
Abstract
Human adenovirus (HAdV) group C are the common etiologic in infants with severe acute respiratory infections (SARI). In the study, we report that a novel recombinant HAdV-C group strain (SH2016) was isolated from an infant with SARI in Shanghai in Feb. 4, 2016. The whole-genome sequence of SH2016 strain was generated and compared to other HAdV genomes publicly available. The strain SH2016 genome contains 35,946 nucleotides and coded 40 putative proteins, which was divided into 11 regions. RDP and phylogenetic analyses of the complete genome showed that the SH2016 strain was arranged into a novel subtype and might be recombined with HAdV-1 and HAdV-2. Our finding indicated that the frequent recombination among the HAdV-C group played an important role in driving force for polymorphism of human HAdV-C group prevalent in Shanghai, China. Further epidemiological surveillance of HAdV-C group is necessary to explore whether the novel HAdV-C group will maintain long-term stability. And the pathogenicity and clinical characteristics of the novel HAdV-C group member should be done more.
Similar content being viewed by others

Introduction
The international committee on taxonomy of viruses had divided Adenoviridae into 5 genera, Atadenovirus, Aviadenovirus, Mastadenovirus, Siadenovirus, and Ichtadenovirus. Through more than 6 decades, since the first characterizations of human adenoviruses (HAdVs)1, all of HAdVs falled within the genus Mastadenovirus. And HAdVs were classified into 7 groups (HAdV-A to HAdV-G), including 52 serotypes and 90 human HAdV genotypes2,3,4,5,6, which were recognized by Human Adenovirus Working Group, July, 2018 Update (http://hadvwg.gmu.edu/). Over the past 30 years, recombinant adenovirus-vectors based on the HAdV-C group had also been developed and extensively used in preclinical and clinical studies7. Among of these, members of the HAdV-B group (types 3, 7, 11, 14, 16, 21, 34, 35, 50 and 55) and HAdV-C group (types 1, 2, 5, 6 and 57) cause a variety of typically acute respiratory diseases. Especially, HAdV-C group could cause severe bronchiolitis or pneumonia in the early childhood8,9.
Three recombinant HAdV-C strains (BJ04, BJ09 and CBJ113), classified within HAdV-2 (P1H2F2), isolated from infants with acute respiratory infection in Beijing in 2009–2013 by labs in China CDC10,11. Among three strains, CBJ113 was characterized by a recombination among HAdV-2, HAdV-6, HAdV-1, HAdV-5, and HAdV-57 sequences. BJ04 recombination event involved parental strains HAdV-1, HAdV-2, whereas BJ09 involved in HAdV-1, HAdV-5 and CBJ113. Therefore, HAdV-1 was involved in recombination of other HAdV-C types.
The National Adenovirus Type Reporting System (NATRS) of the United States described trends in reported HAdV-C group was circulating in the United States after initiation of surveillance in 20148. NATRS also displayed that HAdV-1 was identified as the pathogen responsible for that outbreak8. Interestingly, here, we describe the characterization of a novel type of HAdV-1 isolated from a hospitalized infant with SARI. We found that there was the possibility of intraspecies recombination among HAdV-C group on the whole genome sequence analysis. In order to gain a better understanding of this phenomenon, we determined and analyzed the whole-genome sequence of HAdV-1 strain SH2016.
Results
Isolation and complete genomic characterization of the novel HAdV-C type
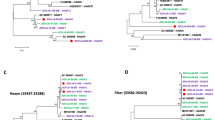
Throat swabs positive for the HAdV, when other viral nucleic acid detection was negative, were used initially for viral isolation. The isolated strain caused a visible CPE on culturing. It was archived as strain “human/China/SH/2016/1[P1H1F1]”, which we referred to as “SH2016” strain. Using next-generation and Sanger sequencing, the full-length genomic sequence of strain SH2016 was determined, and the genomic data, was deposited in GenBank (accession number: MH183293). The genome length of SH2016 strain (35,946 bp) was similar to the length of the prototype strain, human adenovirus type 1 (AC_000017, 36001 bp). The G + C content of the genome was 55.2%, which is similar to G + C content of other members of species group C adenoviruses1, and the plus strand had an overall base composition of 23.23% A, 27.99% C, 27.21% G, and 21.57% T. Similar to the genomes of prototype HAdV-1 reference strain (AC_000017), the genome encoded 40 coding sequences (Table 1) and 35 non-coding motifs (Table 2) were recognized. Whole genome phylogenetic analysis of 43 archived complete HAdV genomes from GenBank illustrated that strain SH2016 were clustered into HAdV-1, but it branched out independently with human/EGY/E13/2001/1[P1H1F1]12 (Fig. 1A). Then, phylogenetic analysis of 3 major antigen genes (penton, hexon and fiber) of the SH2016 showed that the 3 genes were classified to H1, P1, and F1 (Fig. 1B–D).

Neighbor-joining phylogenetic trees based on the open reading frame sequences of the whole genome (A), hexon gene (B), penton gene (C) and fiber gene (D) of SH2016 strain in this study and those of HAdV-C whole genome reference strains from GenBank. Strain SH2016 highlighted with a solid square was characterized in this study. The trees were constructed using the neighbor-joining method of MEGA 6.06 with 1000 bootstrap trials performed to assign confidence to the grouping.
Comparative genome analysis
Compared with the complete group C genome sequences of the 5 prototype strains of HAdV-1 (AC_000017), HAdV-2 (AC_000007), HAdV-5 (AC_000008), HAdV-6 (HQ413315) and HAdV-57 (HQ003817), the SH2016 strain is conserved, sharing the highest nucleotide identity (97.93%) with the prototype strain of HAdV-1 (Table 3). Based on the nucleotide alignment of the different gene sequences, the nucleotide sequences of the penton, hexon and fiber genes showed the highest degree of homology between the prototype strain HAdV-1, with identities of 99.82%, 99.68% and 98.79% respectively. Genomic map of strain SH2016, contained 40 ORFs (rightward ORFs: 33, leftward ORFs: 7), was showed in Fig. 2A. Comparison of the nucleotide sequences of the 11 coding regions (E1A, E1B, E2B, L1 13.6 kDa, pTP, L1 52 kDa, pIIIa, pVII, E3 and E4) showed the highest sequence similarity between strains HAdV-2, HAdV-5, HAdV-6, and HAdV-57, with identities of 98.19~99.75%. On the other hand, HAdV-1 and HAdV-57 showed the greatest similarities to SH2016 in the pIX gene (99.53%), HAdV-6 and HAdV-57 in putative protein U gene (98.19%), HAdV-1, HAdV-5 and HAdV-6 in pX gene (99.59%), respectively. While the Iva2, pV, pX, pVI, DBP, and L4 coding regions displayed the highest similarity with HAdV-1.Through comparative genomics analysis, the novel HAdV-1 type showed limited sequence variation between the HAdV-C group.

Genetic recombinant analyses of the complete genome of the novel strain SH2016. (A) Genomic map of strain SH2016. The l-strand of the genome is represented by a straight line. Rightward (top) and leftward (bottom) ORFs are represented by grey arrows. (B) Recombination events predicted in strain SH2016. Strain SH2016 genome is shown as a thick black line. The likely backbone is shown as a cyan line. Genetic components predicted by RDP4 to be involved in a recombination event are shown as purple line. Likely breakpoint positions are shown below the genome. (C) Similarity analyses of SH2016. SH2016 was used as the query sequence to compare with other 2 representative strains of HAdV-C. The default setting of SimPlot software was used as followed: Window size 200 bp, step size 20 bp, replicates 1000 times and tree model neighbor-joining.
Genomic recombination analysis of strain SH2016
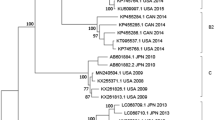
RDP4 package strongly predicted that the strain SH2016 was a highly probable homologous recombinant resulting from HAdV-1 (strain: human/USA/VT2672/2003/1[P1H1F1], GenBank ID: JX173083) and HAdV-2 (stain: T215/Ft Jackson South Carolina USA/2002, GenBank ID: KX384959) with beginning breakpoint located around 28040 (without gaps) of HAdV-1, within the gene coding for putative host modulation protein E3 (early E3 12.5 kDa glycoprotein) and with ending breakpoint located around 31067 (without gaps) of HAdV-1, within the gene coding for fiber protein (Fig. 2B). The similarities with possible major parent strain (HAdV-1) and minor parent strain (HAdV-2) were 99.3% and 98.6%, respectively. Indeed, 7 algorithms (RDP, GENECONV, BootScan, MaxChi, Chimaera, SiScan, 3Seq, LARD, PhylPro (Supplemental Figs S1–7), were utilized to predict potential recombination events between the input sequences) supported this event with p-values ranging from 2.347 × 10−187 to 2.179 × 10−12 (Table 4). Similarity plot analysis using SimPlot software were performed to confirm the consequent of recombination events within the genome of SH2016. As well as, SimPlot analysis indicated that the mosaic structure comprised of the SH2016 genome originated not only from mainly circulating viral strain: prototype HAdV-1 basically, but also from a small quantity of HAdV-2 (Fig. 2C). The results coincided with phylogenetic analyses, which indicated that both of the left region of recombinant point (5′-end, 1–28039) and the right region of recombinant point (3′-end, 31067–35946) of SH2016 strain were clustered into HAdV-1 group with high confidence (bootstrap value = 100% or 97%, Fig. 3A,B), but the recombinant region was clustered into HAdV-2/6/57 group (bootstrap value = 100%, Fig. 3C). So these findings re-confirmed that SH2016 appeared from potential genetic recombination events, which HAdV-1, and HAdV-2 participated in this process.

Neighbor-joining phylogenetic trees based on the left region (A) of recombinant point (5′-end), the right region (B) of recombinant point (3′-end) and the recombinant frame (C) sequences of the recombinant regionof SH2016 strain in this study and those of HAdV-C whole genome reference strains from GenBank. Strain SH2016 highlighted with a solid square was characterized in this study. The trees were constructed using the neighbor-joining method of MEGA 6.06 with 1000 bootstrap trials performed to assign confidence to the grouping.
Discussion
In order to ensure the accuracy of the results, the phylogenetic trees were also constructed by maximum likelihood (ML) method implemented in IQ-TREE 1.6.7.113 under the most suitable nucleotide substitution models respectively, which were selected by jModeltest14 [Supplemental Figs S8–14]. The frameworks of all neighbor-joining (NJ) trees in this study were consistent with ML trees. Intriguingly, the penton NJ-tree was not really informative as the 43 sequences did not feature much divergence between each other (Fig. 1C), and the same situation still appeared in maximum likelihood tree (Fig. S10). This showed that SH2016 was convergent evolution with known HAdV-C sequences in the penton region. As the full genome trees had shown, SH2016 was related to AC_000017 which could be considered as the backbone of the prototype HAdV-1 genome (Fig. 1A, Supplemental Fig. S8). However, SH2016 genome sequence was showing some divergence at the E3 region and putative protein U region of the genome, both of which were located in reconstituted area. The E3 region and putative protein U region of the SH2016 genome (major areas of recombination) were more divergent than the rest of the genome, which had only 82.79% and 85.50% identities with the prototype HAdV-1 (Table 3), respectively.
In summary, the complete genome sequence of the novel recombinant HAdV-1 strain (SH2016) was determined and characterized, isolated in Shanghai, China. Phylogenetic and SimPlot analyses both displayed that the novel subtype of HAdV-1 (SH2016) was a recombinant event involving HAdV-1 and HAdV-2 (Figs 2B,C and 3). And The recombination area was located between 28040 and 31067, which including most of E3, whole U and few of L5 (Fig. 2A,B). However, the process of intratypic recombination incident is not clear in its evolutionary history, only in the case that more sequences were needed to investigate the spatiotemporal relationships of the novel HAdV-C group all over the world.
Comparison of the amino acid sequences of the fiber, hexon and penton of strain SH2016 with other type HAdV-1 fibers, hexons and pentons, only the fiber of strain SH2016 has three mutations. According to the protein structure, the fiber of SH2016 strain could also be divided into three components including an N-terminal tail (FNPVYPYD)2,15,16, two repeat/shaft regions and a C-terminal globular knob17,18,19,20. One (A71T) of mutations occurred in the first repeat/shaft region, and two (V432I and H470N) other mutations occurred in C-terminal globular knob, which is typically responsible for interaction with the cell receptors. Whether the mutations at these sites lead to antigenic drift need to be experimentally validated.
In conclusion, we propose that the SH2016 strain is a novel intratypic HAdV-C strain and may be an etiological agent of SARI. On the basis of their complete genome sequences, it arose through the recombination of two HAdV genotypes, HAdV-1 and HAdV-2, which frequently cause respiratory infection9,21,22,23. Whether the emergence of recombination strain might increase virulence, thereby posing a new global challenge with regard to acute respiratory diseases in the near future, warrants further investigation. So, epidemiological and virological surveillance of this uninvestigated respiratory disease pathogen should be strengthened.
Material and Methods
Specimen collection and identification
Throat swab specimens were collected from the outpatients with respiratory tract infection for surveillance subjects at designated intervals by trained medical staff of Xinhua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine in this study. SH2016 was collected in February 4 at outpatient. The patient was more than two years old and clinically diagnosed with bronchitis and upper respiratory tract infection. The patient was diagnosed with human adenovirus infection and ruled out other possible common viral infections using our previous diagnostic methods24. After 3 days of antiviral treatment, the patient recovered.
Cell culture and virus isolation
HEp-2 cells (from American Type Culture Collection, ATCC Number CCL-23, Manassas, VA, USA) were maintained in complete DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 µg/mL streptomycin (Invitrogen, Carlsbad, CA, USA) at 37 °C with 5% CO2. For the virus culture, DMEM with 2% FBS and antibiotics was used. Cells inoculated with clinical samples, which were filtered by the 0.22 m filter (Millipore, Merch, Germany), were incubated at 37 °C for 7 days. If no cytopathic effect (CPE) was observed, the culture supernatants were used to inoculate fresh cells for 2 additional passages. And if the adenovirus-like CPE were appeared, the cultures were passaged again to confirm the presence of the viruses. Virus-infected cells and supernatant were collected and used for subsequent detection and genome sequencing.
DNA extraction, PCR strategy and sequencing
Strain SH2016 was isolated from throat swab and underwent three passages in HEp-2 cells to obtain high-tilter stocks. The viral DNA was extracted using a QIAamp MinElute Virus Spin Kit (Qiagen, Germany) following the manufacturer’s instructions. The primer pairs (Supplementary Table 1) used to amplify complete genome was designed based primarily on of human mastadenovirus C strain CBJ113 (KR699642), human mastadenovirus C isolates human/CHN/BJ04/2012/[P1/H2/F2] (MF315028), human/CHN/BJ09/2012/[P1/H2/F2] (MF315029) and human adenovirus C strain human/EGY/E13/2001/1[P1H1F1] (JX173080)12, respectively.
Twenty four overlapping PCR fragments covering the entire genome were amplified by using the Platinum™ Taq DNA (Invitrogen, Thermo Fisher, CA, USA) according to the manufacturer’s protocol. PCR amplification was carried out at 95 °C for 5 min for one cycle to denature, and followed by 40 cycles for amplification at 95 °C for 30 s, 55 °C for 30 s, 72 °C for 180 s. At the end of the cycling, an additional extension period of 72 °C for 10 min was included, after which the samples were stored at 4 °C. For the 5′/3′-terminal genome sequences, the covalent junction between the purified DNA template and the terminal protein (TP) was broken by the addition of 0.4 N NaOH as described in Xu’s protocol2. The PCR products were separated by electrophoresis on 1.5% agarose gels and visualized under UV light. The PCR amplicon was then inserted into pGEM-T Easy Vector using TA cloning. The recombinant plasmid were identified by amplification primer pairs respectively, and was confirmed via sequencing using M13 forward and M13 reverse primers as sequencing primers. The recombinant plasmids were directly sequenced on an ABI 3730XL automatic DNA analyzer using an ABI Prism BigDye Terminator cycle sequencing kit 3.1 (Applied Biosystems). Either bracketing PCR or internal primers were used as sequencing primers to obtain overlapping and complementary sequences and a minimum twofold coverage. Whole genome sequences were obtained from 24 overlapping sequences assembled in ContigExpress Progect (Vector NTI).
Nucleotide sequence accession number
Annotated genome sequence of SH2016 was submitted to GenBank database under the following accession number MH183293.
Genome annotation
The BLASTn program (National Center for Biotechnology Information, Bethesda, MD, USA) was used to identify the homologous nucleotide sequences in the GenBank database (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM = blastn&PAGE_TYPE = BlastSearch&LINK_LOC = blasthome). The SH2016 genome sequence was annotated based on the previous annotation of HAdV-C strain (human/EGY/E13/2001/1[P1H1F1]). The DNA and protein sequence alignments were created by using BioEdit sequence alignment editor software (version BioEdit v7.1.3; Tom Hall, Ibis BioSciences, CA).
Phylogenetic analysis
Phylogenetic trees were generated with MEGA6.06 using the neighbor-joining (NJ) method with the maximum composite likelihood nucleotide substitution model and bootstrap test of phylogeny with replicates set to 1000 to assign confidence to the grouping. The maximum likelihood (ML) phylogenetic tree were reconstructed by the ML method implemented in IQ-TREE 1.6.7.113 based on the different models. Additional, the optimal evolutionary models were identified with the aid of the computer program jModelTest 2.1.714. The resulting ML trees were created and edited using FigTree (http://tree.bio.ed.ac.uk/software/figtree/). Strain SH2016 highlighted with a solid square in NJ trees or with red fond in ML trees were characterized in this study, respectively.
Recombination analysis
The aligned sequences of the SH2016 sequence available from GenBank were subjected to recombination analysis. The Recombination Detection Program (RDP) package Beta 4.96 was used for identifcation of recombinant sequences. Multiple methods in its default mode, such as RDP, GENECONV, BootScan, MaxChi, Chimaera, SiScan, 3Seq, LARD, PhylPro, were utilized to predict potential recombination events between the input sequences. Only those recombination events were taken into considerations which were supported by at least 4 methods to avoid misidentifcation using only a single methodology. The best signals for recombination are associated with the lowest P-values; the highest acceptable P-value was set to 0.05. Recombination events detected with RDP Beta 4.96 were confirmed and visualized with SimPlot Version 3.5.1. Bootscan analysis in the SimPlot package version 3.5.1 was used to test potential recombination events. Bootscan analysis in the SimPlot package version 3.5.1 was used to test potential recombination events. Similarity was calculated in each window of 200 bp by the Kimura (2-parameter) distance model with a transition-transversion ratio of 2.0. The window was successively advanced along the genome alignment in 20 bp increments. For bootscan analysis, the neighbor-joining algorithm was run with 1000 bootstrap replicates. A threshold of 70% or more of the observed permuted trees indicated potential recombination events. Potential genomic components were identified based on genetic distances and phylogenetic analyses.
Amino acid analysis
SH2016 ORFs were compared to 5 prototype sequences and the 38 remaining HAdV-C whole genome sequences from GenBank. The complete genome of SH2016 strain was annotated using AC_000017 (HAdV-1 prototype strain) as template.
Ethics statement
This study was reviewed and approved by the human Research Ethics Committee Ethics Review Committee of the Shanghai Public Health Clinical Center. All methods used in this study were performed in accordance with the relevant guidelines. Written informed consent for the collection of throat swabs for pathogenic identification was obtained from the participants involved in this study.
References
Adeyemi, O. A., Yeldandi, A. V. & Ison, M. G. Fatal adenovirus pneumonia in a person with AIDS and Burkitt lymphoma: a case report and review of the literature. AIDS Read 18, 196–198, 201–202, 206–207 (2008).
Yang, Z. H. et al. Genomic Analyses of Recombinant Adenovirus Type 11a in China. J Clin Microbiol 47, 3082–3290 (2009).
Lauer, K. P. et al. Natural variation among human adenoviruses: genome sequence and annotation of human adenovirus serotype 1. J Gen Virol 85, 2615–2625 (2004).
Buescher, E. L. Respiratory disease and the adenoviruses. Med Clin North Am 51, 769–779 (1967).
Hillerman, M. R. & Werner, J. R. Recovery of new agent from patients with acute respiratory illness. Proc Sac Exp Biol Med 85, 183–188 (1954).
Ison, M. G. & Hayden, R. T. Adenovirus. Microbiol Spectrum 4, DMIH2-0020–2015 (2016).
Zhang, W. & Ehrhardt, A. Getting genetic access to natural adenovirus genomes to explore vector diversity. Virus Genes 53, 675–683 (2017).
Binder, A. M. et al. Human Adenovirus Surveillance - United States, 2003–2016. MMWR Morb Mortal Wkly Rep 66, 1039–1042 (2017).
Ma, G. et al. Species C is Predominant in Chinese children with acute respiratory adenovirus infection. Pediatr Infect Dis J 34, 9 (2015).
Wang, Y. et al. Phylogenetic evidence for intratypic recombinant events in a novel human adenovirus C that causes severe acute respiratory infection in children. Sci Rep 6, 23014 (2016).
Mao, N. et al. Whole genomic analysis of two potential recombinant strains within Human mastadenovirus species C previously found in Beijing, China. Sci Rep 7, 15380 (2017).
Ismail, A. et al. Genomic analysis of a large set of currently—and historically—important human adenovirus pathogens. Emerg Microbes Infect 7, 10 (2018).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32, 268–74 (2015).
Darriba, D. et al. jModelTest 2: more models, new heuristics and parallel computing. Nature Methods 9, 772 (2012).
Reddy, P. S. et al. Complete sequence and organization of the human adenovirus serotype 46 genome. Virus Res 116, 119–128 (2006).
Caillet-Boudin, M. L. Complementary peptide sequences in partner proteins of the adenovirus capsid. J Mol Biol 208, 195–198 (1983).
van Raaij, M. J., Mitraki, A., Lavigne, G. & Cusack, S. A triple beta-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 401, 935–938 (1999).
Xia, D., Henry, L. J., Gerard, R. D. & Deisenhofer, J. Crystal structure of the receptor-binding domain of adenovirus type 5 fiber protein at 1.7 A resolution. Structure 2, 1259–1270 (1994).
Kidd, A. H. & Erasmus, M. J. Sequence characterization of the adenovirus 40 fiber gene. Virology 172, 134–144 (1989).
Marchler-Bauer, A. et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res 45(D1), D200–D203 (2017).
Liu, C. et al. Adenovirus infection in children with acute lower respiratory tract infections in Beijing, China, 2007 to 2012. BMC Infect Dis 15, 408 (2015).
Chehadeh, W. et al. Adenovirus types associated with severe respiratory diseases: A retrospective 4-year study in Kuwait. J Med Virol 90, 1033–1039 (2018).
Walsh, M. P. et al. Computational analysis of two species C human adenoviruses provides evidence of a novel virus. J Clin Microbiol 49, 3482–3490 (2011).
Zhang, W. et al. Study on the application of multiplex PCR method and liquid chip technology in the detection of respiratory viruses. Chinese Journal of Experimental and Clinical Virology 28, 302–304 (2014).
Acknowledgements
This work was sponsored by Natural Science Foundation of Shanghai (18ZR1431900) and supported by 81102283 grant from National Natural Science Foundation of China and grant KY-GW-2018-14 from Shanghai Public Health Clinical Center.
Author information
Authors and Affiliations
Contributions
Wanju Zhang and Lisu Huang conceived and designed the study. Wanju Zhang analyzed the data and wrote the manuscript. Wanju Zhang and Lisu Huang coordinated the study and edited the manuscript. Wanju Zhang performed the experiments. Lisu Huang treated the patient and supplied the clinical specimens.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, W., Huang, L. Genome Analysis of A Novel Recombinant Human Adenovirus Type 1 in China. Sci Rep 9, 4298 (2019). https://doi.org/10.1038/s41598-018-37756-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37756-4
This article is cited by
-
Identification of functional pathways and potential genes associated with interferon signaling during human adenovirus type 7 infection by weighted gene coexpression network analysis
Archives of Virology (2023)
-
Genomic analyses of human adenoviruses unravel novel recombinant genotypes associated with severe infections in pediatric patients
Scientific Reports (2021)
-
A streamlined clinical metagenomic sequencing protocol for rapid pathogen identification
Scientific Reports (2021)
-
Whole genomic analysis of a potential recombinant human adenovirus type 1 in Qinghai plateau, China
Virology Journal (2020)
-
Human adenovirus species C recombinant virus continuously circulated in China
Scientific Reports (2019)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
