plant-pathogen interactions. Nat Rev Genet 11 (2010).
Monaghan, J. & Zipfel, C. Plant pattern recognition receptor complexes at the plasma membrane. Curr Opin Plant Biol 15, 349–357 (2012).
Rosli, H. et al. Transcriptomics-based screen for genes induced by flagellin and repressed by pathogen effectors identifies a cell wall-associated kinase involved in plant immunity. Genome Biol 14, R139 (2013).
Pombo, M. A. et al. Transcriptomic analysis reveals tomato genes whose expression is induced specifically during effector-triggered immunity and identifies the Epk1 protein kinase which is required for the host response to three bacterial effector proteins. Genome Biol 15, 492 (2014).
Feng, F. & Zhou, J. M. Plant-bacterial pathogen interactions mediated by type III effectors. Curr Opin Plant Biol 15, 469–476 (2012).
Macho, A. P. Subversion of plant cellular functions by bacterial type-III effectors: beyond suppression of immunity. New Phytol 210, 51–57 (2016).
Toruño, T. Y., Stergiopoulos, I. & Coaker, G. Plant pathogen effectors: cellular probes interfering with plant defenses in spatial and temporal manners. Annu Rev Phytopathol 54, https://doi.org/10.1146/annurev-phyto-080615-100204 (2016).
Moffett, P. Mechanisms of recognition in dominant R gene mediated resistance. Adv Virus Res 75, 1–33 (2009).
Maekawa, T., Kufer, T. A. & Schulze-Lefert, P. NLR functions in plant and animal immune systems: so far and yet so close. Nature Immunol 12, 817–826 (2011).
Tsuda, K., Sato, M., Stoddard, T., Glazebrook, J. & Katagiri, F. Network properties of robust immunity in plants. PLoS Genet 5, e1000772, https://doi.org/10.1371/journal.pgen.1000772 (2009).
Wei, H.-L., Zhang, W. & Collmer, A. Modular study of the type III effector repertoire in Pseudomonas syringae pv. tomato DC3000 reveals a matrix of effector interplay in pathogenesis. Cell Rep 23, 1630–1638 (2018).
Jones, J. D. G., Vance, R. E. & Dangl, J. L. Intracellular innate immune surveillance devices in plants and animals. Science 354, https://doi.org/10.1126/science.aaf6395 (2016).
Xin, X.-F. & He, S. Y. Pseudomonas syringae pv. tomato DC3000: a model pathogen for probing disease susceptibility and hormone signaling in plants. Annu Rev Phytopathol 51, 473–498 (2013).
Todesco, M. & de Felippes, F. F. Why benthamiana went viral. Trends Plant Sci 21, 4–6 (2016).
Goodin, M. M., Zaitlin, D., Naidu, R. A. & Lommel, S. A. Nicotiana benthamiana: its history and future as a model for plant-pathogen interactions. Mol Plant-Microbe Interact 21, 1015–1026 (2008).
Bombarely, A. et al. A draft genome sequence of Nicotiana benthamiana to enhance molecular plant-microbe biology research. Mol Plant-Microbe Interact 25, 1523–1530 (2012).
Bustin, S. Quantification of mRNA using real-time reverse transcription PCR (RT-PCR): trends and problems. J Mol Endocrinol 29, 23–39 (2002).
Bustin, S. A. et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin Chem 55, 611–622 (2009).
Derveaux, S., Vandesompele, J. & Hellemans, J. How to do successful gene expression analysis using real-time PCR. Methods 50, 227–230 (2010).
Huggett, J., Dheda, K., Bustin, S. & Zumla, A. Real-time RT-PCR normalisation; strategies and considerations. Genes Immun 6, 279–284 (2005).
Liu, D. et al. Validation of reference genes for gene expression studies in virus-infected Nicotiana benthamiana using quantitative real-time PCR. PLoS ONE 7, e46451, https://doi.org/10.1371/journal.pone.0046451 (2012).
Rotenberg, D., Thompson, T. S., German, T. L. & Willis, D. K. Methods for effective real-time RT-PCR analysis of virus-induced gene silencing. J Virol Methods 138, 49–59 (2006).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Rev 10, 57–63 (2009).
Ansorge, W. J. Next genenration DNA sequencing techniques. New Biotechnol 25, 195–203 (2009).
Haas, B. J. & Zody, M. C. Advancing RNA-Seq analysis. Nature Biotechnol 28, 421–423 (2010).
Wang, L., Li, P. & Brutnell, T. P. Exploring plant transcriptome using ultra high-throughput sequencing. Brief Funt Genomics 9, 118–128 (2010).
Pombo, M. A., Zheng, Y., Fei, Z., Martin, G. B. & Rosli, H. G. Use of RNA-seq data to identify and validate RT-qPCR reference genes for studying the tomato-Pseudomonas pathosystem. Sci Rep 7, 44905, https://doi.org/10.1038/srep44905 (2017).
Huang, H. C. et al. Molecular cloning of a Pseudomonas syringae pv. syringae gene cluster that enables Pseudomonas fluorescens to elicit the hypersensitive response in tobacco plants. J Bacteriol 170, 4748–4756 (1988).
Cuppels, D. A. Generation and characterization of Tn5 insertion mutations in Pseudomonas syringae pv. tomato. Appl Environ Microbiol 51, 323–327 (1986).
Wei, C. F. et al. A Pseudomonas syringae pv. tomato DC3000 mutant lacking the type III effector HopQ1-1 is able to cause disease in the model plant Nicotiana benthamiana. Plant J 51, 32–46 (2007).
Kozera, B. & Rapacz, M. Reference genes in real-time PCR. J Appl Genet 54, 391–406 (2013).
Vandesompele, J. et al. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3, 1–12, https://doi.org/10.1186/gb-2002-3-7-research0034 (2002).
Hellemans, J., Mortier, G., De Paepe, A., Speleman, F. & Vandesompele, J. qBase relative quantification framework and software for management and automated analysis of real-time quantitative PCR data. Genome Biol 8, R19, https://doi.org/10.1186/gb-2007-8-2-r19 (2007).
Andersen, C. L., Jensen, J. L. & Ørntoft, T. F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64, 5245–5250 (2004).
Pfaffl, M. W., Tichopad, A., Prgomet, C. & Neuvians, T. P. Determination of stable housekeeping genes, differentially regulated target genes and sample integrity: BestKeeper – Excel-based tool using pair-wise correlations. Biotechnol Lett 26, 509–515 (2004).
Xie, F., Xiao, P., Chen, D., Xu, L. & Zhang, B. miRDeepFinder: a miRNA analysis tool for deep sequencing of plant small RNAs. Plant Mol Biol 80, 75–84 (2012).
Lacerda, A. L. M. et al. Reference gene selection for qPCR analysis in tomato-bipartite Begomovirus interaction and validation in additional tomato-virus pathosystems. PLoS ONE 10, e0136820, https://doi.org/10.1371/journal.pone.0136820 (2015).
Wang, Q. et al. Stability of endogenous reference genes in postmortem human brains for normalization of quantitative real-time PCR data: comprehensive evaluation using geNorm, NormFinder, and BestKeeper. Int J Legal Med 126, 943–952 (2012).
Robledo, D. et al. Analysis of qPCR reference gene stability determination methods and a practical approach for efficiency calculation on a turbot (Scophthalmus maximus) gonad dataset. BMC Genom 15, 648 (2014).
Zhang, W.-X. et al. Selection of suitable reference genes for quantitative real-time PCR normalization in three types of rat adipose tissue. Int J Mol Sci 17, 968 (2016).
Scholtz, J. J. & Visser, B. Reference gene selection for qPCR gene expression analysis of rust-infected wheat. Physiol Mol Plant Pathol 81, 22–25 (2013).
Miranda, Vd. J. et al. Validation of reference genes aiming accurate normalization of qPCR data in soybean upon nematode parasitism and insect attack. BMC Res Notes 6, 196, https://doi.org/10.1186/1756-0500-6-196 (2013).
Fang, P. et al. Assessment of reference gene stability in Rice stripe virus and Rice black streaked dwarf virus infection rice by quantitative Real-time PCR. Virol J 12, 175, https://doi.org/10.1186/s12985-015-0405-2 (2015).
Rosli, H. G. & Martin, G. B. Functional genomics of tomato for the study of plant immunity. Brief Funct Genomics 14, 291–301 (2015).
González-Agüero, M. et al. Identification of two putative reference genes from grapevine suitable for gene expression analysis in berry and related tissues derived from RNA-Seq data. BMC Genom 14, 1–12 (2013).
Yim, A. K.-Y. et al. Using RNA-seq data to evaluate reference genes suitable for gene expression studies in soybean. PLoS ONE 10, e0136343, https://doi.org/10.1371/journal.pone.0136343 (2015).
Mariot, R. F. et al. Selection of reference genes for transcriptional analysis of edible tubers of potato (Solanum tuberosum L.). PLOS ONE 10, e0120854, https://doi.org/10.1371/journal.pone.0120854 (2015).
Ma, R., Xu, S., Zhao, Y., Xia, B. & Wang, R. Selection and validation of appropriate reference genes for quantitative real-time PCR analysis of gene expression in Lycoris aurea. Front Plant Sci 7, 536, https://doi.org/10.3389/fpls.2016.00536 (2016).
Muller, O. A. et al. Genome-wide identification and validation of reference genes in infected tomato leaves for quantitative RT-PCR analyses. PLoS ONE 10, e0136499, https://doi.org/10.1371/journal.pone.0136499 (2015).
Fernandez-Pozo, N. et al. The Sol Genomics Network (SGN)—from genotype to phenotype to breeding. Nucleic Acids Res 43, D1036–D1041, https://doi.org/10.1093/nar/gku1195 (2014).
Nguyen, H. P. et al. Methods to study PAMP-triggered immunity using tomato and Nicotiana benthamiana. Mol Plant-Microbe Interact 23, 991–999 (2010).
Acknowledgements
We thank Mauro Bartolozzi and Santiago Martínez Alonso for their assistance in plant growth and maintenance. This research was supported by National Science Foundation grant (IOS-1546625, GBM), Agencia Nacional de Promoción Científica y Técnica - Argentina (PICT2014-1589, HGR), Consejo Nacional de Investigaciones Científicas y Técnicas - Argentina (PIP2014-0314, MAP).
Author information
Authors and Affiliations
Contributions
M.A.P., G.B.M. and H.G.R. designed the research, M.A.P., R.N.R. and H.G.R. performed the research, M.A.P., Y.Z., Z.F., G.B.M. and H.G.R. analyzed the data, M.A.P., G.B.M. and H.G.R. wrote the paper. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pombo, M.A., Ramos, R.N., Zheng, Y. et al. Transcriptome-based identification and validation of reference genes for plant-bacteria interaction studies using Nicotiana benthamiana. Sci Rep 9, 1632 (2019). https://doi.org/10.1038/s41598-018-38247-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-38247-2
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
