
Abstract
The understanding of black tea quality and percent relative water content (%RWC) traits in tea (Camellia sinensis) by a quantitative trait loci (QTL) approach can be useful in elucidation and identification of candidate genes underlying the QTL which has remained to be difficult. The objective of the study was to identify putative QTL controlling black tea quality and percent relative water traits in two tea populations and their F1 progeny. A total of 1,421 DArTseq markers derived from the linkage map identified 53 DArTseq markers to be linked to black tea quality and %RWC. All 53 DArTseq markers with unique best hits were identified in the tea genome. A total of 5,592 unigenes were assigned gene ontology (GO) terms, 56% comprised biological processes, cellular component (29%) and molecular functions (15%), respectively. A total of 84 unigenes in 15 LGs were assigned to 25 different Kyoto Encyclopedia of Genes and Genomes (KEGG) database pathways based on categories of secondary metabolite biosynthesis. The three major enzymes identified were transferases (38.9%), hydrolases (29%) and oxidoreductases (18.3%). The putative candidate proteins identified were involved in flavonoid biosynthesis, alkaloid biosynthesis, ATPase family proteins related to abiotic/biotic stress response. The functional annotation of putative QTL identified in this current study will shed more light on the proteins associated with caffeine and catechins biosynthesis and % RWC. This study may help breeders in selection of parents with desirable DArTseq markers for development of new tea cultivars with desirable traits.
Similar content being viewed by others

Introduction
The identification of putative candidate genes underlying QTLs in plants is often complicated and time-consuming. This is even further complicated by genotype-environment interactions, which influence the growth and development of plants. However, using recently unveiled tea genome sequence1, it was possible to annotate candidate genes and enzymes involved in the biosynthesis of phenolic compounds and drought tolerance. The phenolic compounds in tea are closely associated to the classical flavour of tea infusions and to the health benefits ascribed to tea consumption2,3. Catechins (flavan-3-ols), flavonols, and their derivatives (galloylated catechins and flavonol 3-O-glycosides) are among the major biomolecules in tea with important bioactivities. The polyphenols in tea have been shown to contribute to the astringency in black tea infusions4. Especially, galloylated catechins have been found to confer astringent and bitter tastes in tea5 while flavonol 3-O-glycosides have been found to confer velvety, mouth-drying, and mouth-coating sensations6. During the production of black tea, the enzymatic oxidation of the leaf catechins by polyphenol oxidase enzyme leads to the formation of theaflavins (TF) and thearubigins (TR)7. Theaflavins which are responsible for the taste, brightness and contribute to the colour of black teas. Thearubigins are responsible for thickness and colour of both the liquors and infusion8. The other plain tea quality parameters or attributes, include black tea brightness, astringency, colour, briskiness and aroma9. Indeed, theaflavins have become a critical parameter in estimating the quality and the price of black teas10. Caffeine is important in cream formation and the briskness of tea and therefore, has been used in predicting black tea quality11,12. Polyphenols and caffeine biosynthesis are dependent on structural and regulatory genes; structural genes encode enzymes catalyzing the biosynthesis, while regulatory genes control the expression of the genes.
Drought is a major factor which significantly influences the growth and production of tea13,14. Hence, breeding drought-tolerant tea cultivars that have high recoverability potential is an important target for tea breeders. Although, the morphological and physiological mechanisms of drought tolerance in tea plant has been reported, only few reports are available on the changes of genes related to catechin biosynthesis in response to environmental stresses15,16. This is because drought tolerance is a complex multigenic trait, and various regulatory and functional genes are involved. In tea plant17, reported that polyphenols can be used as a potential indicator for drought tolerance. In another study, results suggested epicatechin and epigallocatechin as potential indicators in predicting drought tolerance in tea plant18. Recently, transcriptomic analysis of drought susceptible and drought-tolerant tea plants during drought stress has been reported15,19,20. The molecular responses of plants to drought stress include perception, signal transduction, gene expression, and ultimately metabolic changes that lead to stress tolerance. The induced drought stress lead to gene expression which participate in the generation of hormones like abscisic acid (ABA), salicylic acid, and ethylene. For example, ABA has been shown to play crucial roles in regulating the drought response in many plants, and its metabolic pathway involves multiple steps and genes21. The level of ABA in plants increases rapidly during drought stress, leading to stomatal closure and induction of stress genes to cope with the stress22. Drought stress also results in accumulation of proline, mannitol, sorbitol, formation of radical scavenging compounds (ascorbate, glutathione, tocopherol), and synthesis of protein23,24. Proline acts as an osmoprotectant and its accumulation is induced to tolerance of tea to water deficit stress, as this helps in maintaining water relations, prevents membrane distortion, and also acts as a hydroxyl radical scavenger24. The increase in generation of reactive oxygen species (ROS) such as superoxide radicals (O2), hydrogen peroxide (H2O2), hydroxyl radical (.OH), alkoxyl radical (.RO), and singlet oxygen (1O2) within the cell, leads to lipid peroxidation, protein degradation, and enzyme inactivation and affects nucleic acid leading to cell death23,24. The objective of the current study was to annotate and assign biosynthetic pathway functions to putative QTLs linked to caffeine content, catechins fractions, theaflavins fractions, tea taster scores and %RWC.
Materials and Methods
Preparation of FASTA files
The sequences of DArTseq- based markers were derived from the tag sequence associated with each DArTseq marker as explained in the previous work9 and were generated by Diversity Arrays Technology Pty. Ltd. (Canberra, Australia). Marker sequences were arranged in FASTA format by starting with a single-line description of the sequence, followed by sequence data. The single-line description was distinguished from the sequence data by placing a symbol “ > ” in front of the description.
BLAST search
A total of 1,421 DArTseq markers on LG1 to LG15 were subjected to a BLAST search. The marker tag sequences (FASTA format) derived from the DArTseq map were searched using the non-reductant BLASTN program against the assembled draft tea genome (Camellia sinensis var assamica)1. The best hit was then selected based on E-value and % identity. The E-value served as a measure of the number of hits one can “expect” to see by chance when searching a database with small E-value indicating homology. The % identity showed the percentage of identical residues between query sequences and hit sequences from a database, with longer stretches of homology more likely to indicate a genuine match.
Functional annotation and pathway assignment
Functional annotation of all 1,421 DArTseq markers was performed on BLASTX search against the non-redundant GenBank protein sequence database with a threshold E-value of 10-625,26. Functional annotation and mapping of gene ontology (GO) terms was done with the Blast2GO program (Blast2GO v3.2)27. Each BLAST hit associated with the GO terms were retrieved and annotated using the following annotation score parameters; E-Value Hit Filter (default = 1.0E-6), GO-Weight (default = 5), Hsp-Hit Coverage Cut Off (default = 0), Annotation Cut-Off (90). The contig sequences were also queried for conserved domains/motifs using InterProScan, which is an on-line sequence search plug-in within the Blast2GO program. The Kyoto Encyclopedia of Genes and Genomes (KEGG) mapping was used to determine metabolic pathways. The sequences with corresponding evidence code (EC) numbers obtained from Blast2GO were mapped to the KEGG metabolic pathway database.
Results
Functional annotation and pathway assignment
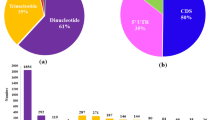
The identification of candidate genes for tea was conducted in all the 15 LGs derived from the DArTseq maps. Sequences of each marker on all the LGs were subjected to a BLAST search against tea genome BLAST database in an effort to compare the homology and identify the location of the gene of interest within the tea genome. The results of BLAST searches for markers derived from the DArTseq map are presented in Table 1. Of 1,421 DArTseq markers derived from DArTseq map, 53 markers were identified to be link to black tea quality and %RWC. A total of seven markers were dominant in TRFK 303/577 parent while 9 markers were dominant in GW Ejulu parent, respectively. All the 53 markers with unique best hits were identified in the tea genome. The 23 markers showed locations across ten chromosomes in tea, which were LG1 (1), LG3 (1), LG4 (8), LG6 (1), LG9 (1), LG10 (2), LG12 (2), LG13 (4) LG14 (2) and LG15 (1). A total of 14 markers were dominant, 8 were dominant in GW Ejulu and six were dominant in TRFK 303/577, respectively. The dominant markers in GW Ejulu were for caffeine, catechin, ECG, EGC and EGCG. On the other hand, the dominant markers in TRFK 303/577 were for caffeine, ECG, EGC, TF1 and %RWC. All the 13 dominant markers were identified in the tea genome for the two parents used in this study as reciprocal crosses in the two tea populations (Table 1)
Annotation and mapping of gene ontology
All unigenes in this study were assigned GO terms based on BLASTX searches against the non-redundant (NR) databases (Fig. 1 and 2). Our results showed that 86–100% similarity distribution of the top hits in the non-redundant (NR) database (Fig. 1). The E-value distribution of the mapped sequences with high homologies (smaller than 1e-6) was high as compared to homologous sequences that were greater than 1e-5 (Fig. 2). The E-value distribution of the best hits in the NR database showed that 26% annotated sequences had high identity with their best hits (smaller than between 1e-28 to 1-e26), whereas 15% ranged from 1e-21 to 1e-25 and another 18% ranged from 1e-15 to 1e-20 (Fig. 1). The similarity distribution revealed that 43% of the query sequences had greater than 91–95% similarity, whereas 21% of the query sequences varied in similarity from 86–90%, and the other 3% varied from 81–85% (Fig. 2).

E-value distribution of the best matches for unigenes (E-value ≤ 1.0e-5).

Similarity distribution of the hit BLAST matches for unigenes.
A total of 5,592 unigenes were assigned GO terms, of these, biological processes (56%) comprised the largest category, followed by cellular component (29%) and molecular functions (15%), respectively (Fig. 3). In biological process (BP) category, the majority of unigenes were involved in regulation of biological process (35%), cellular process (16%), metabolic process (12%), response to stimulus (11%), developmental process (9%) and localization and signaling (6%), respectively (Fig. 3). The cellular and metabolic processes were the most highly represented groups in the biological process, which suggests that extensive metabolic activities taking place in the fresh tea flush. The best-represented groups of molecular function (MF) were binding activity (48%), catalytic activity (27%), signal transducer activity (10%), molecular transducer activity (8%) and transporter activity (7%), respectively (Fig. 3). Within the molecular function category, binding and catalytic activities were the most abundant groups. The cellular component (CC) included the cell (21%), cell part (21%), organelle (16%), membrane (11%) organelle part (7%), membrane part (7%), and macromolecular complex (6%), respectively (Fig. 3). The transcripts that corresponded to cell, cell parts, and cell organelles were the most abundant groups within the cellular component category. Among the top 20 functional sub-groups in biological processes, multicellular organism development, response to stress and response to endogenous stimulus represented the largest group (Supplementary Fig. 1). The top 20 functional sub-groups for molecular functions, protein binding, receptor binding and signal transducer activity represented the largest group (Supplementary Fig. 2), while plasma membrane, nucleus and cytoplasm were the largest group that represented the top 20 sub-groups of cellular component (Supplementary Fig. 3).

Function classifications of GO terms of C. sinensis transcripts based on high-score BLASTX matches in NR plant proteins database, a total of 5,592 unigenes were classified into three main GO categories which are biological process (BP), molecular function (MF) and cellular component (CC), respectively.
KEGG pathway mapping: Flavonoid pathway, caffeine pathway and abiotic stress
Pathway-based analysis for the tea flush transcriptome is helpful to further understand the biological functions of genes involved in the tea flavonoid biosynthesis pathway, caffeine pathway and stimulus to response to abiotic stress (drought stress). A total of 53 unigenes linked to putative QTL were assigned two different KEGG database pathways (Supplementary Table 1) while 84 unigenes in 15 linkage groups were assigned to 25 different KEGG database pathways based on categories of secondary metabolite biosynthesis (Supplementary Table 2, Fig. 4). The pathways with most representations were metabolic pathways and biosynthesis of secondary metabolites, which was consistent with the GO categories of the biological process (Fig. 3 and Supplementary Fig. 1). Of these, 52 unigenes were categorized into the metabolism groups, with most of them involved in purine metabolism (11%), cysteine and methionine metabolism (9.6%), pyruvate metabolism (9.6%), alanine, aspartate and glutamate metabolism (7.6%), arginine and proline metabolism (7.6%) and fructose and mannose metabolism (7.6%) and other sub-categories, respectively. The most abundant unigenes were for carbohydrate and amino acids biosynthesis, which are mostly involved in plant hormone signal transduction pathways in response to abiotic stress and biosynthesis of other secondary metabolites such as phenylpropanoid biosynthesis, flavonoid biosynthesis and alkaloid biosynthesis. The unigenes associated with flavone and flavonol biosynthesis, a key regulatory pathways in the metabolism of the main chemical components in C. sinensis leaves were present (Fig. 4). Unigenes associated with purine metabolism, also a key regulatory pathway which is involved in metabolism of adenine and guanosine nucleotides were present.

KEGG ontology (KO) analysis of differently expressed genes (DEGs) related to secondary metabolism in C. sinensis.
Also, major enzymes that are involved in different metabolic pathways were further grouped into the six different categories; oxidoreductases, transferases, hydrolases, lyases, isomerases and ligases. The distribution of the three abundant enzymes was also consistent with the GO categories of molecular function (Fig. 3 and Supplementary Fig. 2). The three major enzymes distribution included; transferase enzymes (38.9%), which are a class of enzymes that catalyze the transfer of specific functional groups (a methyl, or glycosyl group, or galloyl group) from a donor to acceptor molecules, hydrolase enzymes (29%), which catalyze the hydrolysis of compounds and oxidoreductase enzymes (18.3%), which catalyze the oxidation-reduction reactions (Fig. 5 and Supplementary Fig. 3). The three other enzymes that were less abundant in distribution included; lyase enzymes (8.4%), which cleave carbon-carbon, carbon-oxygen, phosphorous-oxygen and carbon-nitrogen bonds without hydrolysis or oxidation reactions, isomerase enzymes (3.1%), convert a molecule from one isomer to another while ligase enzymes (2.3%), catalyze the formation of a new chemical bond by joining of two large molecules (Fig. 5).

KEGG ontology (KO) distribution of major enzymes code related to secondary metabolism in C. sinensis.
Functional annotation of putative candidate genes 15 linkage groups of C. sinensis
The 28% of the mapped sequences (405 out of 1,471) were annotated. Functional annotations of detected QTL led to the identification of functions of 37 putative candidate genes in parental clones that could be involved in the expression of the targeted traits (Table 1). The putative candidate genes involved in secondary metabolism (phenylpropanoid biosynthesis, flavonoid biosynthesis, alkaloid biosynthesis and volatile compounds biosynthesis) were located in five linkage groups LG04, LG09, LG10 and LG13. The putative candidate genes identified were involved in phenylpropanoid and flavonoid biosynthesis pathways included a group or class of enzymes namely; 2-oxoglutarate/Fe(II)-dependent dioxygenases superfamily (2-ODDs), aminotransferase class I and II, acyltransferase, glycosyl hydrolase family 9. This was in agreement with KEGG pathway mapping where enzymes mapped clustered into six major groups according to different metabolic pathways they are involved with transferases, hydrolases and oxidoreductases as three abundant enzymes. The multidrug and toxic compound extrusion (MATE) transporter was also putatively identified in LG09. BT1 family protein located in LG04 was putatively annotated for qCAFF. The putative QTL and their corresponding putative candidate genes associated with phenylpropanoid biosynthesis, and flavonoid biosynthesis were identified in both parental clones.
Proteins related with oxidative stress and ATPase family proteins or proteins related to abiotic stress were found in the confidence intervals of QTL for drought tolerance traits. Among the 53 examined putative QTL in parental clones (Table 1), 18 putative QTL were related to abiotic stress (ABA signaling, amino acids peptide degradation) and were mostly found on LG04 and LG13. However, the putative QTL related to abiotic stress, including percent RWC was also found on LG01, LG06, LG10, LG12, LG14 and LG09, respectively. The putative candidate genes identified included, pectinesterase, 14-3-3 protein, 2OG-Fe (II) oxygenase superfamily, protein kinase domain, transmembrane amino acid transporter protein, peptidase family M3, histone acetyltransferase subunit NuA4, DnaJ domain and MatE. ATPase family proteins associated with various cellular activities (AAA), and Armadillo/beta-catenin-like repeat protein were found to be associated with the heat stress response. The putative QTL in LG13 that was associated with disease stress response was NB-ARC domain protein. Putative candidate genes related with ABA signaling pathway were found on QTL for qCAT, qEC, qEGCG and qRWC (Table 1), respectively. The putative candidate gene related to degradation of peptides by targeting the amino acids residues that are formed during abiotic stress was found on QTL for qEC. The parental clone TRFK 303/577 which is known to be high yielding and drought-tolerant cultivar, exhibited most putative candidate genes related to drought stress response as compared to parental clone GW- Ejulu.
Discussion
Flavonoid biosynthesis pathway in tea begins with the amino acid phenylalanine, which undergoes deamination to form trans-cinnamic acid28,29. The oxidation process of trans-cinnamic acid yields p-coumaric acid, which undergoes transformation to form p-coumaroyl-CoA. The committed step in flavonoid biosynthesis is the subsequent condensation of p-coumaroyl-CoA and malonyl-CoA catalyzed by chalcone synthase enzyme to form chalcone. The subsequent isomerization of chalcone forms (2S)-flavonones. The identification of putative QTL, qCAT, for 2-ODDs superfamily protein in this study corroborates with other previous reports on the role of 2-ODDs in flavonoid biosynthesis. The 2-ODDs which are large superfamily of non-heme proteins, which facilitate numerous oxidative reactions, including hydroxylation, epimerization, C-C bond cleavage, ring formation and fragmentation. These 2-ODDs superfamily catalyze key oxidative reactions that facilitate the formation of different flavonoid subclasses, for example, the oxidation of (2S)-flavonones. The (2S)-flavonones are converted to dihydroflavonols through hydroxylation process which is then catalyzed by another 2-ODD, flavonone 3β-hydroxylase (F3H). The dihydroflavonols formed is a substrate for flavonol synthase (FNS I), which competes with dihydroflavonol 4-reductase (DR4) to form flavonols, anthocyanidins and procyanidins.
Glycosylation is an important process for the diverse functions of polyphenolic compounds in plants and is known to be able to increase the solubility and stability of hydrophobic flavonoids30. The glycosylation process is regulated by glycoside hydrolases, which are involved in hydrolysis and/or rearrangement of glycosidic bonds. We hypothesize the putative QTL, qBRT linked to glycosyl hydrolase family 9, a family of glycoside hydrolases in the current study to play a role in the synthesis and sequestration of some phenylpropanoids31. In tea, the galloylated catechins formed through the glucosylation activity of UDP-glucosyltransferase, have been found to confer astringent and bitter tastes, while flavonol 3-O-glycosides have been found to induce velvety, mouth-drying, and mouth-coating sensations6. In legume plant, bur clover (Medicago truncatula), epicatechin 3′-O-glucoside, has also been shown to be involved in the biosynthesis of proanthocyanidin in the seed coat32,33. In addition, the glycosylated polyphenols function also as acyl donors in biochemical reactions, for example, β-glucogallin, a glucose ester of gallic acid, functions as an acyl transfer donor in the biosynthesis of both galloylated tannins34 and galloylated catechins35.
Acyltransferases which participates in many secondary metabolism pathways was putatively identified in this current study (qECG) in LG10. Acyltransferases are involved in the biosynthesis of anthocyanidin and other flavonoid groups by transferring acyl groups to the sugar moiety of anthocyanins using acyl-CoA as the donor36. The process of modification of anthocyanin and other flavonoid groups with acyl groups is catalyzed by acyltransferases and is important in maintaining their stability. In LG04, qCAFF, was putatively annotated as BT1 family protein which is has been identified to be involved in export of adenine nucleotides, which are exclusively synthesized in plants plastids, and are precursors for caffeine biosynthesis37. Furthermore, nucleotides, adenine and guanosine are precursors in caffeine biosynthesis in tea plants38.
The 14-3-3 proteins which bind to other proteins to induce target-site specific alteration and conformation of target proteins are important plant signal transduction pathways. Previous reports implicate plant 14-3-3s in key physiological processes, in particular, abiotic and biotic stress responses, metabolism (especially primary carbon and nitrogen metabolism), as well as various aspects of plant growth and development. In the current study, putative QTL, qCAT in LG13 was putatively annotated 14-3-3 protein, which is abiotic or biotic stress protein in tea. This, therefore, is in agreement with previous study by18 where individual catechins were demonstrated as potential indicators in predicting drought tolerance in tea plant. Meanwhile, a number of recent studies in Arabidopsis have linked 14-3-3s to ABA signaling. Some of the main functions ascribed to ABA include response to abiotic/biotic stress.
Drought stress which usually affects the plant photosynthetic process also interferes with plant nutrient availability leading to ion intoxication. The reversible protein phosphorylation by protein kinases at the early and later stage of the signaling pathways is one of the mechanisms that plants use in response to abiotic stress. In addition, to maintain the osmotic homeostasis and tolerance due to drought stress, plants also do regulate gene expression of some specific genes of protein kinases. The presence of a putative QTL, qCAT, which was annotated for protein kinase domain in this study supports the argument of the previous studies on the role of tea catechins as potential indicators of drought tolerance in tea plants. A previous study by39 indicated that catechins were higher in the drought-tolerant cultivars and therefore, corroborated with the presence of qCAT, annotated as protein kinase domain in drought-tolerant parental clone TRFK 303/577.
Transmembrane amino acid transporter protein functions as gateways to permit the transport of amino acids across the biological membrane. Amino acids are not only critical for the synthesis of proteins, but other many essential compounds, which act as signal molecules. For example, in Arabidopsis, water and salt stress induce a strong expression of proline transporter 1 (ProT1) and proline transporter two while the expression of amino acid permease 4 (AAP4) and amino acid permease 6 (AAP6) is repressed40,41. In tea, proline has been shown to act as an osmo-protectant during drought stress23,24 and the presence of a putative candidate gene (transmembrane amino acid protein) in this current study, could hypothesize the functions of transmembrane amino acid transporter for proline presence during drought stress. Furthermore, the presence of transmembrane amino acid transporter proteins could be their involvement of transport of aromatic amino acids phenylalanine, tyrosine and tryptophan, which serve as precursors for a wide range of secondary metabolites (polyphenols) in tea. This is because the two identified putative QTL, qCAT and qEGC in LG13 in both parental clones (TRFK 303/577 and GW Ejulu) are products of shikimate and phenylalanine pathways.
The MATE proteins are a family of secondary active transporters, which utilize electrochemical gradient of membrane maintained by ATPases for their transport activity. MATE transporters are involved in transport of secondary metabolites into vacuoles, which are the major storage site of most of the conjugated flavonoids, which comprise of flavonols, anthocyanins and flavone glycoside42. In the current study, putative QTL, qRWC in LG09 was annotated for MATE proteins, which are involved in the sequestration of flavonoids in vacuoles in response to drought stress43. Secondary metabolites in plants, especially flavonoids play an important role in response to abiotic stress such as drought stress. A member of MATE gene family (TT12) has also been elucidated for sequestration of proanthocyanidins in seed vacuoles, which leads to pigmentation of the seed coat44. In yeast, it was revealed that TT12 proteins can specifically transport glycosidic form like epicatechin 3′-O-glucoside and cyanidin 3-O glucoside45. The MATE proteins from a legume, bur clover (Medicago truncatula) and grapevines (Vitis) have been characterized for transport of flavonoids through the sequestration of epicatechin 3-O-glucoside and cyanidin 3-O glucoside (proathocyanidins) into the vacuole for storage33. In another study, a part from the role of vacuolar sequestration flavonoids, MATE proteins have been recognized in transport of alkaloids into vacuoles in tobacco. Furthermore, MATE proteins have facilitated and modulated abscisic acid efflux and sensitivity in drought-tolerant Arabidopsis46. Since a putative QTL for %RWC annotated as MATE in this current study, it therefore, corroborates with previous reports47 which relied on the percent %RWC to classify tea cultivars as either drought tolerant or drought susceptible based on tea leaves metabolites.
In this current study, putative QTL, qECG in LG06 was putatively annotated DnaJ protein. DnaJ proteins are essential components that contribute to cellular protein homeostasis and complex stabilization of proteins (protein folding, degradation and refolding) under stress conditions in plants48. They function as molecular chaperones either alone or in association with heat-shock protein 70 (Hsp70). In a previous study on the transcriptomic analysis of the effect of drought-induced-stress on tea leaf quality, revealed that the levels of ECG and EGCG increased during drought stress49. This was also reported by39 on the effects of soil moisture stress alterations on tea biomolecules in relation to tea quality. Furthermore, in transgenic tobacco, the overexpression of tomato chloroplast-targeted DnaJ enhanced drought tolerance50 and maintained photosystem II under chilling stress51. Our findings, therefore, suggest that ECG can be used as a potential marker of drought tolerance or cold stress in tea cultivars.
The putative QTL, qEGCG, in LG04 was annotated diacylglycerol kinase catalytic domain protein which we hypothesize as a marker for drought and cold tolerance in tea cultivars. Abiotic stress, including salinity, drought, and osmotic adjustments triggers the production of phosphatidic acid52. Phosphatidic acid is formed by phosphorylation of diacylglycerol by diacylglycerol kinase. Thus, phosphatidic acid rather than diacylglycerol has typically been implicated as a major plant secondary messenger. However, the presence of diacylglycerol is necessary for certain developmental processes and the response to particular environmental stimuli. For example, salinity stress in A. thaliana increased the activity of non-specific phospholipase C, which promoted the production of diacylglycerol. In addition, cold stress has recently been found to stimulate the formation of phosphatidic acid in suspension-cultured Arabidopsis cell53.
Histone acetyltransferases play a critical role in histone acetylation of plant chromatin, which is important in epigenetic control of gene expression. The transfer of acetyl groups to core histone tails by histone acetyltransferases, promote transcription of target genes involved in drought response and absisic acid signaling in several plants, including Arabidopsis54, rice55, and maize56. Furthermore, previous studies reported up-regulation of drought stress responsive genes and found to be correlated with changes in histone modification57,58. The putative QTL, qCAT, qEC and qEGCG in this current study were annotated histone acetyltransferase subunit NuA4 protein in drought-tolerant parental clone TRFK 303/577. Therefore, catechins could act as potential indicators or markers of drought tolerance in tea, which corroborates with previous reports18,39 which investigated the biochemical changes in tea constituents as influenced by varying levels of water stress.
Aminotransferases, also known as transaminases, catalyze amino group transfers from amino donor to amino acceptor compounds. Aminotransferases play a major role in a variety of metabolic pathways, including, amino acid biosynthesis, secondary metabolites biosynthesis and photorespiration. Prephenate aminotransferase, a class of aminotransferase enzyme, catalyze the final step in the synthesis of phenylalanine, which is the central product of shikimic acid pathway and serves as a precursor for the synthesis of flavonoids59. The identification of putative QTL, qEC in the current study annotated as aminotransferase I and II is hypothesized as a marker linked to flavonoids biosynthesis in tea.
Conclusion
This current study is the first attempt to obtain more information on genomic and functional annotation of proteins/ enzymes involved in black tea quality and drought tolerance traits in tea plant based on DArTseq markers. The DArTseq markers used, and putative QTL identified in this current study will shed more light on the proteins/ enzymes associated with the biosynthesis of caffeine, individual catechins in green tea which are converted to individual theaflavins in black tea processing. In addition, the DArTseq markers will provide useful information, in particular for studies of the genetic determinants of black tea quality traits and drought tolerance in tea. Also, the DArTseq markers used in the current study can be useful markers for construction of linkage maps or may be used to discover new functions of genes. We successfully constructed a genetic linkage map using SNPs, but it was impossible to anchor it to other previously constructed linkage maps of tea since no anchoring markers were available. Furthermore, it was also difficult to deduce the relative genetic positions of the SNPs on the previous different linkage maps. Indeed, previous studies used RAPD, AFLP and SSRs markers to construct tea genetic linkage maps, while in our study, we used DArTseq markers. However, the information obtained through the annotation of putative gene functions and the alignment of DArTseq sequences relative to recently published reference tea genome, has set a milestone for a future and further researches on marker-assisted selection and breeding of tea plants. In addition, this work may help breeders in selection of parents with desirable DArTseq markers for development of new tea cultivars with desirable traits.
Data Availability
The DArT sequences have been submitted to NCBI (http://www.ncbi.nlm.nih.gov/). BioProject PRJNA398959.
References
Xia, E. H. et al. The tea tree genome provides insights into tea flavor and independent evolution of caffeine biosynthesis. Mol. Plant. 10, 866–877 (2017).
Preedy, V.R. Tea in health and disease prevention, (Academic Press, 2012).
Wang, K. et al. Comparison of phenolic compounds and taste of Chinese black tea. Food Sci. Technol. Res. 20, 639–646 (2014).
Scharbert, S. & Hofmann, T. Molecular definition of black tea taste by means of quantitative studies, taste reconstitution, and omission experiments. J. Agric. Food Chem. 53, 5377–5384 (2005).
Rossetti, D., Bongaerts, J., Wantling, E., Stokes, J. & Williamson, A.-M. Astringency of tea catechins: more than an oral lubrication tactile percept. Food Hydrocoll. 23, 1984–1992 (2009).
Cui, L. et al. Identification of UDP-glycosyltransferases involved in the biosynthesis of astringent taste compounds in tea (Camellia sinensis). J. Exp. Bot. 67, 2285–2297 (2016).
Obanda, M., Owuor, P. O., Mang’oka, R. & Kavoi, M. M. Changes in thearubigin fractions and theaflavin levels due to variations in processing conditions and their influence on black tea liquor brightness and total colour. Food Chem. 85, 163–173 (2004).
Owuor, P. O., Wachira, F. N. & Ng’etich, W. K. Influence of region of production on relative clonal plain tea quality parameters in Kenya. Food Chem. 119, 1168–1174 (2010).
Koech, R. K. et al. Identification of novel QTL for black Tree Genet. Genomes tea quality traits and drought tolerance in tea plants (Camellia sinensis). Tree Genet. Genomes. 14, 9 (2018).
Owuor, P. O. & Obanda, M. The use of green tea (Camellia sinensis) leaf flavan-3-ol composition in predicting plain black tea quality potential. Food Chem. 100, 873–884 (2007).
Obanda, M., Owuor, P. O. & Taylor, S. J. Flavanol composition and caffeine content of green leaf as quality potential indicators of Kenyan black teas. J. Sci. Food Agric. 74, 209–215 (1997).
Liang, Y., Lu, J. & Zhang, L. Comparative study of cream in infusions of black tea and green tea [Camellia sinensis (L.) O. Kuntze]. Int. J. Food Sci. 37, 627–634 (2002).
Ng’etich, W., Stephens, W. & Othieno, C. Responses of tea to environment in Kenya. 3. Yield and yield distribution. Exp. Agr. 37, 361–372 (2001).
Cheruiyot, E., Mumera, L., Ng’etich, W., Hassanali, A. & Wachira, F. Threshold soil water content for growth of tea [Camellia sinensis (L.) O. Kuntze]. Tea 29, 29–38 (2008).
Liu, S.-C. et al. Transcriptomic analysis of tea plant responding to drought stress and recovery. PloS one 11, e0147306 (2016).
Tony, M. et al. Transcriptome-based identification of water-deficit stress responsive genes in the tea plant, Camellia sinensis. Plant Biotechnol. J. 43, 302–310 (2016).
Cheruiyot, E. K., Mumera, L. M., Ng’etich, W. K., Hassanali, A. & Wachira, F. Polyphenols as potential indicators for drought tolerance in tea (Camellia sinensis L.). Biosci. Biotechnol. Biochem. 71, 2190–2197 (2007).
Cheruiyot, E. K. et al. Shoot epicatechin and epigallocatechin contents respond to water stress in tea [Camellia sinensis (L.) O. Kuntze]. Biosci. Biotechnol. Biochem. 72, 1219–1226 (2008).
Maritim, T. K. et al. Physiological and biochemical response of tea [Camellia sinensis (L.) O. Kuntze] to water-deficit stress. J. Hortic. Sci. Biotechnol. 90, 395–400 (2015).
Zhang, H. B. et al. De novo transcriptome assembly of the wild relative of tea tree (Camellia taliensis) and comparative analysis with tea transcriptome identified putative genes associated with tea quality and stress response. BMC Genomics 16, 298 (2015).
Zhou, L. et al. Exogenous abscisic acid significantly affects proteome in tea plant (Camellia sinensis) exposed to drought stress. Horticulture. Research 1, 14029 (2014).
Sah, S. K., Reddy, K. R. & Li, J. Abscisic acid and abiotic stress tolerance in crop plants. Front. Plant Sci. 7, 571 (2016).
Upadhyaya, H. & Panda, S. K. Abiotic stress responses in tea [Camellia sinensis L (O) Kuntze]: an overview. Reviews in Agricultural Science 1, 1–10 (2013).
Upadhyaya, H. B. K., D. & Panda, S. K. Drought induced physiological and biochemical changes in leaves of developing seedlings of Tea [Camellia sinensis (L) O Kuntze] Cultivars Journal of Tea Science Research 6, 1–11 (2016).
Conesa, A. et al. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676 (2005).
Conesa, A. & Götz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genomics. 2008(2008).
Blake, J. et al. Gene ontology annotations and resources. Nucleic Acids Res. 41 (2013).
Punyasiri, P. et al. Flavonoid biosynthesis in the tea plant Camellia sinensis: properties of enzymes of the prominent epicatechin and catechin pathways. Arch. Biochem. Biophys. 431, 22–30 (2004).
Vankatesh, P. et al. Flavanoid biosynthesis in Camellia sinensis. (2007).
Xu, L., Qi, T., Xu, L., Lu, L. & Xiao, M. Recent progress in the enzymatic glycosylation of phenolic compounds. J. Carbohyd Chem. 35, 1–23 (2016).
Jones, P., Messner, B., Nakajima, J.-I., Schäffner, A. R. & Saito, K. UGT73C6 and UGT78D1, glycosyltransferases involved in flavonol glycoside biosynthesis in Arabidopsis thaliana. J. Biol Chem. 278, 43910–43918 (2003).
Pang, Y., Peel, G. J., Wright, E., Wang, Z. & Dixon, R. A. Early steps in proanthocyanidin biosynthesis in the model legume Medicago truncatula. Plant Physiol. 145, 601–615 (2007).
Zhao, J. & Dixon, R. A. MATE transporters facilitate vacuolar uptake of epicatechin 3′-O-glucoside for proanthocyanidin biosynthesis in Medicago truncatula and Arabidopsis. The Plant Cell 21, 2323–2340 (2009).
Niemetz, R. & Gross, G. G. Enzymology of gallotannin and ellagitannin biosynthesis. Phytochemistry 66, 2001–2011 (2005).
Liu, Y. et al. Purification and characterization of a novel galloyltransferase involved in catechin galloylation in the tea plant (Camellia sinensis). J Biol Chem. 287, 44406–44417 (2012).
Mizutani, M. et al. An acyltransferase involved in biosynthesis of polyacylated anthocyanin, s198 (2006).
Haferkamp, I. The diverse members of the mitochondrial carrier family in plants. FEBS Lett. 581, 2375–2379 (2007).
Negishi, O., Ozawa, T. & Imagawa, H. Biosynthesis of Caffeine from Purine Nucleotides in Tea Plant. Biosci. Biotechnol. Biochem. 56, 499–503 (1992).
Jeyaramraja, P., Pius, P., Raj Kumar, R. & Jayakumar, D. Soil moisture stress‐induced alterations in bioconstituents determining tea quality. J. Sci. Food Agric. 83, 1187–1191 (2003).
Hayat, S. et al. Role of proline under changing environments: a review. Plant Signal. Behav. 7, 1456–1466 (2012).
Hua, X. et al. Arabidopsis AMINO ACID PERMEASE1 contributes to salt stress-induced proline uptake from exogenous sources. Front. Plant Sci. 8, 2182 (2017).
Shitan, N. Secondary metabolites in plants: transport and self-tolerance mechanisms. Biosci. Biotechnol. Biochem. 80, 1283–1293 (2016).
Petrussa, E. et al. Plant flavonoids—biosynthesis, transport and involvement in stress responses. Int. J. Mol. Sci. 14, 14950–14973 (2013).
Gonzalez, A. et al. TTG2 controls the developmental regulation of seed coat tannins in Arabidopsis by regulating vacuolar transport steps in the proanthocyanidin pathway. Dev. Biol. 419, 54–63 (2016).
Marinova, K. et al. The Arabidopsis MATE transporter TT12 acts as a vacuolar flavonoid/H + -antiporter active in proanthocyanidin-accumulating cells of the seed coat. The Plant Cell 19, 2023–2038 (2007).
Zhang, H. et al. A DTX/MATE-type transporter facilitates abscisic acid efflux and modulates ABA sensitivity and drought tolerance in Arabidopsis. Mol. Plant. 7, 1522–1532 (2014).
Nyarukowa, C., Koech, R., Loots, T. & Apostolides, Z. SWAPDT: A method for Short-time Withering Assessment of Probability for Drought Tolerance in Camellia sinensis validated by targeted metabolomics. J. Plant Physiol. 198, 39–48 (2016).
Park, C. J. & Seo, Y. S. Heat shock proteins: a review of the molecular chaperones for plant immunity. Plant Pathol. J. 31, 323 (2015).
Wang, W. et al. Transcriptomic analysis reveals the molecular mechanisms of drought-stress-induced decreases in Camellia sinensis leaf quality. Front. Plant Sci. 7, 385 (2016).
Wang, G. et al. Overexpression of tomato chloroplast-targeted DnaJ protein enhances tolerance to drought stress and resistance to Pseudomonas solanacearum in transgenic tobacco. Plant Physiol. Biochem. 82, 95–104 (2014).
Kong, F. et al. A chloroplast-targeted DnaJ protein contributes to maintenance of photosystem II under chilling stress. J. Exp. Bot. 65, 143–158 (2013).
Zhu, J. K. Abiotic stress signaling and responses in plants. Cell 167, 313–324 (2016).
Arisz, S. A. et al. Rapid phosphatidic acid accumulation in response to low temperature stress in Arabidopsis is generated through diacylglycerol kinase. Front. Plant Sci. 4, 1 (2013).
Kim, J. M. et al. Alterations of lysine modifications on the histone H3 N-tail under drought stress conditions in Arabidopsis thaliana. Plant Cell Physiol. 49, 1580–1588 (2008).
Fang, H., Liu, X., Thorn, G., Duan, J. & Tian, L. Expression analysis of histone acetyltransferases in rice under drought stress. Biochem. Biophys. Res. Commun. 443, 400–405 (2014).
Kapazoglou, A. & Tsaftaris, A. Epigenetic chromatin regulators as mediators of abiotic stress responses in cereals. In Abiotic Stress in Plants-Mechanisms and Adaptations (InTech, 2011).
Kim, J. M., Sasaki, T., Ueda, M., Sako, K. & Seki, M. Chromatin changes in response to drought, salinity, heat, and cold stresses in plants. Front. Plant Sci. 6, 114 (2015).
Asensi-Fabado, M. A., Amtmann, A. & Perrella, G. Plant responses to abiotic stress: The chromatin context of transcriptional regulation. Biochim Biophys Acta (BBA) 1860, 106–122 (2017).
Ververidis, F. et al. Biotechnology of flavonoids and other phenylpropanoid‐derived natural products. Part I: Chemical diversity, impacts on plant biology and human health. Biotechnol. J. 2, 1214–1234 (2007).
Acknowledgements
The authors acknowledge the financial support to conduct this research, and study grants for RK and PM from James Finlay (Kenya) Ltd., George Williamson (Kenya) Ltd., Sotik Tea Company (Kenya) Ltd., Mcleod Russell (Uganda) Ltd., the TRI of Kenya and Southern African Biochemistry and Informatics for Natural Products (SABINA). The C. sinensis cultivars used in this study were provided by the TRI of Kenya. Supplementary funding was provided by, the Technology and Human Resources for Industry Programme (THRIP), an initiative of the Department of Trade and Industries of South Africa (dti), the National Research Foundation (NRF) of South Africa, and the University of Pretoria (South Africa).
Author information
Authors and Affiliations
Contributions
Z.A., S.K. and R.M. were involved with the design of the experiment and plant material used. R.K., P.M. and C.N. were involved in collection of plant material. R.K. performed the experiments. R.K., P.M., C.N., S.K. and Z.A. analyzed samples and interpreted the data. F.J. performed BLASTX and functional annotation. R.K. wrote the manuscript and revised by P.M., C.M., R.M., S.K. and Z.A. The final manuscript was reviewed and approved by all the authors.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Koech, R.K., Malebe, P.M., Nyarukowa, C. et al. Functional annotation of putative QTL associated with black tea quality and drought tolerance traits. Sci Rep 9, 1465 (2019). https://doi.org/10.1038/s41598-018-37688-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37688-z
This article is cited by
-
QTL Mapping and Genetic Map for the Ornamental Sunflower in China
Plant Molecular Biology Reporter (2023)
-
Population structure analysis to explore genetic diversity and geographical distribution characteristics of cultivated-type tea plant in Guizhou Plateau
BMC Plant Biology (2022)
-
Breeding tea for drought tolerance
Euphytica (2021)
-
Construction of a DArT-seq marker–based genetic linkage map and identification of QTLs for yield in tea (Camellia sinensis (L.) O. Kuntze)
Tree Genetics & Genomes (2021)
-
Identification and distribution of a single nucleotide polymorphism responsible for the catechin content in tea plants
Horticulture Research (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
