
Abstract
In the recent years, there is an extensive effort concentrated towards the development of nanoparticles with near-infrared emission within the so called second or third biological windows induced by excitation outside 800–1000 nm range corresponding to the traditional Nd (800 nm) and Yb (980 nm) sensitizers. Here, we present a first report on the near-infrared (900–1700 nm) emission of significant member of cubic sesquioxides, Er-Lu2O3 nanoparticles, measured under both near-infrared up-conversion and low energy X-ray excitations. The nanoparticle compositions are optimized by varying Er concentration and Li addition. It is found that, under ca. 1500 nm up-conversion excitation, the emission is almost monochromatic (>93%) and centered at 980 nm while over 80% of the X-ray induced emission is concentrated around 1500 nm. The mechanisms responsible for the up-conversion emission of Er - Lu2O3 are identified by help of the up-conversion emission and excitation spectra as well as emission decays considering multiple excitation/emission transitions across visible to near-infrared ranges. Comparison between the emission properties of Er-Lu2O3 and Er-Y2O3 induced by optical and X-ray excitation is also presented. Our results suggest that the further optimized Er-doped cubic sesquioxides represent promising candidates for bioimaging and photovoltaic applications.
Similar content being viewed by others

Introduction
Among the rare earth oxides, cubic Lu2O3 is an attractive phosphor host for lanthanide (Ln) activators due to its high mass density (9.4 g cm−3), good phase stability, low thermal expansion, low phonon energy (phonon cutoff ~600 cm−1), and broad optical transparency from the visible to the near infrared (NIR) regions1,2,3,4. The top of the valence-band energy levels of Lu-based oxides are mainly composed of lutetium 4f orbital, whereas in Y-based materials, the corresponding top levels are mostly oxygen 2p orbital. Such a difference was considered to make Lu-based compounds more favorable than the Y-based ones as up conversion hosts5. The smaller cell volume may also enhance the energy transfer efficiency between lanthanide dopants in Lu2O3 in comparison with that in isostructural Y2O3 for the same fractional doping concentration2. There are already a few reports that suggest that the up-conversion emission intensity is up to two order of magnitude stronger in Lu2O3 compared to Y2O3 phosphor3,6,7,8. Besides, several investigations on Ln doped Lu-based compounds, such as Tm–Ho doped LuLiF4, and Tm/Er doped NaLuF4, have been proven to have stronger luminescence and better laser performance than their corresponding Y homologues9,10,11.
Despite that Lu2O3 is regarded, in principle, more advantageous than Y2O3 as an up-conversion host, only 17 articles were found by searching Scopus and ISI database in the last 10 years using “Lu2O3” and “up-conversion” as keywords compared to almost 100 for Y2O3. Among the Ln ions used in the up-conversion studies, Er single doped or co-doped with Yb sensitizer, represents the most investigated activator (12 articles) due to its multiple emission and absorption transitions spanning from ultraviolet (UV) to near infrared (NIR) spectral range.
Vetrone et al. showed6 that up-conversion under excitation (980 nm) of nanocrystalline 2%Er-Lu2O3 is lower than its bulk counterpart. Beside the presence of the ground state absorption (GSA) followed by excited state absorption (ESA) mechanisms, the contribution of the energy transfer up-conversion (ETU) mechanism was confirmed by the emission decays measurements both bulked and nanoparticulate samples. Capobianco et al.3 observed that under down-conversion excitation at 488 nm, the relative integrated intensity of the 4I11/2-4I15/2 (around 980 nm) and 4I13/2-4I15/2 (around 1500 nm) emission transitions is about 3:1 for bulked sample compared to 1:1 in nanocrystalline sample due to the presence of the carbonates and hydroxyl groups on the nanocrystalline surface. In addition, the same authors suggested that the much stronger (up to two orders of magnitude) up-conversion emission intensity measured with Er-Lu2O3 compared to Er-Y2O3 nanoparticles under 980 nm excitation was related to reduced adsorption of CO2 and H2O on the Lu2O3 surface induced by the greater particle size of Lu2O3 (50 nm) than Y2O3 (20 nm).
Recent research has shown that extending fluorescence imaging into shortwave IR (1000–2000 nm) wavelengths can further enhance the advantages of NIR imaging12,13,14,15,16. Low levels of background tissue autofluorescence in the shortwave IR increase imaging sensitivity to a target nanoprobe, and the unique tissue absorption and scattering properties increase contrast of structures at greater penetration depths compared with fluorescence imaging in the NIR17,18,19,20,21. On the other side, it is well known that the efficiency of solar cells is increased by converting the photons with energies below the absorption threshold (λ > 1100 nm) into higher energy photons (λ < 1100 nm)22. Such interesting applications explain the recent growing number of studies on Er based systems that present a relative intense up-conversion emission centered at 980 nm following excitation at 1500 nm, such as Er-CeO223,24, Er-Y2O325 and Er-NaYF426, Er(Yb)-Gd2O2S27,28,29, Er-BaTiO330, Er-BaY2F831 and Er-LiYF432,33. To the best of the authors knowledge, only one study investigated so far, the up-conversion emission of Er-Lu2O3 nanoparticles using ca. 1500 nm excitation8. However, the measured up-conversion emission included only the green emission (520–580 nm), disregarding the NIR emission around 980 nm. Following an extensive literature on Scopus database, we have found that, while in the last 10 years more than a 1000 studies have been published on the up-conversion emission of Er(Yb) doped systems under 980 nm excitation, less than 100 studies concerned 1500 nm as up-conversion excitation wavelength. Of these, less than 20 studies measured the 980 nm emission in Er(Yb) nanoparticulate systems. Finally, to the best of our knowledge, less than 10 studies report on (almost) pure/monochromatic up-conversion emission around 980 nm (Fig. S1).
Different from Yb- and Nd- based nanocrystals excitable only at 980 and 808 nm, respectively, Er-based nanocrystals can be efficiently excited at 980 and 1532 nm that is sought to optimal consideration of detection sensitivity, light penetration, and photothermal effects in the context of in vivo imaging34. During the time our manuscript was under review, additional papers have been published on the so-called new type of Er sensitized upconversion nanoparticles exploiting the upconversion excitation wavelength at 1500 nm35,36,37. Liu et al.35 designed a strategy for up-conversion core-shell nanoparticles using Er (1530 nm) as sensitizer, and Er (980 nm), Ho (1180 nm) and Nd (1060 nm) as activators in the second biological window. Also, they demonstrated the use of such nanoparticles for in vivo dynamic inflammation by use of a microneedle patch sensing at very high resolution (200 × 200 μm). Wang et al.36 developed Er enriched core–shell nanocrystals of NaErF4:20%Yb@NaLuF4 with efficient luminescence at 1525 nm under biocompatible excitation wavelength of 808 nm for bioimaging. Cheng et al.37 demonstrated a new type of up-conversion nanoparticle, sensitized by Er ions and extended the excitation wavelength to 1532 nm. Upon excitation, Er ions can transfer the absorbed energy to codoped activators, resulting in upconversion emission ranging from 400 to 1200 nm. Extending the excitation range of upconversion nanocrystals outside 800–1000 nm corresponding to traditional Nd and Yb sensitizers represents therefore an active and formidable challenge targeting applications such as anticounterfeiting, bioimaging, display, photovoltaics and information storage35,36,37,38,39.
Herein, we report a first and comprehensive description of the up-conversion emission properties of Er-Lu2O3 nanoparticles under ns pulsed excitation around 1500 nm monitored between 500 to 1100 nm. We highlight here the advantage offered by simultaneous use of pulsed laser excitation and time-gated luminescence detection. As such, nanoparticles that exhibit long lifetimes (from µs to ms range) in NIR biological window allow an improved contrast visualization40 by avoiding the tissue autofluorescence that is extended even in near infrared (>1000 nm17). The composition of nanoparticles was optimized by varying the concentrations of both Er activator and optically inert Li. Upconversion emission and indirect/direct decay measurements were performed considering multiple absorption transitions in the visible and near-infrared with specific aim to assess the effect of Li on the emission properties and to elucidate the up-conversion mechanisms. Besides optical excitation, we use low energy X-ray excitation and report the induced emission in an extended range ranging from 400 up to 1700 nm. The present results also reinforce our previous findings with Ln (Ln = Eu, Sm, Tb, Dy and Er) doped Y2O3 that established the role Li as a crystallization enhancer and not as local structure modifier25. A comparison between the optical and X-ray induced emission of Er-Lu2O3 and Er-Y2O3 nanoparticles is also discussed.
Results and Discussion
Summary of structural properties of Er-Lu2O3 nanoparticles
Several synthesis methods have been carefully considered to obtain the doped nanoparticles: hydrothermal synthesis, microemulsion reaction and the citrate complexation method. We disregarded the hydrothermal route due its low reproducibility of the final products and their dependence on the experimental conditions41. Further, the oil in water microemulsion reaction was disregarded due to the product separation process, the numerous washing steps as well as the highly dependence on the oil phase used42. We have selected the sol-gel citrate method25 due to the simplicity of the reaction steps and its excellent reproducibility, which is crucial for a study that compares nanoparticles of different (isostructural) hosts doped with Er and Li in varying concentrations as we propose here. At this stage, the advantage of excellent reproducibility was preferred against the disadvantage of agglomeration effect. Through the text, Er (1 and 7%) and Er(1 and 7%) Li (5%) (co)-doped Lu2O3 calcined at 800 °C are denoted as 1Er-Lu2O3 −800 °C, 7Er-Lu2O3 −800 °C, 1Er, 5Li-Lu2O3 −800 °C and 7Er, 5Li-Lu2O3 −800 °C, respectively. The calcination temperature of 800 °C for Li co-doped samples was imposed by elimination of unwanted lithium nitrate and Li volatility according to literature25,43,44. To compare the effect of improved crystallization triggered by thermal annealing to that induced by Li addition, Li free samples were also calcined at 1000 °C and denoted as reference samples.
Figure 1a illustrates the X-ray diffraction (XRD) patterns of Er(Li)-Lu2O3 nanoparticles that agree with those of cubic Lu2O3 (JCPDS card 86–2475). No additional peaks of other phases have been found, indicating the formation of a homogenous solid solution. The average crystallite sizes of samples calcined at 800 °C were estimated by use of Scherrer equation at around 17–19 nm for 1Er-Lu2O3 and 7Er-Lu2O3 and 41 nm for 7Er, 5Li-Lu2O3. Further calcination at 1000 °C increased the crystallite sizes of 1Er-Lu2O3 and 7Er-Lu2O3 from 17–19 to 40–45 nm.

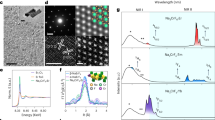
XRD patterns (a), FTIR (b), XRF (Inset: EDX spectra) (c) and Raman spectra excited at 488 nm (d) of Er-Lu2O3 nanoparticles. Highlighted with yellow in (d) is Lu2O3 phonon band.
Figure 1b depicts the Fourier transform infrared (FTIR) spectra of 7Er-Lu2O3 calcined at 800 °C and 7Er, 5Li-Lu2O3 −800 °C. The spectra are dominated by the absorption peaks at around 410, 490 and 580 cm−1 due to Lu–O bond vibration in Lu2O345. It is worth noting that only feeble bands were detected at ~ 1630 cm−1 (deformation vibration) or in the 3000–3600 cm−1 (stretching vibration) region corresponding to O-H bond, indicating the absence of coordinated or adsorbed water, respectively. X-ray fluorescence (XRF) spectra in Fig. 1c and Energy Dispersive X-ray (EDX) (inset of Fig. 1c) spectra evidences the bulk elemental composition given by Lu and Er metals in agreement with the nominal composition without interference from impurity metals. The Raman spectra show the presence of the Fg + Ag vibration mode of the Lu2O3 (393 cm−1)1. The phonon bands are buried into much more intense f-f luminescence of Er corresponding to 4F7/2-4I15/2 emission transition induced by laser excitation at 488 nm.
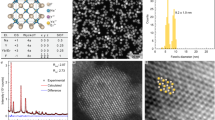
By analyzing the TEM data presented in Fig. 2, we can state that the powder consists from polyhedral nanoparticles that are forming mild to hard agglomerates. The average particle size is ranging from ~22 nm for 7Er-Lu2O3 −800 °C, in agreement with X-ray diffraction data, while the Li addition induces the increase of the particle size to ~ 65 nm, higher than the value obtained from X-ray data calculated using Scherer equation as particle seem to be composed of two-three crystallites. For comparison, in Fig. 2 are presented the TEM images for its isostructural counterpart, Y2O3 powder. In this case, the average particle sizes are slightly higher than those of Lu2O3, ranging from ~31 nm for 7Er-Y2O3 −800 °C to ~80 nm for 7Er, 5Li-Y2O3 −800 °C nanoparticles.

TEM Micrographs obtained on (a) 7Er-Lu2O3 −800 °C, (b) 7Er, 5Li-Lu2O3 −800 °C, (c) 7Er-Y2O3 −800 °C and (d) 7Er, 5Li-Y2O3 −800 °C nanoparticles.
Carlos et al.46 extensively studied the effect of co-doping Er, Yb-Y2O3 with Li on the nanoparticle structure by use of X-ray diffraction and Rietveld refinement as well as TEM analysis, suggesting that Li additions shrinks the lattice constant and increases the nanoparticle size without substituting the Y cation, probably residing interstitially. We observe the same trend here, that is, the lattice constant shrinks from 10.48 (7Er-Lu2O3 −800 °C) to 10.44 Å (7Er, 5Li-Lu2O3 −800 °C) while the nanoparticles size increases from 22 nm to 65 nm (Fig. 2).
Up-conversion of Er-Lu2O3 nanoparticles upon excitation around 1500 nm
Composition optimization
The up-conversion brightness of Er based materials is closely related to the balance between the absorption of the incident light and the quantum efficiency47. The up-conversion quantum efficiency is direct related with Er-Er distance via Er concentration and host selection/manipulation48,49. The probability of energy transfer in Er-Er pairs considering the most common dipole-dipole mechanism can be written as \({\rho }_{Er-Er}=\frac{1}{{\tau }_{Er}}{(\frac{{R}_{0}}{R})}^{6}\)where τEr is the actual lifetime of the Er excited state, including multiphonon radiative decay, and R0 is the critical transfer distance for which excitation transfer and spontaneous deactivation of the Er dopant have equal probability47. We assume that Er doping does not affect the lattice parameter of Lu2O3 and use thus as lattice parameter, a, the value of 10.48 Å for 7Er and 10.43 Å for 1Er (calculated from XRD data, Fig. 1a). Lu2O3 crystallizes in a space group in the C-type metal oxide structure, \(Ia\bar{3}\) space group and its unit cell correspond to the structured cell formula Lu32O48. This means that there is Z = 32 Lu ions in each unit cell. The distribution of Er dopant is considered to be randomly on the low symmetry, C2 and high-symmetry, S6/C3i sites with a relative contributions of 3:14. The average Er-Er distance R, can be approximated: \({R}_{L{u}_{2}{O}_{3}}={(\frac{{a}^{3}\sqrt{3}/2}{32x})}^{\frac{1}{3}}\), where x is the Er doping percentage. According to this equation, the average Er-Er in distance in 1Er and 7Er-Lu2O3 samples can be estimated around 16.39 and 8.57 Å, respectively. The optimum average Er-Er distance obtained for the most emissive sample in Y2O3, 7%Er25 was estimated at 8.67 Å (using 10.602 Å as lattice parameter4). The Er-Er distance obtained in 7Er-Lu2O3 nanoparticles is only 2% smaller than obtained for its Y2O3 homologue. Therefore, we assume a similar optimum concentration of Er of 7% in Lu2O3 further sustained by preliminary measurements on varying Er concentrations. The values of Er-Er distances for the optimum upconversion intensity come close to those reported for β-NaYF4 and Gd2O2S hosts with optimum average Er−Er distances of 8.8 and 8.3 Å, respectively50.
Another well-known strategy to enhance the up-conversion emission intensity is by host lattice manipulation, that is, modification of the local structure around lanthanide dopants by co-doping with optically inert ions, such as monovalent Li43,44,49,51,52,53,54, monovalent K55, divalent Zn56, Cu57 or Mg58,59. Li is the smallest metallic ion in the periodic table with an ionic radius of 0.9 Å. As such, Li is considered to be easily incorporated into various host lattices, including oxides49. Carlos et al.46 reports an 10 times increase of the upconversion quantum yield when adding Li on Y2O3:Yb, Er nanoparticles mainly due to the particle size increment and lattice parameter decrease. Also, they indicated the number of OH− groups does not change significantly with increasing Li content while the role of carbonate groups as upconversion emission quencher was ruled out. This report confirm our previous findings, that is, Li enhances the upconversion emission by improving the crystallization rather than distorting the local structure around the activator25.
Overview of the up-conversion emission spectra
Figure 3a illustrates the main emission/excitation transitions of Er-Lu2O3 under ~1500 nm excitation. The dependence of the up-conversion emission intensity under 1533 nm excitation with Er concentration (1–10%) and Li addition (0, 5 and 10%) is presented in Fig. 3b,c. The optimum composition yielding the highest up-conversion intensity is 7% and 5% for Er and Li concentration, respectively. Figure 3d gathers the up-conversion emission spectra of optimized 7Er-Lu2O3 series: 7Er-Lu2O3 −800 °C; 7Er, 5Li-Lu2O3 −800 °C and 7Er-Lu2O3 −1000 °C, the reference sample. Upon ~1500 nm excitation, Er display typical emission bands assigned to 2H11/2, 4S3/2–4I15/2 (green emission); 4F9/2–4I15/2 (red emission); 4I9/2–4I15/2 and 2H11/2, 4S3/2–4I13/2 (790–850 nm) and 4I11/2-4I15/2 (980 nm) emission transitions. The 980 nm based emission represents 89–93% (compared to almost 99% for 1Er-Lu2O3; data not presented) of the total up-conversion emission as observed previously for its Y2O3 counterpart25.

(a) Schematic representation of the relevant absorption and emission transitions and the possible mechanisms responsible for the up-conversion emission of Er-Lu2O3 following excitation at 1533 nm. Curved arrows indicate multiphonon relaxation; coloured lines correspond to the emission transitions and dashed lines indicate the non-radiative energy transfer/cross relaxation processes. With “*” are labelled the energy levels selected in measurement of the emission decays. Dependence of up-conversion emission intensity of Er-Lu2O3 upon 1533 nm excitation with Er (b) and Li (c) concentration (d) Up-conversion emission spectra of 7Er-Lu2O3 −800 °C, 7Er, 5Li-Lu2O3 −800 °C and, for comparison, 7Er-Lu2O3 −1000 °C (reference sample). All samples were placed in a standard sample holder (14 mm × 7 mm). The digital images were obtain in the dark room light conditions by use of Canon EOS 60D under exposure time of 1 s with 400 ISO. The energy of the excitation is around 1.85 mJ (average power density of ~65 mW/cm2).
As shown in Fig. 3d codoping with Li or increasing the anneal temperature to 1000 °C induce a similar increase by a factor of 10–13 of the up-conversion emission intensity. Since both samples present a similar crystallite sizes around 40–45 nm, such increase of up-conversion emission intensity by Li addition is explained by improved crystallization. Moreover, more than 40% of the 980 emission intensity falls above 1000 nm, outside the water absorption biological media17. To the best of our knowledge, less than 10 studies report so far in the literature on (almost) pure/monochromatic up-conversion emission around 980 nm under up-conversion excitation in Er(Yb) based nanosized systems.
Finally, taking advantage of our OPO tunable excitation laser, we compare the up-conversion emission shapes defined by red to green emission ratio (RGR)60, measured under 1533 and 980 nm excitation. As shown in Fig. S2, the two types of emission spectra display rather close colors, reddish yellow (excitation at 1533 nm) and yellowish orange (excitation at 980 nm) likely due to similar up-conversion energy transfer mechanisms responsible for populating the green and red emission levels (2H11/2, 4S3/2 and 4F9/2, respectively) for 7Er-Lu2O3 nanoparticles.
Up-conversion mechanisms
As proposed by Auzel47, the two main up-conversion mechanisms are ground state absorptions followed by excited state absorptions (GSA/ESA) and energy transfer up-conversion mechanism (ETU) (see also Fig. 3a). To characterize the up-conversion mechanisms under 1533 nm excitation, we have measured (i) dependence of up-conversion emission intensity on the laser pulse energy, (ii) up-conversion emission decays and comparison with down-conversion (indirect and direct excitations) and (iii) up-conversion excitation spectra.

The dependence of up-conversion emission intensity monitored at 565, 675 and 980 nm on the laser pulse energy upon 1533 nm excitation in log-log scale illustrated for 7Er-Lu2O3 −800 °C (a); 7Er, 5Li-Lu2O3 −800 °C (b) and, for comparison, 7Er-Lu2O3 −1000 °C (reference sample) (c).
Dependence of up-conversion emission intensity on the laser pulse energy: As shown in Fig. 4, slope values of 1.8–2 (2H11/2, 4S3/2 emission monitored at 565 nm) or 1.54–1.77 (4F9/2 emission monitored at 675 nm) and 1.1–1.3 (4I11/2 emission monitored at 980 nm) were estimated for 7Er-Lu2O3–800 °C; 7Er, 5Li-Lu2O3 −800 °C and reference sample (7Er-Lu2O3 −1000 °C) suggesting at first glance a one and two photon process for these transitions61. All values are below the theoretical values of 3 and 2, respectively, due to the high-power density excitation29,62 and the influence of up-conversion over linear decay for the depletion of corresponding intermediate excited states63. For high power density, a slope of one is expected for a two-photon ETU process62. This reduction of the slope confirms that our experimental conditions correspond to a high-power regime (~106 W/cm2) during the excitation pulses (5 ns). Therefore, due to the saturation of the dependence of the up-conversion emission intensity on the laser pulse energy in high-power regime, we cannot depict the true number of photons needed to populate the emitting levels. A similar behavior was observed by Martín-Rodríguez et al.29 for Er doped Gd2O2S under similar excitation conditions using an OPO laser system with 2 mJ pulse energy for 10 ns pulse width and 20 Hz repetition rate. Using a cw high power excitation source, Pollnau et al.61 and Suvyer et al.62 observed saturation of the dependence of the up-conversion emission intensity on the laser pump power. Finally, the dependencies in Fig. 4a,b show that addition of Li does not change the mechanisms of UPC emission established in Li free sample.
Up-conversion emission decays: It is well-known that, using a pulsed excitation the GSA/ESA and ETU mechanisms can be readily differentiated by comparing the emission decays measured upon down- and up-conversion excitations47. Up-conversion by energy transfer mechanism typically leads to a decay curve for the up-conversion emission that is longer than that measured in down-conversion or direct excitation mode since the emission level is fed continuously after the excitation pulse by a long-lived intermediate state population (e.g. 4I13/2) that is also decaying47,64. Figure 5 presents the comparison of emission decays corresponding 4I11/2 (NIR emission) and 4F9/2 (red emission) level for 7Er-Lu2O3 −800 °C; 7Er, 5Li-Lu2O3 −800 °C and, for comparison, 7Er-Lu2O3 −1000 °C under direct/down- and up-conversion excitations at 960/492 nm and 1533 nm, respectively.

Comparison between the direct/down- and up-conversion emission decays of 7Er-Lu2O3 −800 °C (a,d); 7Er, 5Li-Lu2O3 −800 °C (b,e) and, for comparison, 7Er-Lu2O3 −1000 °C (reference sample) (c,f). (a–c) The emission was monitored around 980 nm under direct (960 nm) and up-conversion excitation (1533 nm). (d–f) The emission was monitored around 675 nm under down- (492 nm) and up-conversion excitation (1533 nm).
The lengthening of the NIR emission decays under up-conversion compared to down-direct excitation (see also Figure 5 and Table 1) confirms the presence of energy transfer mechanisms that fuels the 4I11/2 level under up-conversion excitation at 1533 nm (see also Scheme from Fig. 3a) through long lived 4I13/2 energy level. Due to the power density of the excitation laser and looping energy transfer up-conversion mechanisms that affects the 4I11/2 emission65 (see also Scheme for Fig. 3a), the up-conversion emission decay is not greatly lengthened compared to the direct excitation emission decay (Fig. 5a–c and Table 1) as is the case of 4F9/2 up-conversion emission decays (Fig. 5d–f) while their respective rise times are of few microseconds. A similar effect was observed by Martín-Rodríguez et al. for Er doped Gd2O2S (microparticles)29 and by Chen et al. for Er doped LiYF433. Also, for higher pump laser energy density regime (peak power >105 W/cm2) the emission decay rise time shortens29 due to the saturation of the initial ground state absorption, leading to a maximum of the subsequent (4I13/2, 4I13/2) → (4I15/2, 4I9/2) energy transfer up-conversion rate.
It is further observed that upon Li addition we observe similar effects on up-conversion emission intensity and decay as increasing the calcination temperature from 800 to 1000 °C. As such, the up-conversion emission intensity is enhanced by a factor of 10–13 (Fig. 3c) and the average decay times corresponding to red (675 nm) and NIR emission (980 nm) transitions are enhanced by a factor of 2–3 (Table 1).
Up-conversion excitation spectra: The comparison of the up-conversion excitation spectra of 7Er, 5Li-Lu2O3 −800 °C that monitor the emissions at 565, 675 and 980 nm is presented in Fig. 6. With increasing the monitored emission wavelength, the lines get narrower due to the greater number of photons required to populate the green and red emitting levels (theoretically three photons), whilst only two photons (theoretically value) are necessary for the ~980 nm emission corresponding to the 4I11/2-4I15/2 emission transition22. Altogether, the shapes of the up-conversion excitation spectra and their resemblance to the diffuse reflectance absorption spectrum (see the diffuse reflectance spectrum illustrated with black line in Fig. 6) from ground state (4I15/2) to initial excited state (4I13/2) confirm the predominance of ETU mechanism47. Because otherwise, new lines would emerge corresponding to the convolution of all involved intermediary states due to the GSA/ESA up-conversion mechanism processes47. As a Kramers lanthanide, the maximum theoretical number of lines corresponding to 4I15/2-4I13/2 transition of Er assuming that only the fundamental level is populated at room -temperature is 16. The number observed is definitely larger (including well resolved lines but also the so -called spectral shoulders) which means that (at least) the next level to the fundamental one is also populated.

Comparison of the up-conversion excitation spectra of 7Er, 5Li-Lu2O3 −800 °C monitored at 565, 675 and 980 nm with diffuse reflectance spectrum. All excitation spectra were normalized at the maximum absorption intensity (1533 nm). Similar excitation spectra were recorded for 7Er-Lu2O3 −800 and 1000 °C.
Considering the dependence of up-conversion emission intensity on the laser pulse energy, up-conversion emission decays and up-conversion excitation spectra described above, we discuss the mechanisms responsible for the up-conversion emission as following (see also Fig. 3a). Upon excitation into the long – lived 4I13/2 level (~0.6 ms as measured for 7Er, 5Li-Lu2O3 −800 °C that is significantly shortened compared to 1Er-Lu2O3 −800 °C around 1.5 ms), Er can be excited to the 4I9/2 energy level which decays mostly non-radiatively on the 4I11/2 energy level that can lead to a strong 980 nm based up-conversion emission. From the excited 4I11/2 energy level, the 4F9/2 state is populated via the (4I11/24, I9/2) → (4F9/24, I13/2) energy transfer mechanism giving rise up-conversion emission in red spectral range via a three-photon process. Also, there are secondary cross relaxations energy transfer mechanisms that favors the 4I13/2 emission such as 4F9/2-H11/24, S3/2 ↔ 4I11/2-4I13/2 and 4I11/2-4F9/2 ↔ 4I9/2-4I13/2 as observed in Fig. S3 when increasing the Er concentration from 1% to 7%. The lengthening of the up-conversion emission decays compared to down-conversion emission decays (Fig. 5 and Table 1) is a clear indicator of the efficient contribution of up-conversion mechanisms to the overall emission47 besides GSA/ESA mechanisms as confirmed further by the analysis of up-conversion excitation spectra (Fig. 6).
Compared to Y2O3, Lu2O3 has a smaller lattice parameter (10.393 Å compared to 10.604 Å), that will lead to slightly different crystal field and thus to a spectral shift of their emission spectra8. Although the Er-Y2O3 and Er-Lu2O3 nanoparticles were synthesized by using the same procedure, the resulted nanoparticles are of distinct size (Fig. 2), Lu2O3 being smaller (~22 nm) than Y2O3 (~31 nm) in size for the same Er concentration (7%) and thermal treatment (800 °C). This indicates that the mechanism of nucleation and growth of nanoparticles in solution is different for the two oxides having consequently the formation of crystallite with different sizes. This phenomenon can be described through the La Mer burst nucleation66,67 that depends strongly on the reaction conditions: even a slight change in condition, such as pH or solution concentration, can lead to a completely different mechanism. For example, in the Ostwald ripening process for growing, due to the high solubility and the surface energy of smaller particles within solution, these redissolve and in turn allow the larger particles to grow even more; higher is the surface energy, bigger crystallite will be formed. This could be one explanation for the different crystallite size, due to the fact that, indeed, the two hydroxides present different free Gibbs energies68. Analyzing the effect of Li addition, the effect is similar for both powders. Thus, for the Lu2O3 nanoparticles, the Li addition induces a particle growth by 2.9 times while in the case of Y2O3 induces a growth of 2.6 times. Another effect, in what concerns the nanoparticle size distribution in the case of Li addition is that it induces a tendency of bimodal distribution of nanoparticles, as we can see in the insets of each sample in Fig. 2. The increase of crystallinity leads to a different emission lifetime, the greater the nanoparticle, the lower the surface to volume ratio leading to longer emission decay69. Differences exist also in emission decays monitoring the 980 and 1533 nm under direct excitation at 960 and 1470 nm, respectively (Fig. S4). In this case, although the average lifetimes are roughly similar, the decay shapes are obviously different, suggesting that a different balance of radiative and non-radiative processes govern the Er emission in the two isostructural hosts. Finally, the comparison of the up-conversion emission intensity of 7Er(5Li)-Lu2O3 −800 °C and 7Er(5Li)-Y2O3 −800 °C measured under the same experimental conditions under 1533 nm excitation, reveal that Y2O3 renders brighter up-conversion emission by a factor of 1.3 for 7Er, 5Li and a factor of 4.7 for 7Er samples (Fig. S5). Similar trends were measured using 980 nm as up-conversion excitation wavelength (using both pulsed OPO and cw laser diode). This observation contradicts the two order of magnitude enhancement observed for 1%Er-Lu2O3 relative to 1%Er-Y2O3 under 980 nm cw excitation3
X-ray induced emission on Er-Lu2O3 nanoparticles
We further present the emission properties of Er-Lu2O3 nanoparticles under X-ray excitation measured from visible to NIR by use of X-ray source with energy and flux in the medical diagnostic range (Mo target, up to 2 Gy/s). According to literature, the X-ray induced emission intensity of Eu/Tb doped Lu2O3 and commercially available Eu/Tb: Gd2O2S70,71,72 and CsI:Tl70 and CaWO473 are comparable, or even brighter for some X-ray energies (such as: 50–100 kVp for Lu2O3:Eu compared to Gd2O2S:Eu71). Eu doped Lu2O3 was reported to be a suitable candidate for X-ray tomography screens as thin film73,74, ceramics70 and nanopowder71,72. However, to the best of our knowledge, no reports on X-ray induced emission for Er-doped Lu2O3 were found.
Figure 7 shows the X-ray induced luminescence spectra of the most emissive samples, 1Er, 5Li-Lu2O3 −800 °C and, for comparison, 1Er-Lu2O3 −1000 °C in the extended range from UV to NIR (400–1700 nm). Under X-ray radiation, Er luminescence is achieved via three main processes: (i) the absorption of high energy photons, (ii) generation of electron -hole pairs and (iii) the recombination of some of these pairs leading to energy transfer to Er75. Thus, under X-ray radiation, Er exhibits emission from UV to NIR region as follow: 410 nm (2H9/2–4I15/2); 480 nm (4F7/2–4I15/2); 565 nm (2H11/2, 4S3/2–4I15/2); 660 nm (4F9/2–4I15/2); 870 nm (4S3/2–4I13/2), 980 nm (4I11/2-4I15/2) and 1500 nm (4I13/2-4I15/2). As previously observed for Y2O3 nanoparticles25, the strongest emission under X-ray radiation is obtained with Er concentration of 1% preserving the characteristic emission spectrum.

(left) X-ray induced luminescence emission spectra of 1Er, 5Li-Lu2O3 −800 °C and, for comparison, 1Er-Lu2O3 −1000 °C (reference sample) in the extended range (400–1700 nm). (right) Schematic representation of the comparison of Y and Lu K edge in respect to the Mo X-ray source energy output.
According to Fig. 7, the X-ray induced luminescence of 1Er-Lu2O3 −1000 °C is dominated by the NIR emission at 1500 nm (4I13/2-4I15/2) representing ~80% of the total emission intensity.
To assess the changes in the emission shape and intensity of 7Er-Lu2O3 nanoparticles under X-ray excitation induced by Li co-doping, we further measured the X-ray induced luminescence of 7Er-Lu2O3 −800 °C; 7Er, 5Li-Lu2O3 −800 °C and for comparison, 7Er-Lu2O3 −1000 °C from 400 to 1100 nm range (Fig. 8).

Comparison of X-ray induced luminescence emission spectra for 7Er-Lu2O3 −800 °C; 7Er, 5Li-Lu2O3 −800 °C and, for comparison, 7Er-Lu2O3 −1000 °C (reference sample) measured in the 400 to 1100 nm range. The spectra are normalized at 565 nm for comparison purposes. The inset compares the integrated emission intensity of 7Er-Lu2O3 −800 °C; 7Er, 5Li-Lu2O3 −800 °C and 7Er-Lu2O3 −1000 °C (reference sample). The emission in the near infrared range (1100–1700 nm) has a very low signal to noise ratio (is too weak to be clearly observed) and is not represented.
The red to green ratio changes from 1.2 for 7Er(5Li)-Lu2O3 −800 °C to 1.65 for 7Er-Lu2O3 −1000 °C and the near infrared to green ratio, from 2.7 for 7Er-Lu2O3 −800 °C to 3.43 for 7Er, 5Li-Lu2O3 −800 °C and to 8 for 7Er-Lu2O3 −1000 °C. The 980 nm emission represents 47% of the total emission for 7Er-Lu2O3 −800 °C and increases up to 80% for 7Er-Lu2O3 −1000 °C. The integrated luminescence emission increases by a factor of 5 and 10 with Li addition and extending the anneal temperature up to 1000 °C, respectively (Fig. 8). As shown in Fig. 8, the X-ray induced luminescence of Er is highly similar to optical induced emission (Fig. S6).
As concerns the comparison of X-ray induced emission properties of Er-Lu2O3 and Er-Y2O3, we note that Y K-edge is 17.0 keV, within the diagnostic range (<45 keV) of the Mo source, while the Lu K-edge at 63.3 keV falls outside the energy range of the Mo target (Scheme in Fig. 7). Still, the X-ray induced emissions are comparable in intensity likely due to the higher atomic number of Lu (Z = 71) compared with Y (Z = 39). The upconversion emission properties are expected to be similar across cubic sesquioxides due to the same physical-chemical properties: maximum phonon band (Fg + Ag maximum phonon band at around 400 cm−1 and cut-off phonon at <600 cm−11), same random distribution in C2 and C3i, same local environment/crystal-field. In contrast, the X-ray luminescence intensity is dependent on the cations K-edge (Sc −4.486 keV; La −38.894 keV; Pr −41.958 keV; Nd −43.538 keV and from Sm −46.801 keV to Lu −63.311 keV) and X-ray energy. X-ray activated near-infrared luminescence benefits from deep penetration in both excitation (Mo X-ray source, less than 5 cm penetration depth76) and emission (around 1 cm penetration depth for 1500–1550 nm)17. Keeping in mind the obvious advantages offered for deep penetration bio-imaging applications, targeted studies using X-ray induced NIR emission in biocompatible nanoparticles are emerging (e.g.77,78).
Conclusions
In summary, we have investigated the properties of Er-Lu2O3 nanoparticles synthesized by citrate complexation method under near-infrared up-conversion (1500 nm) and X-ray excitations in the 400 to 1700 nm range. The composition was varied in terms of Er concentration (1 to 7%) and Li addition (5 and 10%), being optimum for 7%Er and 5% Li. Upconversion emission and indirect/direct decay measurements were performed considering multiple absorption transitions in the visible and near-infrared with specific aim to assess the effect of Li on the emission properties and to elucidate the up-conversion mechanisms. Under 1500 nm excitation, the nanoparticles present an almost near infrared monochromatic up-conversion emission centered at 980 nm while over 80% of the X-ray induced emission is concentrated around 1500 nm. Li enhancement of up conversion emission (up to one order of magnitude) is related to improved crystallization which confirm our previous findings and recently published literature. Although the comparison between the emission properties of Er-Lu2O3 and Er-Y2O3 nanoparticles of similar composition, synthesized by a similar method and measured under similar experimental conditions is not straightforward, we evidence that Lu2O3 is not exceeding the efficiency of Y2O3 as upconversion host as commonly sought. We believe that use of Er sensitized (under 1500 nm excitation) emission of near infrared emissions of upconversion activators (Er at 980 nm, Ho at 1200 nm and Nd at 1060 nm) in optimized cubic sesquioxides will lead to the development of novel near-infrared nanoprobes as recently anticipated in the literature.
Materials and Methods
Synthesis
Er(1–10%) and Er(1 and 7%) Li (5 and 10%) (co)-doped Lu2O3 were prepared using the citrate complexation method described elsewhere79. The Er, Li-Lu2O3 samples were calcined in air at 800 °C for 4 hours at a heating rate of 5 °C/min to ensure the complete elimination of the unwanted nitrate species which vaporizes at 600 °C (CSID:8305408). Also, this temperature was chosen for Li containing samples, because of the Li component which is volatile at elevated temperature. Therefore, only the Li free, Er-Lu2O3 were also calcined at 1000 °C for 4 hours to follow the influence of the crystallite size and compare it with Li addition.
Characterization
Powder X-ray diffraction (XRD) patterns were recorded on a Bruker-AXS D8 Advance diffractometer equipped with a one-dimensional detector (LynxEye type) using Cu-Kα radiation (0.154178 nm) at a scanning speed of 0.10 degrees min−1 in the 15–90 degrees 2θ range. Lattice parameters are calculated using the Bragg’s law. The following equation can be used to obtain the interplanar distance of a system: \(n\lambda =2dsin\theta \), where n is considered to be 1, λ is 1.54178 Å. By using the interplanar distance, the lattice constant (a) for a cubic system is calculated as follows: \(a=\sqrt{{d}^{2}({h}^{2}+{k}^{2}+{l}^{2})}\).
Raman spectra were measured by use of a Horiba Jobin Yvon-Labram Raman Microscope Spectrometer selecting the 488 nm excitation wavelength. Microbeam X-ray fluorescence (micro-XRF) spectra were measure by use of a custom-made instrument80. Attenuated total reflection-Fourier transform infrared (ATR-FTIR) analysis were carried out on a Spectrum Two, PerkinElmer spectrometer with a 4 cm−1 nominal resolution. The Transmission Electron micrographs were obtained using a high-resolution Titan THEMIS transmission electron Microscope operated at 200 kV.
Luminescence measurements
The emission spectra were recorded using a wavelength tunable NT340 Series EKSPLA OPO (Optical Parametric Oscillator) operated at 10 Hz, with a pulse duration of 4 ns and laser spectral width of 5 cm−1. An intensified CCD (iCCD) camera (Andor Technology, iStar iCCD DH720) coupled to a spectrograph (Shamrock 303i, Andor) was used as detection system. An additional InGaAs photodiode array (Andor Technology, iDus DU490A) for monitoring the emission spectra in the 800–1700 nm range was used. The energy of the laser pulse was modified using neutral density filters and measured with a Coherent Energy Max Laser Energy Sensor (J-10MB-HE Energy Max Sensor). The energy of the laser pulse at 1533 nm varied from 0.5 up to 3.2 mJ. The X-ray induced luminescence spectra were measured by use of AvaSpec-HS1024x58/122TEC and AvaSpec-NIR256-1.7TEC fiber optic spectrometers. X-ray excitation source consists of an X-ray tube (Oxford Instruments, Apogee 5011, Mo target, max. high voltage −50 kV, max current −1 mA (<2 Gy/s). The digital photos were obtain by use of a Canon EOS 60D was used. For the near-infrared emission decay measurements, we used a NIR PMT module (H10330B-75, Hamamatsu) as detector coupled to a spectrograph/monochromator (Acton SP2758, Princeton Instruments) and a PCIe TCSPC card TimeHarp 260 NANO (PicoQuant) for acquisition. All lifetime estimations were calculated by integrating the area of the normalized at maximum emission decays.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author (CT) on reasonable request.
References
Yu, J. et al. Raman spectra of RE2O3 (RE=Eu, Gd, Dy, Ho, Er, Tm, Yb, Lu, Sc and Y): laser-excited luminescence and trace impurity analysis. Journal of Rare Earths 32, 1–4, https://doi.org/10.1016/S1002-0721(14)60025-9 (2014).
Li, Y. et al. Color control and white light generation of upconversion luminescence by operating dopant concentrations and pump densities in Yb3+, Er3+ and Tm3+ tri-doped Lu2O3 nanocrystals. Journal of Materials Chemistry 21, 2895–2900, https://doi.org/10.1039/c0jm03394d (2011).
Capobianco, J. A., Vetrone, F., Boyer, J. C., Speghini, A. & Bettinelli, M. Visible upconversion of Er3+ doped nanocrystalline and bulk Lu2O3. Optical Materials 19, 259–268 (2002).
Wyckoff, R. W. G. Crystal Structures-Volume 2. (Interscience Publishers, 1964).
Guillot-Noel, O., Bellamy, B., Viana, B. & Gourier, D. Correlation between rare-earth oscillator strengths and rare-earth-valence-band interactions in neodymium-doped YMO4 (M = V, P, As), Y3Al5O12, and LiYF4 matrices. Physical Review B 60, 1668–1677 (1999).
Vetrone, F., Boyer, J., Capobianco, J., Speghini, A. & Bettinelli, M. NIR to visible upconversion in nanocrystalline and bulk Lu2O3: Er3+. Journal of Physical Chemistry B 106, 5622–5628 (2002).
Wang, N., Zhang, X. & Wang, P. Tartaric acid-assisted co-precipitation synthesis of Er3+ -doped Lu2O3 nanopowders. Chinese Chemical Letters 23, 1299–1302 (2012).
Antić, Ž., Krsmanović, R. M., Nikolić, M. G., Đorđević, V. & Dramićanin, M. D. Processing and characterization of up-converting Er3+ doped (Lu0.5Y0.5)2O3 nanophosphor. International Journal of Materials Research 104, 216–221 (2013).
Jing, F. et al. Growth and Properties of a Thulium, Holmium‐Codoped LuLiF4 Single Crystal. Journal of the American Ceramic Society 91, 296–298 (2017).
Shi, F., Wang, J., Zhai, X., Zhao, D. & Qin, W. Facile synthesis of β-NaLuF4: Yb/Tm hexagonal nanoplates with intense ultraviolet upconversion luminescence. CrystEngComm 13, 3782–3787 (2011).
Liu, Q. et al. Sub-10 nm Hexagonal Lanthanide-Doped NaLuF4 Upconversion Nanocrystals for Sensitive Bioimaging in Vivo. Journal of the American Chemical Society 133, 17122–17125 (2011).
Hong, G. et al. Multifunctional in vivo vascular imaging using near-infrared II fluorescence. Nature Medicine 18, 1841 (2012).
Hong, G. et al. Through-skull fluorescence imaging of the brain in a new near-infrared window. Nature Photonics 8, 723–730 (2014).
Diao, S. et al. Fluorescence Imaging In Vivo at Wavelengths beyond 1500 nm. Angewandte Chemie-International Edition 54, 14758–14762 (2015).
Bruns, O. et al. Next-generation in vivo optical imaging with short-wave infrared quantum dots. Nature Biomedical Engineering 1 (2017).
Sheng, Y., Liao, L., Thakor, N. & Tan, M. Rare-Earth Doped Particles as Dual-Modality Contrast Agent for Minimally-Invasive Luminescence and Dual-Wavelength Photoacoustic Imaging. Scientific Reports 4 (2014).
Hemmer, E., Benayas, A., Légaré, F. & Vetrone, F. Exploiting the biological windows: current perspectives on fluorescent bioprobes emitting above 1000 nm. Nanoscale Horizons (2016).
del Rosal, B. et al. Overcoming Autofluorescence: Long‐Lifetime Infrared Nanoparticles for Time‐Gated In Vivo Imaging. Advanced Materials 28, 10188–10193 (2016).
Liu, T., Conde, J., Lipinski, T., Bednarkiewicz, A. & Huang, C. Revisiting the classification of NIR-absorbing/emitting nanomaterials for in vivo bioapplications. Npg Asia Materials 8 (2016).
Bashkatov, A., Genina, E., Kochubey, V. & Tuchin, V. Optical properties of human skin, subcutaneous and mucous tissues in the wavelength range from 400 to 2000 nm. Journal of Physics D-Applied Physics 38, 2543–2555 (2005).
Diao, S. et al. Biological imaging without autofluorescence in the second near-infrared region. Nano Research 8, 3027–3034 (2015).
Goldschmidt, J. C. & Fischer, S. Upconversion for Photovoltaics–a Review of Materials, Devices and Concepts for Performance Enhancement. Advanced Optical Materials 3, 510–535 (2015).
Florea, M. et al. Defect induced tunable near infrared emission of Er–CeO2 by heterovalent co-dopants. Physical Chemistry Chemical Physics 18, 18268–18277 (2016).
Porosnicu, I., Avram, D., Cojocaru, B., Florea, M. & Tiseanu, C. Up-conversion luminescence of Er(Yb)-CeO2: Status and new results. Journal of Alloys and Compounds 711, 627–636 (2017).
Avram, D., Cojocaru, B., Tiseanu, I., Florea, M. & Tiseanu, C. Down-/Up-Conversion Emission Enhancement by Li Addition: Improved Crystallization or Local Structure Distortion? Journal of Physical Chemistry C 121, 14274–14284 (2017).
Fischer, S., Fröhlich, B., Steinkemper, H., Krämer, K. W. & Goldschmidt, J. C. Absolute upconversion quantum yield of β-NaYF4 doped with Er3+ and external quantum efficiency of upconverter solar cell devices under broad-band excitation considering spectral mismatch corrections. Solar Energy Materials and Solar Cells 122, 197–207 (2014).
Martin-Rodriguez, R. et al. Highly Efficient IR to NIR Upconversion in Gd2O2S: Er3+ for Photovoltaic Applications. Chemistry of Materials 25, 1912–1921 (2013).
Avram, D. & Tiseanu, C. Thermometry properties of Er, Yb–Gd2O2S microparticles: dependence on the excitation mode (cw versus pulsed excitation) and excitation wavelength (980 nm versus 1500 nm). Methods and Applications in Fluorescence (2017).
Martín‐Rodríguez, R., Rabouw, F. T., Trevisani, M., Bettinelli, M. & Meijerink, A. Upconversion Dynamics in Er3+‐Doped Gd2O2S: Influence of Excitation Power, Er3+ Concentration, and Defects. Advanced Optical Materials 3, 558–567 (2015).
Vega, M., Fuentes, S., Martín, I. R. & Llanos, J. Up-conversion photoluminescence of BaTiO3 doped with Er3+ under excitation at 1500 nm. Materials Research Bulletin 86, 95–100 (2017).
Fischer, S., Favilla, E., Tonelli, M. & Goldschmidt, J. C. Record efficient upconverter solar cell devices with optimized bifacial silicon solar cells and monocrystalline BaY2F8:30% Er3+ upconverter. Solar Energy Materials and Solar Cells 136, 127–134 (2015).
Favilla, E. et al. Comparative analysis of upconversion efficiencies in fluoride materials for photovoltaic application. Solar Energy Materials and Solar Cells 157, 415–421 (2016).
Chen, G., Ohulchanskyy, T., Kachynski, A. & Agren, H. & Prasad, P. Intense Visible and Near-Infrared Upconversion Photoluminescence in Colloidal LiYF4:Er3+ Nanocrystals under Excitation at 1490 nm. Acs Nano 5, 4981–4986 (2011).
Chen, Q. et al. Confining Excitation Energy in Er3+ -Sensitized Upconversion Nanocrystals through Tm3+ -Mediated Transient Energy Trapping. Angewandte Chemie-International Edition 56, 7605–7609 (2017).
Liu, L. et al. Er3+ Sensitized 1530 nm to 1180 nm Second Near‐Infrared Window Upconversion Nanocrystals for In Vivo Biosensing-Liu-2018-Angewandte Chemie-Wiley Online Library. Angewande Chemie 130, 7640–7644 (2018).
Wang, X. et al. Efficient Erbium‐Sensitized Core/Shell Nanocrystals for Short Wave Infrared Bioimaging. Advanced Optical Materials, 1800690 (2018).
Cheng, X. et al. Er3+ Sensitized Photon Upconversion Nanocrystals. Advanced Functional Materials 28, 1800208 (2018).
Seyed Shariatdoust, M. et al. Harvesting Dual-Wavelength Excitation with Plasmon-Enhanced Emission from Upconverting Nanoparticles. ACS Photonics (2018).
Yan, L. et al. Self-sensitization induced upconversion of Er3+ in core-shell nanoparticles. Nanoscale (2018).
Zheng, X. et al. High-Contrast Visualization of Upconversion Luminescence in Mice Using Time-Gating Approach. Analytical Chemistry 88, 3449–3454 (2016).
Adschiri, T., Kanazawa, K. & Arai, K. Rapid and Continuous Hydrothermal Synthesis of Boehmite Particles in Subcritical And Supercritical Water. Journal of the American Ceramic Society 75, 2615–2618 (1992).
Sanchez-Dominguez, M. et al. Synthesis of CeO2, ZrO2, Ce0.5Zr0.5O2, and TiO2 nanoparticles by a novel oil-in-water microemulsion reaction method and their use as catalyst support for CO oxidation. Catalysis Today 158, 35–43 (2010).
Li, D. et al. Effect of Li+ ions on enhancement of near-infrared upconversion emission in Y2O3:Tm3+/Yb3+ nanocrystals. Journal of Applied Physics 112, 094701 (2012).
Chen, G., Liu, H., Liang, H., Somesfalean, G. & Zhang, Z. Upconversion Emission Enhancement in Yb3+/Er3+-Codoped Y2O3 Nanocrystals by Tridoping with Li+ Ions. The Journal of Physical Chemistry C 112, 12030–12036 (2008).
Xu, M. et al. Preparation and characterization of optical spectroscopy of Lu2O3: Eu nanocrystals. Journal of Solid State Chemistry 178, 477–482 (2005).
Debasu, M. L., Riedl, J. C., Rocha, J. & Carlos, L. D. The role of Li+ in the upconversion emission enhancement of (YYbEr)2O3 nanoparticles. Nanoscale 10, 15799–15808 (2018).
Auzel, F. Upconversion and anti-stokes processes with f and d ions in solids. Chemical Reviews 104, 139–173 (2004).
Nadort, A., Zhao, J. & Goldys, E. Lanthanide upconversion luminescence at the nanoscale: fundamentals and optical properties. Nanoscale 8, 13099–13130 (2016).
Han, S., Deng, R., Xie, X. & Liu, X. Enhancing Luminescence in Lanthanide‐Doped Upconversion Nanoparticles. Angewandte Chemie International Edition 53, 11702–11715 (2014).
Fischer, S., Frohlich, B., Kramer, K. & Goldschmidt, J. Relation between Excitation Power Density and Er3+ Doping Yielding the Highest Absolute Upconversion Quantum Yield. Journal of Physical Chemistry C 118, 30106–30114 (2014).
Singh, A. K., Singh, S. K. & Rai, S. B. Role of Li+ ion in the luminescence enhancement of lanthanide ions: favorable modifications in host matrices. RSC Advances 4, 27039–27061 (2014).
Li, A. et al. A novel anion doping strategy to enhance upconversion luminescence in NaGd(MoO4)(2): Yb3+/Er3+ nanophosphors. Physical Chemistry Chemical Physics 19, 15693–15700 (2017).
Singh, B. P. et al. Enhanced up-conversion and temperature-sensing behaviour of Er3+ and Yb3+ co-doped Y2Ti2O7 by incorporation of Li+ ions. Physical Chemistry Chemical Physics 16, 22665–22676 (2014).
Lin, H. et al. Morphology evolution and pure red upconversion mechanism of beta-NaLuF4crystals. Scientific Reports 6 (2016).
Luitel, H., Chand, R., Torikai, T., Yada, M. & Watari, T. Highly efficient NIR-NIR upconversion in potassium substituted CaMoO4:Tm3+, Yb3+ phosphor for potential biomedical applications. Rsc Advances 5, 17034–17040 (2015).
Mahata, M. et al. Incorporation of Zn2+ ions into BaTiO3:Er3+/Yb3+ nanophosphor: an effective way to enhance upconversion, defect luminescence and temperature sensing. Physical Chemistry Chemical Physics 17, 20741–20753 (2015).
Du, K. et al. Enhanced upconversion luminescence and controllable phase/shape of NaYF4:Yb/Er crystals through Cu2+ ion doping. Crystengcomm 20, 1945–1953 (2018).
Xing, L., Yang, W., Lin, J., Huang, M. & Xue, Y. Enhanced and Stable Upconverted White-light Emission in Ho3+/Yb3+/Tm3+ -doped LiNbO3 Single Crystal via Mg2+ Ion Doping. Scientific Reports 7, https://doi.org/10.1038/s41598-017-15367-9 (2017).
Wang, X. et al. Influence of Doping and Excitation Powers on Optical Thermometry in Yb3+-Er3+ doped CaWO4. Scientific Reports 7 (2017).
Liu, F., Ma, E., Chen, D., Yu, Y. & Wang, Y. Tunable red-green upconversion luminescence in novel transparent glass ceramics containing Er: NaYF4 nanocrystals. Journal of Physical Chemistry B 110, 20843–20846 (2006).
Pollnau, M., Gamelin, D. R., Lüthi, S. R., Güdel, H. U. & Hehlen, M. P. Power dependence of upconversion luminescence in lanthanide and transition-metal-ion systems. Physical Review B 61, 3337 (2000).
Suyver, J., Aebischer, A., Garcia-Revilla, S., Gerner, P. & Gudel, H. Anomalous power dependence of sensitized upconversion luminescence. Physical Review B 71 (2005).
Li, Y. et al. Near-Infrared to Visible Upconversion in Er3+ and Yb3+ Codoped Lu2O3 Nanocrystals: Enhanced Red Color Upconversion and Three-Photon Process in Green Color Upconversion. Journal of Physical Chemistry C 113, 4413–4418 (2009).
Luthi, S., Pollnau, M., Gudel, H. & Hehlen, M. Near-infrared to visible upconversion in Er3+ -doped Cs3Lu2Cl9, Cs3Lu2Br9, and Cs3Y2I9 excited at 1.54 mu m. Physical Review B 60, 162–178 (1999).
Lu, H., Gillin, W. & Hernandez, I. Concentration dependence of the up- and down-conversion emission colours of Er3+ -doped Y2O3: a time-resolved spectroscopy analysis. Physical Chemistry Chemical Physics 16, 20957–20963 (2014).
LaMer, V. K. & Dinegar, R. H. Theory, Production and Mechanism of Formation of Monodispersed Hydrosols. Journal of the American Chemical Society 72, 4847–4854 (1950).
La Mer, V. K. Nucleation inPhase Transitions. Industrial & Engineering Chemistry 44, 1270–1277 (1952).
Brookins, D. G. In Eh-pH Diagrams for Geochemistry Ch. Tellurium, 20–21 (Springer, 1988).
Zheng, K., Song, W., Lv, C., Liu, Z. & Qin, W. Controllable synthesis and size-dependent upconversion luminescence properties of Lu2O3:Yb3+/Er3+ nanospheres. Crystengcomm 16, 4329–4337 (2014).
Lempicki, A. et al. A new lutetia-based ceramic scintillator for X-ray imaging. Nuclear Instruments & Methods in Physics Research Section a-Accelerators Spectrometers Detectors and Associated Equipment 488, 579–590 (2002).
Seferis, I. et al. Light emission efficiency and imaging performance of Lu2O3:Eu nanophosphor under X-ray radiography conditions: Comparison with Gd2O2S:Eu. Journal of Luminescence 151, 229–234 (2014).
Seferis, I. E. et al. In Medical Imaging 2013: Physics of Medical Imaging. 86683W-86681 (2013).
Sengupta, D. et al. Bright Lu2O3:Eu Thin-Film Scintillators for High-Resolution Radioluminescence Microscopy. Advanced Healthcare Materials 4, 2064–2070 (2015).
Marton, Z. et al. High efficiency microcolumnar Lu2O3:Eu scintillator thin film for hard X-ray microtomography-IOPscience. Journal of Physics: Conference Series 425, 062016 (2013).
Dujardin, C. et al. Luminescence and Scintillation Properties at the Nanoscale. Ieee Transactions on Nuclear Science 57, 1348–1354 (2010).
Boke, A. Linear attenuation coefficients of tissues from 1 keV to 150 keV. Radiation Physics and Chemistry 102, 49–59 (2014).
Naczynski, D. J. et al. X-ray-Induced Shortwave Infrared Biomedical Imaging Using Rare-Earth Nanoprobes. Nano Letters 15, 96–102.
Karnkaew, A., Chen, F., Zhan, Y., Majewski, R. & Cai, W. Scintillating Nanoparticles as Energy Mediators for Enhanced Photodynamic Therapy. Acs Nano 10, 3918–3935 (2016).
Avram, D. et al. Heavily impregnated ceria nanoparticles with europium oxide: spectroscopic evidences for homogenous solid solutions and intrinsic structure of Eu3+ -oxygen environments. Journal of Materials Science 49, 2117–2126 (2014).
Lungu, M. et al. Mixed Film Coatings Analyzed By Micro X-Ray Fluorescence Method. Digest Journal of Nanomaterials and Biostructures 9, 899–906 (2014).
Acknowledgements
DA, IT and CT acknowledge CNCS-UEFISCDI, the program NUCLEU, contract 4N/2016 for the financial support. MF acknowledges the grant of Ministry of Research and Innovation, CNCS-UEFISCDI, project number PN-III-P4-ID-PCE-2016-0692. BV acknowledges the EU-funding grant POSCCE-A2O2.2.1-2013-1, Project No. 638/12.03.2014, SMIS-CSNR code 48652. The authors acknowledge CETAL infrastructure for the near-infrared emission decay measurements.
Author information
Authors and Affiliations
Contributions
M.F. and D.A. performed structural and luminescence experiments and processed the data, B.S. performed TEM investigations, I.T. designed the X-ray induced experiments and C.T. designed the studies and wrote the manuscript. All authors contributed to the discussion of the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Avram, D., Tiseanu, I., Vasile, B.S. et al. Near infrared emission properties of Er doped cubic sesquioxides in the second/third biological windows. Sci Rep 8, 18033 (2018). https://doi.org/10.1038/s41598-018-36639-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-36639-y
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
