
Abstract
Potential hybridization between wolves and dogs has fueled the sensitive conservation and political debate underlying the recovery of the grey wolf throughout Europe. Here we provide the first genetic analysis of wolf-dog admixture in an area entirely recolonized, the northwestern Alps. As part of a long-term monitoring program, we performed genetic screening of thousands of non-invasive samples collected in Switzerland and adjacent territories since the return of the wolf in the mid-1990s. We identified a total of 115 individuals, only 2 of them showing significant signs of admixture stemming from past interbreeding with dogs, followed by backcrossing. This low rate of introgression (<2% accounting for all wolves ever detected over 1998–2017) parallels those from other European populations, especially in Western Europe (<7%). Despite potential hybridization with stray dogs, few founders and strong anthropogenic pressures, the genetic integrity of the Alpine population has remained intact throughout the entire recolonization process. In a context of widespread misinformation, this finding should reduce conflicts among the different actors involved and facilitate wolf conservation. Real-time genetic monitoring will be necessary to identify potential hybrids and support an effective management of this emblematic population.
Similar content being viewed by others


Introduction
Anthropogenic hybridization between wild species and their domesticated counterparts is a central concern in conservation biology. Introgression of artificially selected alleles may be beneficial, by bringing adaptive genetic variation into natural gene pools1. Alternatively, it may threaten wild populations due to genetic homogenization, incompatibilities and/or disruption of locally-adapted allelic combinations2,3,4,5. In any case, crossbreeding and subsequent introgression jeopardizes the genetic integrity of wild populations, on which legal actions are enforced by institutional stakeholders.
The grey wolf (Canis lupus) and its domesticated form, the dog (C. l. familiaris), make one of the most emblematic and complex example of this issue. After their nearly complete eradication in large parts of their original distribution over the last centuries, wolves are now recovering and recolonizing parts of their former ranges6, raising serious concerns regarding threats to livestock to the point that their management has become a political debate. Wolf-dog hybridization is a central aspect of this debate. Hybrids have been reported throughout Europe (Table 1), essentially from asymmetric crosses (male dogs × female wolves), with backcrossed individuals subsequently integrated into wolf packs7,8,9,10,11 (but see12). While wolf is under strict protection by several international treaties, although with national derogations to reduce predation and conflicts with local husbandry, crossbred individuals fall into a legal gap. Recommendations include their removal to protect the integrity of wild populations13,14,15,16. Hence, characterizing the genetic nature of expanding wolves is a major aspect of their conservation, even more so since morphological criteria potentially diagnostic of hybrids (i.e. black coat, white claws and spur on the hind legs) are unreliable to properly identify admixed individuals17,18,19.
In the recent years, a wide array of studies have comprehensively monitored the rate and modality of wolf-dog hybridization in expanding European populations (summarized in Table 1), which appears to depend on their level of disturbance (see below), in link with the abundance of feral (returned to the wild state) and stray (free-ranging) dogs. Accordingly, admixture remains low in Western Europe (i.e. 0 to 6.5% across the Italian Apennines and Iberia, Table 1), being locally higher in anthropogenic environments where stray dogs are common19,20, and where residing packs are introgressed21. The figures are expectedly higher in eastern countries (10–14% in Georgia, Bulgaria and nearby Greece), where free-ranging large-size guarding dogs are an issue22. All studies, however, have focused on recovering populations that never faced full eradication. In contrast, the risk of crossbreeding with dogs is expected to be significantly higher in newly recolonized areas. This is firstly due to the low number of founders involved: hybridization in the initial stages of colonization could lead to widespread introgression as the population expands. Second, the areas where the wolf formerly vanished are usually human-dominated, with low acceptance by locals. These conditions, and notably the potentially high rate of poaching, may disrupt their social structure, in turn favoring crossbreeding with stray dogs. For instance, in Croatia and Italy, many hybrids are found near human settlements with a high rate of human-caused mortality19,20.
The Alpine wolf population remains a major gap in the European wolf-dog hybridization literature. Exterminated in the French and Swiss Alps in the late 19th century, and in the Italian Alps during the early 20th century, wolves have been re-establishing across this range from relict Italian Apennine pockets ever since the early 1990s23,24,25,26. Their formerly high mitochondrial DNA diversity has now essentially been replaced by a single Control Region (CR) haplotype (thereafter the Italian haplotype), diagnostic of these regions and never found in dogs nor wolves elsewhere24,27,28,29,30. This new population thus makes an ideal system to test whether hybridization with dogs had stronger genetic consequences in recolonized areas. Listed as “endangered” (based on data from 200731,), the Alpine wolf population is constantly increasing, forming about 65 packs in the last update (2015–2016), mostly located in the Western parts between France and Italy32. In addition to the lack of suitable habitat and persecution, potential hybridization with dogs is considered as a major threat33. In Switzerland, only three established packs occur, but tens of itinerant individuals migrate throughout the country and neighboring regions. Wolf presence is highly controversial in this country: despite its strictly protected status, the species is under very stringent management through selective removal of stock-raiding individuals (when predation impact is considered too high), but is facing strong pressure by livestock breeders, hunters and local communities34, which conveys public rumors such as that wolf-dog hybrids are widespread. Hence, inferring the rate of wolf-dog introgressive hybridization in the Alpine population is also crucial for its future management and social perception.
In this study, we conducted a genetic survey of all the wolves detected in the Swiss and adjacent territories, as part of an ongoing non-invasive genetic monitoring since the early stages of the recolonization process started more than 20 years ago24,25. We genotyped thousands of non-invasive DNA samples and performed admixture analyses of multilocus genotypes of putative wolves and reference dogs, in order to estimate the rate of hybridization and admixture in the newly colonized Alpine population.
Results
Genetic screening
We sequenced the left domain of the hypervariable mitochondrial (mtDNA) Control Region (CR) in a total of 3,463 non-invasive unidentified samples and 23 tissues from dead wolves (359–360 bp or 235–236 bp, see Methods). A total of 1,645 samples could be assigned to the private Italian wolf haplotype (47.2%), 409 to typical dog haplotypes (C. l. familiaris; 11.7%), 709 to red foxes (Vulpes vulpes; 20.3%), 361 to other mammals (10.4%) and 362 (10.4%) could not be either PCR-amplified or sequenced (due to contamination by several taxa, e.g. saliva samples on preys). The 68 reference dogs did not carry the Italian wolf CR haplotype, but only typical dog ones (Table S2).
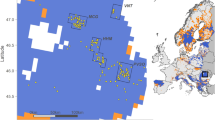
Reliable microsatellite genotyping (11 loci) according to the multitube approach was successful in 874 out of 1,645 putative wolf samples (53%; scat: 60%; saliva: 50%; hair: 22%; urine: 22%; regurgitate: 100%; blood: 43%; tissue: 100%), identifying 115 different individuals from 1998 to 2017 (79 males, 36 females), with their number of detections ranging from 1 to 62 (average: 7.6; Table S1). Allelic dropout and false allele rate across loci averaged 5.8% and 0.9% respectively. The number of wolves increased over years, males first (Fig. 1 and Table S1). The maximum consecutive detection of a single individual reached at least seven years (2011–2017), and 21 wolves were ultimately found dead or legally removed from the population. An additional two died in Germany.

Evolution of the numbers of detected wolves in Switzerland in space and time. Place of last genetic detection (circles) or death (triangles) is shown on the map. Colors distinguish pure (green) from admixed wolves (orange). Approximate locations of the three residing packs are encircled in white. Wolf management compartments as defined by the FOEN are delineated in red. The bottom left panel frames the study area (red) and the main recolonization pathway presumably taken by the wolf from the Apennine population (green arrows) according to24,25,26.
Population genetic and admixture analyses
The Swiss wolves met Hardy-Weinberg Equilibrium (HWE) and appear panmictic among the three mainly occupied management compartments defined for Switzerland (n = 73, non-significant pairwise Fst); note that this analysis excluded presumptive cubs from the three packs, known from field observations and confirmed by their genetic profiles. We found similar levels of diversity between these compartments (observed heterozygosity, He = 0.62–0.64; Allelic richness, Ar ~ 2.4, scaled on two individuals). This lack of genetic structure is also supported by the frequent movement of individuals between Swiss cantons (i.e. states; Table S1). The full wolf dataset had a probability of identity (PID) of 2.2 × 10–8 and a PIDsibs of 3.8 × 10−4.
Bayesian clustering analyses of the 115 putative wolves and 68 reference dogs (obtained from Switzerland) by STRUCTURE35 unambiguously discriminated between the two groups (K = 2, ΔK = 1485.0) (Fig. 2A). Principal Component Analysis (PCA) on individual genotypes also recover the two corresponding clusters along the first axis (Fig. 3). At least four individuals, however, were not perfectly assigned to their corresponding group, i.e. STRUCTURE admixture coefficients (Q) below 1.0 and intermediate positions on the first axis of the PCA (Fig. 3). One challenge is to interpret whether such pattern arises from recent gene flow or imperfect clustering due to shared polymorphism (i.e. false positive), which is bounded by the number and informativeness of the genetic marker used19,36. To address this issue, we simulated different classes of hybrids, using genotypes that were correctly assigned to their clusters (“pure” wolves and dogs, Q > 0.99) as parental references, on which we conducted similar analyses (Fig. S1). F1s, F2s and wolf first generation backcrosses received average wolf ancestry Qw values of 0.48 (95% CI: 0.28–0.72), 0.50 (95% CI: 0.26–0.79), 0.73 (95% CI: 0.50–0.94) respectively, and thus marginally encompass those of pure individuals (95% CI: Qw = 0.90–1.0; Fig. S1). However, the distinction becomes difficult with second-generation backcrosses (average Qw = 0.89, 95% CI: 0.61–0.96; Fig. S1). Following the methodology of previous studies19,36, we used the minimum Qw value of pure individuals (0.86) as an ad-hoc threshold to consider individuals as admixed.

Bayesian clustering of individual wolf and dog genotypes (A) with STRUCTURE into two groups, and (B) with NewHybrids into eight genotype classes, including six hybrid and two parental classes (n = 99 pure wolves and 55 pure dogs). Individuals are arranged by their time of first detection. The dotted line show the threshold computed from analyses of simulated hybrids (Fig. S1). The two individuals above this threshold are shown.

Principal Component Analysis (PCA) on individual wolf (green) and dog (red) genotypes. The two admixed wolves identified by the admixture analyses are shown by red frames. Ellipses correspond to the 80% inertia of each group.
From the empirical dataset, only two putative wolves (out of 115; 1.7%) were assigned with a probability lower than Qw = 0.86 and hence were most likely introgressed by dogs (Fig. 2): individuals M51 (Qw = 0.72; male, 9% of missing data) and F16 (Qw = 0.79; female, no missing data). Inspection of the 90% confidence intervals for Qw did not encompass 1.0 for M51 (0.40–0.96), confirming its admixed nature, but it did for F16 (0.54–1.0). Two additional individuals received low Qw ( < 0.95), i.e. M83 (Qw = 0.88; male), M80 (Qw = 0.93; male), although these genotypes respectively featured 31% and 18% of missing data.
Moreover, Bayesian assignment of empirical genotypes to hybrid classes (F1, F2, first and second backcross generations in each direction) with NewHybrids37 confirmed that almost all Alpine wolves had the best probabilities to be purebred animals, except for M51 and F16, which shared an equal probability to be first or second generation backcrosses (Fig. 2B). Five additional individuals had cumulated probabilities above 0.1 to be backcrosses, although there were always primarily assigned to the parental wolf class, rather than to any other class.
Discussion
Our long-term genetic monitoring supports very limited wolf-dog hybridization and introgression in the Swiss Alps and surroundings, confirming the genetic integrity of this recently established wolf population throughout time. Over the past two decades, no F1 hybrid was detected and only two wolves (out of 115) featured significant signs of introgression by dogs, likely stemming from backcrossing. There were accordingly assigned as first or second generation wolf backcrosses. We cannot exclude that other individuals descend from older hybridization events, with the little dog ancestry they hypothetically retained confounding with the standard variation shared by the two parental genomes at our limited set of loci. Because these crossbreeding events are very rare, the wolf gene pool is rapidly restored due to gene flow from pure individuals. While our resolution seems sufficient to detect the first backcrosses (as in other studies, Table 1), false positives could arise if individual genotypes are uninformative or incomplete17,19. Here, the two individuals M80 and M83, with Qw < 0.95, were among the very few with > 15% of missing data, and hence unreliable estimates (Table S1 and Methods).
The anecdotal rate of admixture in the Alpine wolves ever since their very first establishment (<2% considering all individuals since 1998) is in line with other European populations, although these were mainly studied over shorter timeframes (Table 1). It is also supported from a recent assessment in the Italian Alps, which did not identify any hybrid (www.lifewolfalps.eu). Feral and stray dogs are supposedly absent in Switzerland, and wolves usually avoid the immediate vicinity of human settlements. All dogs detected here are doubtlessly pets, hunting or guarding dogs, the Swiss mountains being extensively hiked and exploited for pastoralism. Although local crossbreeding cannot be ruled out, these rare hybridization events may have rather occurred in the Apennines, the source population of Alpine wolves24,25. About a million of free-ranging dogs has been reported over Italy by perhaps outdated estimates (10% of feral individuals38,39). While the overall dog introgression rate of Italian wolves still remains low (<7%, Table 1), some localized areas host introgressed packs21. Among them, western Tuscany, at the northwestern edge of the Apennine population, appears to be a hotspot of wolf × dog hybridization20, near the corridor connecting the Apennines and the Alps. Alpine wolves thus seems to have avoided gene flow with dogs and admixed individuals, which is remarkable given the low numbers of effective founders that arrived in this human-dominated region (~1525). The high mortality rate in Switzerland (21 out of the 115 individuals analyzed) could have also hampered the rapid establishment of stable social structures, promoting mating with dogs instead (as shown in wolf-coyote hybridization40); the first Swiss pack settled >16 years after the species’ arrival. Therefore, and despite these suboptimal initial conditions, the now established Alpine population has so far resisted the risks of hybridization.
Note that because we identified putative wolves from non-invasive samples based on maternally transmitted mtDNA (i.e. the diagnostic CR Italian haplotype), we only tested admixture resulting from crossbreeding between female wolves and male dogs. Accordingly, all wolf genetic surveys from Italy and the Alps published to date (featuring thousands of samples) failed to detect the reverse (dog mitochondrial introgression in wolf populations; reviewed in41). Potential reasons for this asymmetry are multiple, e.g. less aggressiveness of female wolves towards dogs (while male wolves prey on them), longer partner-seeking activity by female wolves, continuous physiological availability of male dogs for mating, greater survival chances of pups raised by wolf mothers in the wild (12 and references therein). Accordingly, all tissues from dead wolves identified here featured the Italian CR haplotype (n = 23). As populations are reconnecting, other wolf haplotypes have recently been found in the eastern Alps (originating from the Balkans and Central Europe42,43) and even southern Italy44,45, but so far have not reached our study area30,41. The absence of nuclear differentiation between Swiss wolves also confirms that the recolonization of the Alpine arc stems from a single source, as previously suggested24,25. Moreover, it indicates that the environmental conditions in Switzerland are favorable enough that itinerant and resident wolves remain panmictic, implying a good connectivity, as also suggested by the migration patterns of individuals between cantons (Table S1).
The idea sometimes propagated that Alpine wolves are “hybrids”, largely based on unconvincing and unpublished evidence, is thus not supported by our genetic data. This misconception is confusing the public debate between the various actors involved, e.g. livestock breeders, hunters and institutional stakeholders. Wolves and dogs obviously share a common evolutionary history, intertwined by recurrent crossbreeding ever since the earliest stages of domestication, between 35,000 and 11,000 years ago46. As a result, ancient dog breeds feature traces of admixture with wolves and, reciprocally, most Eurasian wolf populations acquired dog alleles through past introgression events, which can confound with ancestral polymorphisms maintained across these recently diverged taxa47,48,49. While this should not be confused with contemporary hybridization, it conditions why detecting wolf-dog hybrids for management purposes is far from trivial, given the limitation of resources (i.e. number and nature of analyzable genetic loci) imposed by genetic profiling of wild individuals from low-quality non-invasive samples. New methodologies and technologies (e.g. high-throughput microsatellite genotyping50; SNPs51,52) will allow better resolution to detect hybrid backcrosses from pure individuals in future genetic screening.
In Switzerland, the recolonizing wolves have thus retained their genetic integrity. The two confirmed backcrossed individuals are no longer in the country. Female F16 arrived and settled in the Central Alps from June 2014 to February 2017, when it was ultimately poached (Table S1). Male M51 was only detected at nine instances from February to August 2015 in the eastern and southern parts of the country. None of the parents, and by extension their offspring, of the three resident wolf packs feature signs of dog introgression. In order to protect this population, we stress the need to prevent dog vagrancy in the species’ expansion range, as well as to legally remove any F1 hybrids as soon as they are detected by real-time genetic screening and/or suspicion from morphological characters. In the presumed absence of dogs, these F1 hybrids, potentially migrating from Italy, are the proximate cause of subsequent dog introgression into the wolf gene pool, and should be the main target of legal regulations. In contrast, managing admixed individuals beyond F1s seems neither relevant nor efficient, since the wolf genome is being restored, and because effectively tracing such level of admixture remains challenging. External criteria are indeed unreliable to identify backcrosses, even from dead animals: despite its dog introgression, no question was raised regarding female F16. Reciprocally, this questions the validity of so-called “reference” wolves identified by morphology; putative wolves should only be selected based on molecular analyses (e.g.21,36,53,54, this study). Finally, we emphasize the crucial role played by such genetic monitoring, which provides an empirical basis to move the public debate forward, and hopefully improve the tense relationship between the many actors discussing the fate of large predators claiming back their former ranges.
Methods
Study area and sample collection
The study was conducted in Switzerland and neighboring territories (nearby France and Italy), covering an area of about 45,000 km2, as a part of a long-term non-invasive genetic monitoring of the species since its recolonization of the Alpine range in the 1990s. A total of 3,486 samples were analyzed. These include 3,463 unidentified samples, comprising 1,191 scats (34.4%), 2,007 saliva swabs collected on preys (58.0%), 106 hair (3.1%), 119 urine samples (3.4%), 7 regurgitates (0.2%) and 33 blood samples (0.9%), which were non-invasively collected in the field from November 1998 to December 2017 by trained rangers and wardens from Swiss regional wildlife offices and researchers. In addition, tissues from 23 dead wolves (accidentally, illegally or legally killed) were also analyzed. The geographic origin of samples and their number of detections are given in Table S1. Sites for non-invasive sampling were chosen opportunistically based on known or presumed wolf presence, documented prey kills or random direct observations. Tissue, scat and hair samples were stored in 80–95% ethanol at +4 or −20 °C. Swab samples were stored dried and immediately processed after arrival in the laboratory. Regurgitate and urine samples were stored at −20 °C.
No ethics approval was necessary to work with non-invasive samples or tissues from dead animals. Fieldwork procedures were specifically approved by regional wildlife offices and the Federal Office for the Environment (FOEN) as a part of national wolf monitoring activities.
DNA extraction, sequencing and genotyping
DNA from scat samples was extracted with the QIAamp® DNA Stool or Fast DNA Stool Mini kit (Qiagen), while all other samples except urine were extracted with the QIAamp® Tissue or DNeasy® Blood & Tissue kit (Qiagen), following manufacturer’s instructions. DNA from urine samples collected in snow was extracted according to55. Species identification was assessed by sequencing a 359–360 bp portion of the left domain of the mtDNA Control Region (CR; primers and methods in24), except for saliva samples for which we used a newly-designed ungulate-unspecific internal primer (HW3: 5′-GCCCTTATTGGACTAAGTG-3′; 235–236 bp amplified) in order to avoid co-amplification of undesirable prey DNA. DNA sequencing was performed on an ABI3100 platform (Applied Biosystems), after purification with the QIAquick PCR Purification kit (Qiagen) or Wizard® SV Gel and PCR Clean-Up System (Promega). All DNA extractions and pre-PCR setups occurred in a physically-separated laboratory exclusively devoted to the analysis of low copy number DNA samples. Negative controls were employed during all extraction and amplification experiments to monitor contaminations.
Eleven microsatellite (i.e. STR) loci (FH2054, FH2140, FH2161, FH2096, FH2137, FH2088, FH2001, FH201056, PEZ17, PEZ157, CPH558) and a Y-chromosome sex marker (Sryw f: 5′- GCCGAGTCCTCTCCTGTA-3′/Sryw r: 5′-TTGTATGAACCATCATTGTGA-3′) were amplified, following the multiplex preamplification method59, with some modifications. The principle of this two-step PCR approach is that an initial large-volume PCR is carried out including primers for all 12 markers to be genotyped. A post-amplification aliquot from this PCR is then used as template in separate amplifications for each marker to genotype individuals. This procedure was repeated eight times (four times for tissues) to obtain a consensus genotype, following the multi-tube approach60. Multiplex amplifications were performed in 50 µl reactions containing 2.5 mM MgCl2, 0.1 mM of each dNTP, 0.01 µmol of each primer, 0.2 mg/ml BSA, 1 x PCR Buffer and 1 U of AmpliTaq® Gold DNA polymerase (Applied Biosystems), and 20 µl of extract. For multiplex reactions we used an annealing temperature of 50–55 °C and 25 PCR cycles. The second PCR step was performed in 20 µl reactions containing 2.5–5.0 mM MgCl2, 0.1 mM of each dNTP, 0.2–0.5 µmol of each primer, 0.2 mg/ml BSA, 1 x PCR Buffer and 1 U of AmpliTaq® Gold DNA polymerase (Applied Biosystems), and 2.0 µl of extract. We used an annealing temperature of 50–57 °C and 40 PCR cycles. One primer of each pair was synthesized with a 5′-end fluorescent dye (ATTO532, ATTO550, Dyomics 630, FAM and HEX) to allow detection and sizing of fragments on an ABI Prism 3100 DNA sequencer (Applied Biosystems). Alleles were scored using GeneMapper 4.0 (Applied Biosystems). Consensus genotypes, allelic dropout and rates of false alleles were computed manually as follows: heterozygous at one locus were accepted if both alleles appeared at least four times among the eight replicates, and homozygous were accepted if the genotype was observed in five independent PCRs. If neither of those cases occurred, the alleles were treated as missing data (NA). In addition, we genotyped saliva samples of dogs (n = 68) from 35 different breeds, obtained in Switzerland through veterinary practice, to be used as a reference (Table S2). The final dataset (n = 183) included 131 individuals with no NA (72%), 41 with 0–10% NA (22%), 7 with 10–20% NA (4%), and 4 with 20–45% NA (2%) (Details in Tables S1 and S2).
Standard Genetic Analyses
We assigned individuals to the management compartments defined by the FOEN for this species (Fig. 1), based on the area they spent most of their time according to our genetic detections. Individuals sampled in neighboring France and Italy where assigned to the closest compartment. For the three main occupied compartments (n = 7, 35 and 31 for compartments III, IV and V, respectively, not-including cubs from packs) we computed observed heterozygosity (He), allelic richness (Ar), pairwise Fst (with significance tested by 1,000 permutations), and performed test for Hardy-Weinberg Equilibrium (HWE) in FSTAT61. Probabilities of identity (PID) were estimated with GenAlEx 6.562.
Admixture analyses
We assigned putative wolves (n = 115) and dogs (n = 68) to groups with the Bayesian clustering algorithm of STRUCTURE35. We used the admixture model and ran 10 replicates for K from 1 to 11, each including 100′000 iterations after a burnin of 10′000. Replicates were combined with CLUMP63 and admixture proportions Q were visualized with DISTRUCT64. The ΔK statistic65 was computed to infer the number of K best explaining the data (STRUCTURE HARVESTER66). In parallel, we conducted a PCA to assess the genetic variance between individuals (ade4 and adegenet R package67).
In order to infer the nature of individuals that featured intermediate STRUCTURE Q values, we used the software NewHybrids37 to compute their probability of belonging to the six following hybrid classes: F1, F2, wolf first generation backcross, wolf second generation backcross, dog first generation backcross and dog second generation backcross. We pre-assigned pure individuals (Q > 0.99 in the STRUCTURE analyses) as parental references (n = 99 wolves and 55 dogs). The chain was ran for 100′000 iterations, which was sufficient to reach stationarity.
We further implemented a simulation approach to evaluate the power to discriminate between pure and introgressed individuals from our dataset. To this end, we generated F1, F2 and backcrossed hybrids (100 for each category) from the reference individuals selected above, using the hybridize function of adegenet. These simulated and pure genotypes were then analyzed by STRUCTURE (K = 2) and PCA, in the same way as the empirical dataset (see above). This allowed to obtained the distribution of Qw (the wolf ancestry of individual) for each hybrid class, and calculate the threshold below which individuals can be considered admixed without confusion with pure ones19,36.
Data Availability Statement
Microsatellite genotypes are available from the Dryad Digital Repository (https://doi.org/10.5061/dryad.7g2g68d).
References
Hedrick, P. W. Wolf of a different colour. Heredity 103, 435–436 (2009).
Rhymer, J. M. & Simberloff, D. Extinction by hybridization and introgression. Annu Rev Ecol Evol Syst. 27, 83–109 (1996).
Allendorf, F., Leary, R., Srpuel, P. S. & Wenburg, J. K. The problems with hybrids: setting conservation guidelines. Trends Ecol. Evol. 16, 613–622 (2001).
Randi, E. Detecting hybridization between wild species and their domesticated relatives. Mol. Ecol. 17, 285–293 (2008).
Dufresnes, C., Dubey, S., Ghali, K., Canestrelli, D. & Perrin, N. Introgressive hybridization of threatened European tree frogs (Hyla arborea) by introduced H. intermedia in Western Switzerland. Conserv. Genet. 16, 1507–1513 (2015).
Chapron, G. et al. Recovery of large carnivores in Europe’s modern human-dominated landscapes. Science 346, 1517–1519 (2014).
Randi, E. & Lucchini, V. Detecting rare introgression of domestic dog genes into wild wolf (Canis lupus) populations by Bayesian admixture analyses of microsatellite variation. Conserv. Genet. 3, 31–45 (2002).
Andersone, Z., Lucchini, V., Randi, E. & Ozolins, J. Hybridization between wolves and dogs in Latvia as documented using mitochondrial and microsatellite DNA markers. Mamm. Biol. 67, 79–90 (2002).
Verardi, A., Lucchini, V. & Randi, E. Detecting introgressive hybridization between free-ranging domestic dogs and wild wolves (Canis lupus) by admixture linkage disequilibrium analysis. Mol. Ecol. 15, 2845–2855 (2006).
Vilà, C. et al. Combined use of maternal, paternal and bi-parental genetic markers for the identification of wolf-dog hybrids. Heredity 90, 17–24 (2003).
Godinho, R. et al. Genetic evidence for multiple events of hybridization between wolves and domestic dogs in the Iberian Peninsula. Mol. Ecol. 20, 5154–5166 (2011).
Hindrikson, M., Männil, P., Ozolins, J., Krzywinski, A. & Saarma, U. Bucking the trend in wolf-dog hybridization: first evidence from Europe of hybridization between female dogs and male wolves. PLoS ONE 7, e46465 (2012).
Trouwborst, A. Managing the Carnivore Comeback: International and EU Species Protection Law and the Return of Lynx, Wolf and Bear to WesternEurope. . J. Env. Law 22, 347–372 (2010).
Trouwborst, A. Exploring the Legal Status of Wolf-Dog Hybrids and Other DubiousAnimals: International and EU Law and the Wildlife Conservation Problem of Hybridization with Domestic and Alien Species. RECIEL 23, 111–124 (2014).
Schnidrig R., et al (Eds). Wolf in the Alps: Recommendations for an internationally coordinated management. RowAlps Report Objective 3. KORA Bericht Nr. 72. KORA, Muri bei Bern, Switzerland, and BAFU, Ittigen, Switzerland, 70 pp. (2016)
Bern Convention on the Conservation of European Wildlife and Natural Habitats. Recommendation No. 173 of the Standing Committee, adopted on 5 December 2014, on hybridization between wild grey wolves (Canis lupus) and domestic dogs (Canis lupus familiaris). https://rm.coe.int/1680746b83. (2014)
Randi, E. et al. Multilocus detection of wolf × dog hybridization in Italy, and guidelines for marker selection. PLoS ONE 9, e86409 (2014).
Galaverni, M. et al. Disentangling timing of admixture, patterns of introgression, and phenotypic indicators in a hybridizing wolf population. Mol. Biol. Evol. 34, 2324–2339 (2017).
Kusak, J. et al. Wolf-dog hybridization in Croatia. Vet. Arhiv 88, 375–395 (2018).
Caniglia, R. et al. Black coats in an admixed wolf × dog pack is melanism an indicator of hybridization in wolves? Eur. J. Wildl. Res. 59, 543–555 (2013).
Bassi, E. et al. Trophic overlap between wolves and free-ranging wolf × dog hybrids in the Apennine Mountains, Italy. Global Ecol. Conserv. 9, 39–49 (2017).
Kopaliani, N., Shakarashvili, M., Gurielidze, Z., Qurkhuli, T. & Tarkhnishvili, D. Gene flow between wolf and shepherd dog population in Georgia (Caucasus). J. Hered. 105, 345–353 (2014).
Breitenmoser, U. Large predators in the Alps: the fall and rise of man’s competitors. Biol. Conserv. 83, 279–289 (1998).
Valière, N. et al. Long-distance wolf recolonization of France and Switzerland inferred from non-invasive genetic sampling over a period of 10 years. Anim. Conserv. 6, 83–92 (2003).
Fabbri, E. et al. From the Apennines to the Alps: colonization genetics of the naturally expanding Italian wolf (Canis lupus) population. Mol. Ecol. 16, 1661–1671 (2007).
Fabbri, E. et al. Genetic structure of expanding wolf (Canis lupus) populations in Italy and Croatia, and the early steps of the recolonization of the Eastern Alps. Mamm. Biol. 79, 138–148 (2014).
Vilà, C. et al. Multiple and ancient origins of the domestic dog. Science 276, 1687–1689 (1997).
Randi, E. et al. Mitochondrial DNA variability in Italian and East European Wolves: Detecting the consequences of small population size and hybridization. Conserv. Biol. 14, 464–473 (2000).
Pilot, M. et al. Phylogeographic history of grey wolves in Europe. BMC Evol. Biol. 10, 104 (2010).
Dufresnes, C. et al. Howling from the past: historical phylogeography and diversity losses in European grey wolves. Proc. R. Soc. B. 285, 20181148 (2018).
IUCN 2018. The IUCN Red List of Threatened Species. Version 2018-1. < http://www.iucnredlist.org >. Downloaded on 05 July 2018.
Wolf Alpine Group (2018): Wolf population status in the Alps: pack distribution and trend up to 2016, with focus on year 2015-2016. Available at http://www.lcie.org. 2018 March.
Boitani, L., et al. Key actions for Large Carnivore populations in Europe. Institute of Applied Ecology (Rome, Italy). Report to DG Environment, European Commission, Bruxelles. Contract no. 07. 0307/2013/654446/SER/B3. 119 pp. (2015)
Breitenmoser, U., et al. The recovery of wolf Canis lupus and lynx Lynx lynx in the Alps: Biological and ecological parameters and wildlife management systems. RowAlps Report Objective 1. KORA Bericht Nr. 70. KORA, Muri bei Bern, Switzerland. 276 pp. (2016)
Pritchard, J. K., Stephens, M. & Donelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Pacheco, C. et al. Spatial assessment of wolf-dog hybridization in a single breeding period. Sci. Rep. 7, 42475 (2017).
Anderson, E. C. & Thompson, E. A. A model-based method for identifying species hybrids using multilocus genetic data. Genetics. 160, 1217–1229 (2002).
Boitani, L. & Fabbri, M. L. Censimento dei cani in Italia con particolare riguardo al fenomeno del randagismo. Ricerche di Biologia della Selvaggina, INBS, Bologna 73, 1–51 (1983).
Genovesi, P. & Dupre, E. Strategia nazionale di conservazione del lupo (Canis lupus): indagine sulla presenza e la gestione dei cani vaganti in Italia. Biol. Cons. Fauna (I.N.F.S.) 104, 1–36 (2000).
Rutledge, L. Y., White, B. N., Row, J. R. & Patterson, B. R. Intense harvesting of eastern wolves facilitated hybridization with coyotes. Ecol. Evol. 2, 19–33 (2012).
Hindrikson, M. et al. Wolf population genetics in Europe: a systematic review, meta-analysis and suggestions for conservation and management. Biol. Rev. 92, 1601–1629 (2016).
Rauer, G., Knauer, F., & Fumagalli, L. Austria - meeting place of three wolf populations: melting pot or black hole? In Potocnik, H. et al. (eds) Wolf Conservation in a Human Dominated Landscape, SloWolf Project (University of Ljubljana, Slovenia, 2013).
Razen, N. et al. Long-distance dispersal connects Dinaric-Balkan and Alpine grey wolf (Canis lupus) populations. Eur. J. Wildl. Res. 62, 137–142 (2016).
Boggiano, F. et al. Detection of an East European wolf haplotype puzzles mitochondrial DNA monomorphism of the Italian wolf population. Mamm. Biol. 78, 374–378 (2013).
Montana, L., Caniglia, R., Galaverni, M., Fabbri, E. & Randi, E. A new mitochondrial haplotype confirms the distinctiveness of the Italian wolf (Canis lupus). Mamm. Biol. 84, 30–34 (2017).
Freedman, A. H. & Wayne, R. K. Deciphering the origin of dogs: From fossils to genomes. Annual Review of Animal Biosciences 5, 281–307, https://doi.org/10.1146/annurev-animal-022114-110937 (2017).
Vonholdt, B. M. et al. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 8, 464 (2010).
Fan, Z. et al. Worldwide patterns of genomic variation and admixture in gray wolves. Genome Res. 26, 163–173 (2016).
Pilot, M. et al. Widespread, long-term admixture between grey wolves and domestic dogs across Eurasia and its implication for the conservation status of hybrids. Evol. Appl. 2018, 1–19 (2018).
De Barba, M. et al. High‐throughput microsatellite genotyping in ecology: improved accuracy, efficiency, standardization and success with low‐quantity and degraded DNA. Mol. Ecol. Res. 15, 492–507 (2017).
Kraus, R. H. S. et al. A single-nucleotide polymorphism-based approach for rapid and cost effective genetic wolf monitoring in Europe based on noninvasively collected samples. Mol. Ecol. Res. 15, 295–305 (2014).
von Thaden, A. et al. Assessing SNP genotyping of noninvasively collected wildlife samples using microfluidic arrays. Sci. Rep. 7, 10768 (2017).
Lorenzini, R., Fanelli, R., Grifoni, G., Scholl, F. & Fico, R. Wolf-dog crossbreeding: “Smelling” a hybrid may not be easy. Mamm. Biol. 79, 149–156 (2014).
Godinho, R. et al. Real-time assessment of hybridization between wolves and dogs: combining noninvasive samples with ancestry informative markers. Mol. Ecol. 15, 317–328 (2015).
Valière, N. & Taberlet, P. Urine collected in the field as a source of DNA for species and individual identification. Mol. Ecol. 9, 2150–2152 (2000).
Francisco, L. V., Langston, A. A., Mellersh, C. S., Neal, C. L. & Ostrander, E. A. A class of highly polymorphic tetranucleotide repeats for canine genetic mapping. Mamm. Genome 7, 359–362 (1996).
Neff, M. W. et al. A second-generation linkage map of the domestic dog. Canis familiaris. Genetics 151, 803–820 (1999).
Fredholm, M. & Winterø, A. K. Variation of short tandem repeats within and between species belonging to the Canidae family. Mamm. Genome 6, 11–18 (1995).
Piggott, M. P., Bellemain, E., Taberlet, P. & Taylor, A. C. A multiplex pre-amplification method that significantly improves microsatellite amplification and error rates for faecal DNA in limiting conditions. Conserv. Genet. 5, 417–420 (2004).
Taberlet, P. et al. Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res. 24, 3189–3194 (1996).
Goudet, J. FSTAT (version 1.2): a computer program to calculate F-Statistics. J. Hered. 86, 485–486 (1995).
Peakall, R. & Smouse, P. E. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research - an update. Bioinformatics 28, 2537–2539 (2012).
Jakobsson, M. et al. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806 (2007).
Rosenberg, N. A. DISTRUCT: a program for the graphical display of population structure. Mol. Ecol. Notes 4, 137–138 (2003).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software Structure: a simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Earl, D. A. & Vonholdt, B. M. Structure Harvester: a website and program for visualizing Structure output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361 (2012).
Jombart, T. Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405 (2008).
Caniglia, R., Fabbri, E., Galaverni, M., Milanesi, P. & Randi, E. Noninvasive sampling and genetic variability, pack structure, and dynamics in an expanding wolf population. J. Mammal. 95, 41–59 (2014).
Ramirez, O. et al. Genetic assessment of the Iberian wolf Canis lupus signatus captive breeding program. Conserv. Genet. 7, 861–878 (2006).
Torres, R. T., Ferreira, E., Rocha, R. G. & Fonseca, C. Hybridization between wolf and domestic dog: first evidence from an endangered population in central Portugal. Mamm. Biol. 86, 70–74 (2017).
Moura, A. E. et al. Unregulated hunting and genetic recovery from a severe population decline: the cautionary case of Bulgarian wolves. Conserv. Genet. 15, 405–417 (2014).
Ciucci, P., Lucchini, V., Boitani, L. & Randi, E. Dewclaws in wolves as evidence of admixed ancestry with dogs. Can. J. Zool. 81, 2077–2081 (2003).
Khosravi, R., Rezaei, H. R. & Kaboli, M. Detecting hybridization between Iranian wild wolf (Canis lupus pallipes) and free-ranging domestic dog (Canis familiaris) by analysis of microsatellite markers. Zool. Sci. 30, 287–34 (2013).
Acknowledgements
We thank all rangers and game wardens from Swiss regional wildlife offices and collaborators who participated to sampling. We also thank C. Jan, C. Ossola, K. Parker and Y. Ulrich for help in the laboratory; P. Taberlet and C. Miquel (Univ. of Grenoble, France) for general advices and for providing the sex-linked marker; H. J. Blankenhorn, R. Schnidrig, C. Jaeggi, C. Nienhuis and M. Pewsner from the Federal Office for the Environment (FOEN); Dr L. Gagnebin who provided the reference dog samples. This study was supported by the Federal Office for the Environment (FOEN). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
L.F. designed the study and supervised molecular work. N.R., C.S. and L.F. performed the lab work. C.D. and L.F. analyzed the genetic data. R.M. and J.M.W. coordinated field monitoring and sample collection. C.D. and L.F. wrote the manuscript. All authors discussed and commented on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dufresnes, C., Remollino, N., Stoffel, C. et al. Two decades of non-invasive genetic monitoring of the grey wolves recolonizing the Alps support very limited dog introgression. Sci Rep 9, 148 (2019). https://doi.org/10.1038/s41598-018-37331-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-37331-x
This article is cited by
-
A decade of genetic monitoring reveals increased inbreeding for the Endangered western leopard toad, Sclerophrys pantherina
Conservation Genetics (2022)
-
Eyes, ears, or nose? Comparison of three non-invasive methods to survey wolf recolonisation
Mammalian Biology (2021)
-
Lifelong non-invasive genetic monitoring of a philopatric female wolf in the Tuscan Apennines, Italy
European Journal of Wildlife Research (2021)
-
Reliable wolf-dog hybrid detection in Europe using a reduced SNP panel developed for non-invasively collected samples
BMC Genomics (2021)
-
A standardized approach to empirically define reliable assignment thresholds and appropriate management categories in deeply introgressed populations
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
