
Abstract
FliG, which is composed of three distinctive domains, N-terminal (N), middle (M), and C-terminal (C), is an essential rotor component that generates torque and determines rotational direction. To determine the role of FliG in determining flagellar rotational direction, we prepared rotational biased mutants of fliG in Vibrio alginolyticus. The E144D mutant, whose residue is belonging to the EHPQR-motif in FliGM, exhibited an increased number of switching events. This phenotype generated a response similar to the phenol-repellent response in chemotaxis. To clarify the effect of E144D mutation on the rotational switching, we combined the mutation with other che mutations (G214S, G215A and A282T) in FliG. Two of the double mutants suppressed the rotational biased phenotype. To gain structural insight into the mutations, we performed molecular dynamic simulations of the FliGMC domain, based on the crystal structure of Thermotoga maritima FliG and nuclear magnetic resonance analysis. Furthermore, we examined the swimming behavior of the fliG mutants lacking CheY. The results suggested that the conformation of FliG in E144D mutant was similar to that in the wild type. However, that of G214S and G215A caused a steric hindrance in FliG. The conformational change in FliGM triggered by binding CheY may lead to a rapid change of direction and may occur in both directional states.
Similar content being viewed by others


Introduction
Bacteria move in various environments by rotating flagellar filaments in either counterclockwise (CCW) or clockwise (CW) direction. The flagellar motor, which is present at the base of the filament and is embedded in the membrane, can rotate by conversion of the electrochemical potential across the membrane. It is known that Escherichia coli and Salmonella enterica have H+-driven lateral (or peritrichous) flagella, and Vibrio alginolyticus has an Na+-driven polar flagellum as well as H+-driven lateral flagella1. The motor is composed of two parts, the stator and rotor, and torque is generated by interaction between the stator and rotor. The stator functions as an ion channel to convert the ion flux into mechanical forces.
In V. alginolyticus, the Na+-driven stator is composed of two membrane proteins, PomA and PomB, which assemble in a 4 PomA: 2 PomB stoichiometry2. It is known that PomA and PomB are orthologs of MotA and MotB, respectively, in the H+-driven motor of E. coli and S. enterica3,4,5. The stators can be activated by interacting with the C ring composed of cytoplasmic rotor components6,7,8. The C ring consists of three proteins (FliG, FliM, and FliN) and is important for torque generation and determination of the rotational direction9. FliM and/or FliN interact with CheY, one of the chemotaxis signaling factors, and switch the flagellar rotational direction. The phosphorylated CheY (CheY-P) can bind FliM and/or FliN to switch the rotational direction of flagellar motor from the default CCW to CW rotation. CheY-P is automatically dephosphorylated by CheZ, which is the other component of chemotactic signal factor, and CheY dissociates from FliM and/or FliN10,11.
The full-length structure of FliG was described in Aquifex aeolicus12. There are three distinctive domains in FliG: N-terminal domain (FliGN), middle domain (FliGM), and C-terminal domain (FliGC). Each domain has an Armadillo repeat motif (ARM): ARMN, ARMM and ARMC, respectively12,13,14. The domains have tandem repeats of several hydrophobic residues in α-helices. Several ARMs were also found in the crystal structure of β-catenin, suggesting that it is important for intermolecular interaction15. The hydrophobic residues of ARM in FliG are more conserved than those of β-catenin; moreover, the crystal structure of β-catenin, ARMM and ARMC were well aligned14. FliGN works as the anchor of the C ring by interacting with the MS ring, which is composed of FliF and is buried in the inner membrane16. It has been shown that ARMN is important for intermolecular interaction with FliGN to properly assemble the MS-C ring17,18. FliGM interacts with FliM, which receives the chemotaxis signals transmitted by CheY binding19,20. Conserved charged residues in FliGC electrostatically interact with those in cytoplasmic loops of PomA (MotA), thereby generating torque21,22,23. It has been suggested that the conformation of the linker region between FliGM and FliGC, called HelixMC, is important for determining the rotational direction or switching. A three-amino-acid deletion mutant (ΔPAA mutant) in HelixMC of Salmonella drove CW-locked rotation. It was deduced that the mutation stabilized the intramolecular interaction between FliGM and FliGC, and induced a conformational change in the HelixMC region to affect the CheY binding to FliM13,24,25. In contrast, the three-amino-acid deletion mutant (ΔPSA) of V. alginolyticus FliG corresponding to the Salmonella ΔPAA mutant conferred no flagellation phenotype, presumably because of defects in flagellin transport26. The C ring is also a transport machinery for flagellin. The change in the HelixMC region affects the interaction between the two ARMs of FliG: ARMM and ARMC. The interaction between ARMM and ARMC is similar to the intermolecular interaction with ARMN17,18. To change the C ring state, ARMM interacts with intramolecular or intermolecular ARMC, and FliM affects the FliG structure of HelixMC12,19,27. When HelixMC is disordered, FliG folds into the compact structure by the interaction between ARMM and ARMC in CW rotation. In contrast, because of the stable α helix, ARMM of the neighbor molecule interacts with ARMC so that the distance between FliGM and FliGC is extended in CCW rotation. However, the correlation between the structure of FliG and rotational direction is still unclear.
We have isolated and characterized five rotational mutants of the fliG gene of V. alginolyticus: two CCW-biased, two CW-biased, and one CW-locked28. Among the mutants, the G214S and G215A mutants showed CCW-biased and CW-locked rotations, respectively, and these mutations were located in the hinge linker between FliGM and FliGC, called Gly-Gly flexible linker (Fig. 1). It was speculated that the bending at Gly-Gly flexible linker depends on the conformation of the HelixMC. The G214 and G215 residues, which were located next to each other, produced the opposite phenotypes for the rotational bias. This indicated that the orientation of the FliGC domain was determined by the state of the Gly-Gly flexible linker. The A282T mutation in FliGC conferred the CW-biased phenotype. The structure of FliGC should be fixed to the CW conformation stabilized by the interaction between helix α1 and helix α3 of FliGC as suggested by NMR experiments and molecular dynamics (MD) simulation29.

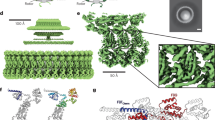
A structural model of the mutations in this study. (A) The structure of the FliG-FliM complex from Thermotoga maritima (PDB code 4FHR). FliG shows two distinct domains, FliGM and FliGC. The ARMM and ARMC domains are a part of FliGM and FliGC, respectively. The HelixMC and the Gly-Gly flexible linker region connect between FliGM and FliGC. The structures of FliGM, FliGC, HelixMC, ARMM, ARMC and FliM are shown in green, gray, white, cyan, orange and magenta, respectively, as a ribbon model. The atoms of blue, red, and magenta balls show Gly-Gly flexible linker (G214 and G215 residues), residues at the EHPQR motif (E144, H145, P146, Q147, and R179) and the residues of other point mutations, respectively, in Vibrio alginolyticus. The hydrophobic residues of the ARMM and ARMC were determined by the alignment of the structure and amino acid sequence between β-catenin, FliGM and FliGC, respectively15. (B) The architecture of the FliG protein is divided into two domains: FliGM and FliGC. The FliGMC1 domain (D113 through M235 of T. maritima) was used for the molecular dynamics simulation. FliGM, FliGC and HelixMC are colored green, orange, and yellow, respectively. In this study, the C-terminal region and N-terminal region of HelixMC are called LC and LN, respectively. E144, G214, and G215 mutation residues of V. alginolyticus (E126, G196, and G197 of T. maritima, respectively) are shown as spheres in magenta.
In this study, we isolated the E144D mutant that increased the switching frequency of the rotational direction (Figs 1 and S1). E144 residue is highly conserved among the species and belonging to the EHPQR-motif which is the FliM binding site in FliGM. To characterize this mutant, we combined 3 mutations (the G214S, G215A, and A282T mutations that showed CCW-biased, CW-locked, and CW-biased rotations, respectively) with the E144D mutation. To elucidate how the conformational change occurred in the 3 mutants (E144D, G214S, and G215A), we performed MD simulation using a co-crystal structure of FliG and FliM in Thermotoga maritima and NMR data was obtained from 2D 1H-15N TROSY HSQC spectra of the Vibrio FliGMC fragment. We observed the rotational direction and the rotational switching of the Vibrio fliG mutants in the absence of CheY to investigate the effect of CheY on fliG mutants. Here, we discuss the association of the conformational change of FliGC and interaction between FliM and FliGM.
Results
Analysis of point mutants of Vibrio fliG
In previous studies, we generated various point mutants of fliG in Vibrio8,28 based on the phenotype of E. coli and S. enterica mutants30,31,32. We generated additional six mutants of Vibrio FliG: E144D, P146L, P188L, A218V, E231V, and E231K; where these residues are highly conserved among the species. The E144D, P188L, A218V and E231K mutants showed CCW-biased rotation, whereas P146L and E231V mutants showed CW-biased rotation in the corresponding mutants of E. coli and S. enterica. We investigated the rotational direction and switching events (Fig. 2A). The wild-type (WT)-FliG had a CW: CCW ratio of 25: 75 and switching events of 10 times per 10 sec as previously reported28. P146L and E231K mutants showed phenotypes similar to that of WT. The E231V mutant showed CW-biased rotation (CW: CCW ratio of 70: 30, P > 0.01) and seven instances of direction-switching per 10 sec; however, the CW-biased rotation was not as significant as that in the Q147H and G215A mutants. P188L and A218V mutants showed strong CCW-biased rotation (CW: CCW ratio of 5: 95 (P > 0.01) and 5: 95 (P > 0.01), respectively) and decreased switching events (three times and two times per 10 sec, respectively (P > 0.01)). The E144D mutant showed the same rotational bias as that of WT, but the switching frequency was twice that of the switching frequency in the WT (i.e., 20 times per 10 sec, P > 0.01). Such frequent rotational switching was observed when the repellent (phenol) was added to WT Vibrio33.

The biased directional rotation of fliG mutants. The fliG/pomA/pomB mutant (NMB301) harboring pNT1 with a fliG mutation and pYA303 with a wild-type (WT)-pomA and pomB was grown and observed by high-intensity dark-field microscopy. (A) The ratio of counterclockwise (CCW) rotation to CW rotation of the fliG mutants is shown in the column. All the experiments were repeated at least 6 times, and average values with standard deviation (SD) are shown. (B) The switching frequency in 10 sec was measured. All the experiments were repeated at least 6 times, and average values with SD are shown. The black bars indicate that P < 0.01 (*) for WT-FliG versus the mutants by the Welch’s t-test.
Analysis of the mutants in the fliG EHPQR motif
To further investigate the role of the E144 residue in determining the rotational direction, we generated seven mutations (E144A, E144N, E144S, E144Q, E144W, H145E, and H145W) in the EHPQR motif. The expressions of the FliG protein in all mutants were detected (Fig. S2). We performed the motility assay on soft-agar plate, to evaluate the bacterial motility (Fig. S3A,B). WT-FliG formed a large motility ring, and no rings were observed in the vector control. The small motility ring was observed in the E144D and E144N mutants, but not in the E144A, E144Q, E144S, E144W, H145E, and H145W mutants.
To reveal whether declining or disappearance of motility were because of defects in flagellum formation, we counted polar flagella by immunostaining (Fig. S3C). The ratio of flagellation of WT-FliG was 28% in the total cells but that of the vector control was zero. The ratios of E144D, E144Q, and E144N mutants, which formed a small motile ring, were 14%, 9%, and 6%, respectively. The ratios of E144A, E144S, E144W, and H145E mutants, which formed no rings, were 7%, 6%, 8%, and 5%, respectively. We could not detect flagella in the H145W mutants. These results indicated that those mutations of the EHPQR motif only partially inhibited flagellar formation.
We observed the motile fraction by dark-field microscopy and measured the ratio of motile cells and non-motile cells (Fig. S3D). The motile fraction in WT was 41%. E144D, E144N, and E144Q mutants had motile fractions of 38%, 4% and 1%, respectively. E144A, E144S, E144W, H145E, and H145W mutants were not motile. The mutations in the EHPQR motif affected motility as well as flagellar formation.
Effects of the E144D mutation on the rotational direction and switching events
To elucidate the effect of the E144D mutation on the conformation of FliG, we generated double mutants of fliG. In our previous report, some fliG mutants showed CW-biased or CCW-biased phenotypes28. We selected three mutations, G214S, G215A, and A282T (Figs 1A and S1), and they showed CCW-biased, CW-locked, and CW-biased rotations respectively. Since the G214S and G215A mutations were located in the Gly-Gly flexible linker, we speculated that the mutations fixed the conformation of FliGC either in CCW state or CW state. In contrast, the A282T mutant tended to change the CW conformation in FliGC, as suggested by MD simulations and NMR experiments29. We combined these mutations with the E144D mutation (E144D/G214S, E144D/G215A, and E144D/A282T mutants) and measured the expression level of FliG. Similar amounts of FliG were detected in all the mutants (Fig. S2). We observed the rotational direction and switching events using high-intensity dark-field microscopy (Fig. 3A,B). E144D/G214S and E144D/A282T mutants showed a rotational direction similar to that of the WT (CW: CCW ratios of 20:80 and 30:70, respectively). For the switching events, the E144D/A282T mutant behaved similarly to the E144D single mutant (20 times in 10 sec, with P > 0.01 versus WT); however, the E144D/G214S mutant behaved similarly to WT (11 times in 10 sec, P > 0.01 versus the E144D mutant). These results indicated that the E144D mutation suppressed the G214S mutation, which conferred the CW state of FliGC. On the other hand, the E144D mutation dominated A282T. The E144D/G215A mutant lost flagellar formation (Fla– phenotype), suggesting that the assembly or transport of flagellar proteins was affected by the double mutation.

Characterizations of the E144D mutant in Vibrio alginolyticus. The fliG/pomA/pomB mutant (NMB301) harboring pNT1 with a fliG mutation and pYA303 with a wild-type (WT)-pomA and pomB and the fliG mutant (NMB198) harboring pNT1 with a fliG-WT or fliG-E144D mutation were grown and observed by high-intensity dark-field microscopy. (A) The ratio of counterclockwise (CCW) rotation and clockwise (CW) rotation of the fliG mutations is shown in the column. All the experiments were repeated at least 6 times, and average values with standard deviation (SD) are shown. (B and C) The switching frequency in 10 sec was measured. All the experiments were repeated at least 6 times, and average values with SD are shown. Data of (A) and (B) for the WT and E144D mutant are from Fig. 2A,B, respectively. The E144D/G215A mutant did not flagellate, so that it showed 0 on (A,B). (C) The effect of serine in the WT-FliG and E144D mutant. To ignore the effect of chemotactic adaptation, the switching events were observed within 3 min after addition of serine. Serine was added to a final concentration of 0.05, 0.1, 0.5, 1.0, 2.0 and 5.0 mM. 0* in the WT with 2.0 mM serine indicates that few cells switched their rotational direction. The WT and E144D mutant without serine are from the WT and E144D mutant of Fig. 2B, respectively. All the experiments were repeated at least 6 times, and average values with SD are shown. The black bars indicate that P < 0.01 for WT-FliG versus the mutants (A,B) or the V buffer background versus the addition of serine with the V buffer (C) by the Welch’s t-test.
To clarify how the E144D mutations affected the state of FliM, we investigated the switching events in 10 sec with serine, an attractant of Vibrio that induced the CCW rotation. We observed the WT and the E144D mutant in the presence of 0.05, 0.1, 0.5, 1.0, 2.0, and 5.0 mM of serine (Fig. 3C). The WT decreased the switching events as the concentration of serine increased, and the switching was not observed above 1 mM serine. On the other hand, the switching of the E144D mutant also significantly decreased with at least 0.5 mM of serine (P > 0.01), but the switching events with 5.0 mM of serine in the E144D mutant were still half of those observed without serine. The E144D mutant seems to be almost insensitive to the chemotactic signal and the motor can still switch the rotational direction under the attractant signal more often compared to the WT.
MD simulation of FliG mutants showed different fluctuations in HelixMC
To gain insight into the molecular mechanisms of how the FliGM mutations affected the rotational directions and switching events at atomic resolution, we performed MD simulations of the structures of FliGMC1 from T. maritima and the three mutants, E126D, G196S and G197A (corresponding to E144D, G214S, and G215A in Vibrio). The mean value of the root mean square deviation (RMSD) of the WT FliGMC1 from the reference during the simulations was 1.9 Å (SD ± 0.4 Å) and showed a slightly broader distribution compared to those of the three mutants (1.9 Å ± 0.2 Å for E126D, 1.6 Å ± 0.2 Å for G196S, and 1.8 Å ± 0.3 Å for G197A) (Fig. 4A). This indicated that all the proteins maintained their native-like structure during the 100 ns simulations.

(A) The root mean square deviation (RMSD) distributions of the wild-type (WT) and mutant FliGMC1 conformations during the simulations compared with the crystal structure of the reference. The horizontal axis shows the RMSD, and the vertical axis shows density of RMSD values. (B) Projections of the conformations of each FliGMC1 protein in the molecular dynamics trajectories upon the PC1 (horizontal) and PC2 (vertical) axes. For each protein, 3,000 conformations were plotted. The densities of conformation on the plane were estimated by the Gaussian kernel density estimation.
Figure 4B shows the projections of the structures of the WT and three mutants during the simulations on to the first (PC1) and second (PC2) components of the principal component analysis (PCA). Compared to the WT, variances for the conformations of E126D mutants along both PC1 and PC2 axes were small, indicating that structural fluctuations in E126D were smaller than those of the WT, even though its major conformation was dispersed from the crystal structure. A similar trend was observed in the G196S mutant, in that, the variances of PC2 of the mutant were relatively smaller than that of the WT. In the G197A mutant, the variance along the PC2 was similar, but the distribution of conformations along the PC1 slightly differed from that of the WT. These results indicated that single amino-acid substitutions appeared to restrict or change the conformational space observed in the WT.
To reveal the amino-acid residues of FliGMC1 that contributed to the difference in the collective motions observed in the PCA, we measured the root mean square fluctuation (RMSF) of each amino-acid residue from the reference structure (Fig. 5A). The regions, which were highly fluctuated were common among the proteins; the HelixMC consisting helix a5 (αM5) and two loops (LN and LC) in between as a linker helix connecting ARMM and ARMC domains20. The degree of fluctuation of the HelixMC differed among the mutants; that of G196S and E126D was smaller, while that of G197A was larger than that in the WT in the LN loop (Fig. 5A). The mutation positions of G196 and G197 corresponding to the Gly-Gly flexible linker were located in the LC of HelixMC. The mutations of G196S and G197A conferred a steric hindrance by increasing side chain volumes and affecting the flexibility of the region. The distributions of main chain dihedral angles of psi at position 195 in G196S mutant and at position 196 in G197A mutant, respectively, were confirmed to be altered compared to that in the WT and E126D (Fig. 6), and these alterations contributed to the different perturbations on HelixMC.

(A) The root mean square fluctuation (RMSF) of the residues in the FliGMC domains of the wild-type (WT) and mutant proteins. The secondary structure of the FliGMC1 domain colored by the domain architecture is shown on the top of the plot. (B) The number of hydrogen bonds formed between LN and LC (upper) and between αΜ5-LC (lower) in each conformation of the WT and mutant proteins. The horizontal axis indicates the number of H-bonds, and vertical axis indicates the number of conformations. For each protein, 3000 conformations were used for the calculation. Colors in this figure indicate WT (black), G196S (G214S) (green), G197A (G215A) (red) and E126D (E144D) (blue), respectively.

Distribution of the main-chain dihedral angles at position 195 (A) and 196 (B) on Thermotoga maritima FliGM. The horizontal axis and vertical axis show the rotatable angles of Cα-C bond (phi, ϕ) and Cα-N bond (psi, ψ), respectively. For each protein, 3,000 conformations were calculated. The densities were calculated by Gaussian kernel density estimation.
We found that the conformational alterations of the HelixMC among the proteins were characterized by the number of hydrogen bonds within the HelixMC, especially in two parts: 1) between LN and LC; 2) αM5 and LC (Fig. 5B). Compared to WT, G196S had many hydrogen bonds in the conformations of LN-LC and αM5-LC, suggesting that an increase in the number of hydrogen bonds within the HelixMC contributed to decreased fluctuation of this region in the mutant protein. On the other hand, G197A had a larger number of hydrogen bonds in LN-LC, but less in αM5- LC than that in the WT, reflecting the increase the structural fluctuations in the LN region in the G197A mutant. Interestingly, E126D showed a similar trend in the number of hydrogen bonds to the G196S mutant, although the mutation position was spatially separated from both LN and LC. The substitution of Glu to Asp, which has a shorter side chain, might decrease the number of contacts with residues on αM5, and simultaneously increase the number of hydrogen bonds between αM5 and LC. These results suggest that the difference in the conformational dynamics of HelixMC induced by mutations might lead to the difference in rotational direction and switching events.
Analysis of 2D 1H-15N TROSY HSQC spectra of Vibrio FliGMC2 fragment by NMR
To support the results of the MD simulation, we obtained 2D 1H-15N TROSY HSQC spectra of Vibrio FliGMC2 fragment, the region from G122 through L351, by NMR. We obtained the spectra in five mutations (E144D, Q147H, G214S, G215A, and A218V) (Figs 7 and S4). The spectra of E144D, Q147H, and A218V were similar to that of the WT, suggesting that the conformation of FliGM and FliGC of the E144D mutant did not largely change compared to that of the WT. In contrast, changes in signal intensities and chemical shift were observed for several amide signals in the G214S and G215A mutations, suggesting that the conformation of FliGM and FliGC of G214S and G215A mutants changed. These results suggested that the in silico model of the conformational change of FliG by MD simulation in T. maritima was supported by the NMR analysis of Vibrio FliGMC2 fragment.

2D 1H-15N TROSY HSQC spectra of Vibrio FliGMC2 fragment of the wild-type (WT)-FliG and the E144D, G214S, and G215A mutants. The concentration and measurement temperature of 15N labeled FliGMC are shown in each spectrum. The mutant phenotype is shown in the parenthesis of each spectrum. All spectra were measured by Avance-III HD 500 spectrometer equipped with a BBO cryogenic probe.
The rotational direction and switching events on fliG mutants in the cheY deletion strain
Since the conformation of FliG depended on CheY binding to FliM, we speculated that the conformational change in fliG mutants also affected the state of FliM. We hypothesized that the E144D mutation affected the interaction with FliM transmitting the chemotaxis signal by the CheY binding. Thus, we constructed a cheY deletion mutant in the ΔfliG strain. We introduced E144D, Q147H, G214S, G215A, and A282T mutations of fliG into this strain and observed the rotational direction and switching events by high-intensity dark-filed microscopy (Fig. S5). Q147H mutants show a CW-biased rotation28. WT-FliG showed a CCW-locked phenotype in the ΔcheY strain. The E144D, Q147H, G214S, and A282T mutants showed CCW-locked rotation in the absence of CheY. These results suggested that the large fluctuation in either CW or CCW state of FliG by the E144D mutation was determined by the interaction between FliG and FliM depending on CheY binding. Furthermore, the CW rotation of these mutants needed the CheY binding. In contrast, G215A mutant showed CW-locked rotation with or without CheY, suggesting that CheY binding to FliM did not affect the flagellar rotation in G215A mutant.
Discussion
In this study, we isolated 6 fliG mutants. Two of them (P188L and A218V) showed CCW-biased flagellar rotation. Surprisingly, although the E144D mutant was similar to the WT in the ratio of the rotational direction, it increased the frequency of switching events from CCW rotation to CW rotation or vice versa. Both the P188L and E144D mutations were expected to confer the CCW-biased rotation of flagella according to a previous mutational analysis of S. enterica, in which the E125D and P169L mutations (corresponding to the residues of E144 and P188 of V. alginolyticus) were used30. We have previously shown that the G204S/A218V mutation conferred the CCW-biased phenotype, and the G204S mutation was similar to the rotational phenotype of WT28. We confirmed that the A218V mutation by itself conferred the CCW-biased rotation. The HelixMC contains 88 residues, and the P188L mutation stabilizes αM5 in HelixMC as α helix. This assumption was supported by the evidence that the ΔPAA mutation of S. enterica conferred the CW-locked phenotype, and this conserved proline residue of P169 in S. enterica corresponded to the P188 residue of V. alginolyticus13. The A218 residue was located on the same side of the Armadillo repeat motifs and the A218V mutation affected the hydrophobic interaction between ARMM and ARMC to stabilize the CCW state structure. The E144D mutation on the EHPQR motif of FliGM was located in the binding site of FliM, affecting both the stability of the α helix in FliGM and the interaction with FliM.
To clarify the effect of the interaction between FliG and FliM on the determination of the flagellar rotation direction, we introduced various mutations in residues on the EHPQR motif of fliG in Vibrio. The Q147H and R179H mutants showed the CW-biased rotation and Fla− phenotype, respectively8,28. We introduced six other mutations on E144 and H145 residues: four of them lacked motility (Mot−), and 2 slightly inhibited motility. Moreover, their flagellation rates were slightly reduced, and the H145W mutant was not flagellated at all. These results suggested that mutational effects on the conformational change between CCW and CW states of FliG and/or the interaction with FliM slightly inhibited the flagellar assembly and torque generation. We speculated that these mutations affected the interaction with FliM rather than the conformational change of FliG. In the studies of E. coli, S. enterica, and Helicobacter pylori, both the rotational biased and Mot− mutants have been isolated, and the mutations were located on the EHPQR motif 31,34,35,36. In some Mot− mutants, the interaction between FliG and FliM was not detected by the pull-down experiments34. It was reported that many fliG mutants exhibited the Mot− phenotype at high temperatures32,37. This may suggest that the structure of FliG is highly mobile and cannot be easily affected by temperature.
In our laboratory collection of Vibrio FliG mutants, the G214S, G215A, and A282T conferred CCW-biased, CW-locked, and CW-biased phenotype, respectively, and these mutations have been suggested to affect the stability of the conformation of FliGC to fix either the CCW or CW state26,27. To investigate the effect of the E144D mutation, we generated three double mutants (E144D/G214S, E144D/G215A, and E144D/A282T). The E144D/G214S mutation conferred a phenotype similar to that of the WT, the E144D/G215A mutant could not form the polar flagellum, and the E144D/A282T mutation conferred the same phenotype of E144D. These results indicated that the effects of the E144D mutation varied among the G214S, G215A, and A282T mutants. The phenotype of E144D/A282T mutant indicated that the conformational change of FliGM, which was induced from the interaction with FliM in a CheY-dependent manner, was counteracted to drive the conformational change of FliGC. The E144D/G215A mutation breaks the structure of FliG, preventing the interaction with FliM or FliG by itself, and, consequently, the ability of flagellar formation is lost. The conformational change in FliGM caused by the E144D mutation dominated the conformational change in FliGC caused by the A282T mutation. This suggests that the CW state of FliGC is canceled by the effect of the E144D mutation. We speculated that the Ala to Thr substitution on the A282 residue affects a local conformation around the α helix of FliGC29. Thus, we can infer that the E144D mutation affects the structure around the α helix.
It is known that serine is the attractant of Vibrio and it decreases the switching frequency of the flagellar motor33,38. Based on the study of the chemotactic signals in E. coli39, it was strongly suggested that serine binding to the chemoreceptor promotes CheY-P dephosphorylation through chemotactic signals, so that CheY dissociates from FliM and induces the CCW rotation. To investigate the CheY binding in the C ring in the E144D mutant, we studied the switching events in the E144D mutant after addition of serine. The switching events decreased in the WT under high concentration of serine (2.0 and 5.0 mM); but did not decrease in the E144D mutant. The C ring containing the E144D mutation is supposed to highly fluctuate either in CW state or CCW state even at the native or low concentration of CheY. We found that the E144D mutant in the absence of CheY showed CCW-locked rotation as WT, indicating that it needed CheY binding to FliM to induce the rotational switching. We speculated that association and dissociation constants of CheY from the C ring increased in the E144D mutant and affected CheY binding to FliM and the structure of FliGM and FliGC; therefore, E144D mutant can switch frequently with low concentration of CheY.
From the MD simulation and analysis of the conformational ensembles of WT and mutants based on the crystal structure from T. maritima, we speculated that the mutations might change the conformation in ARMM and HelixMC on FliGM, but the degree of fluctuations might differ among the mutants. The conformations of FliGC in the G214S and G215A mutants may be fixed either in CCW state or CW state by the mutations of FliGM, but FliGC in the E144D mutant may fix neither CCW state nor CW state. We assumed that the effect of the interaction between FliG and FliM dominantly determined the rotational direction and switching rather than inducing the conformational change of FliGM.
To support the results observed in the phenotype of fliG mutant analysis and MD simulation, we measured 2D 1H-15N TROSY HSQC spectra of the 15N-labeled FliGMC2 (G122-L351) fragments in WT-FliG and 5 mutations (E144D, Q147H, G214S, G215A, and A218V, which showed tumble, CW-biased, CCW-biased, CW-locked, and CCW-biased rotations). The NMR spectra of WT gave many signals, but less than 229 signals (whole number of fragment), therefore, some signals might be weak, and some could not be detected. This suggests that the WT-FliGMC fragment was largely fluctuating between CW and CCW states in solution states. The spectra of the E144D, Q147H, and A218V mutants were similar to those of the WT, suggesting that these mutants are also fluctuating. In contrast, the significant changes in signal intensities and chemical shift were observed for several amide signals in the G214S and G215A mutations. The signals of the G215A mutation were most sensitive in the WT and other mutations. These results suggested that the G214S and G215A mutations at the Gly-Gly flexible linker tightly fixed the conformations of FliGMC, and they supported the results of the MD simulation. In contrast to E144D, the spectra of Q147H and A218V mutant were similar and did not reflect their phenotypes (Fig. S4), suggesting that the conformational changes in FliGMC between the CW state and CCW state were not considerable. We speculated that the Q147H mutation increased the association constant and/or decreased the dissociation constant of between CheY and FliM; thus, the spectrum of the Q147H mutant was similar to that of the WT. We speculated that the structural change caused by the A218V mutation was not so large, but the mutation affected the hydrophobic interaction between ARMM and ARMC, and induced the steric hindrance to next molecules in the ring structure of FliG, because the A218V mutation site was located on the surface, important for the hydrophobic interaction.
We hereby propose a model for determining rotational direction (Fig. 8) based on the information on Salmonella and E. coli, CheY binds FliM to change the state from CCW to CW in the C ring (Fig. 8A). The EHPQR motif forms a binding surface for FliM (Fig. 8B). The CheY binding signal must be transmitted to the EHPQR motif to change the conformation. The EHPQR motif seems to have an intramolecular interaction with the Gly-Gly flexible linker region between FliGn and the neighbor molecule FliGn+1 in the ring structure of the C ring (Fig. 8C). The conformation of the G214S mutation, which is restricted to the CCW state, is relaxed by the E144D mutation. The E144D mutation caused an increase in the switching events of rotation and exhibited a behavior similar to that observed by the repellent treatment of phenol in WT-FliG. We speculated that the conformational change in FliGM with CheY caused the rapid change of direction and is easy to occur to different directional states in the presence of phenol40.

The C ring model of the effects of the mutations in FliG. (A) The model of C ring in Vibrio alginolyticus. Dashed flames of left and right enlarge 1 set of the C ring (containing one molecule of CheY, FliM, FliG, and FliF) and 3 sets, respectively. (B) The mutations in FliG were mapped on 1 set of their components as described in (A). Blue, red, and black balls indicate counterclockwise (CCW)-biased rotation, CW-biased rotation, and tumble phenotype, respectively. FliGC interacts with the stator. (C) The model of the determination of the rotational direction in the C ring. Black arrows show how CheY binding to FliM affects the EHPQR motif in FliGM. Blue arrows show the conformational change in intramolecular FliG. Dashed blue arrows show the effect of the conformational change of FliG on the intermolecular FliG. In our model, CheY binding to FliM induces the conformational change in FliGM. This change is sensed by Gly-Gly flexible linker causing the steric hindrance of the conformation of FliGC. The conformational change of FliGC affects FliG at both side of ring, being transmitted to the whole FliG of the C ring. Interaction surface between FliGC and the stator slides, so that the rotational switching occurs. FliG in the E144D, Q147H, P188L, and A218V mutants affects FliG at both sides. In contrast, FliG in the G214S and G215A mutants causes the steric hindrance of FliGC.
Material and methods
Bacterial strains, medium, and growth condition
Bacterial strains and plasmids used in this study are listed in Table S123,28,41,42,43,44,45,46,47,48,49,50. To introduce the cheY deletion, NMB318 was constructed from NMB198 using pHIDA2, as previous reported7. V. alginolyticus cells were cultured at 30 °C in VC medium (0.5% [wt/vol] polypeptone, 0.5% [wt/vol] yeast extract, 3% [wt/vol] NaCl, 0.4% [wt/vol] K2HPO4, and 0.2% [wt/vol] nglucose) or in VPG medium (1% [wt/vol] polypeptone, 3% [wt/vol] NaCl, 0.4% [wt/vol] K2HPO4, and 0.5% [wt/vol] glycerol). If needed, kanamycin and chloramphenicol were added to a final concentration of 100 μg/mL and 2.5 μg/mL, respectively. E. coli cells were cultured at 37 °C in LB medium (1% [wt/vol] bactotrypeptone, 0.5% [wt/vol] yeast extract, 0.5% [wt/vol] NaCl). If needed, kanamycin and chloramphenicol were added to a final concentration of 50 μg/mL and 25 μg/mL, respectively. E. coli-introduced pColdI-fliGMC for overexpression of FliGMC fragment was cultured in M9 medium (0.7% [wt/vol] Na2HPO4, 0.3% [wt/vol] KH2PO4, 0.05% [wt/vol] NaCl, 0.002% [wt/vol] bases and vitamins (adenosine, guanosine, cytidine, thymine, thiamine, and biotin), 0.67% [wt/vol] cold-glucose, 0.1% [wt/vol] glycerol, 0.1% [wt/vol] 15N-labeled NH4Cl, 0.01 mM FeCl3, 2 mM MgSO4, 0.05 mM MnCl2, and 0.1 mM CaCl2) containing 100 μg/mL of ampicillin.
Mutagenesis
To introduce mutations in the fliG gene on plasmid pNT1 and pColdI-fliGMC, site-directed mutagenesis was performed using the QuikChange method, as described by the manufacturer (Stratagene). All constructs were confirmed by DNA sequencing. Transformation of V. alginolyticus with plasmids pYA303 was performed by electroporation, as described previously51. Transformation of V. alginolyticus with plasmid pNT1 was performed by conjugational transfer from E. coli S17-1, as described previously41. Transformation of E. coli with plasmid pColdI-fliGMC was performed by a conventional method.
Motility assay on soft-agar plate
Cells were grown overnight in VC medium at 30 °C. Approximately 2 μL of cells were spotted on VPG soft-agar containing 0.3% (w/v) agar and incubated for 8 h at 30 °C. We observed the expansion degree of the motility ring.
Analysis of motile fraction in fliG mutants
Cells were grown overnight in VC medium at 30 °C, diluted 1/100 in VPG medium, and incubated for 4 h at 30 °C. Cell suspension (2 μL) was added to 200 μL of V buffer (50 mM Tris-HCl, pH 7.5, 5 mM MgCl2, and 300 mM NaCl), and cells were observed by dark-field microscopy. At least 80 cells were observed per condition, and the experiment was repeated three times, for a total of at least 290 cells.
Investigation of the rotational direction and switching events
The rotational direction and switching events were observed, as described previously28. Cells were grown overnight in VC medium at 30 °C, diluted 1/100 in VPG medium, and incubated for 4 h at 30 °C. Cells were washed twice with V buffer. The cell suspension was diluted (1:1) in fresh V buffer or V buffer containing serine. Serine was added to final concentrations of 0.05, 0.1, 0.5, 1.0, 2.0, and 5.0 mM. Motility was observed by high-intensity dark-field microscopy. To avoid the effect of chemotactic adaptation, we observed the samples within 3 min after addition of serine. We measured the rotational direction and switching events in swimming cells for 10 sec. Flagella rotation was determined from the position of the flagellum and the direction of cell swimming. Flagella push the cell body during CCW rotation and pull it during CW rotation. Experiments were performed at least 6 times. For statistical testing, the Welch’s t-test (unequal variances t-test) was conducted for WT-FliG versus the mutants (Figs 2 and 3A,B) or for the V buffer background versus the addition of serine with the V buffer (Fig. 3C). * was indicated the value of P > 0.01.
Observation of polar flagellum in cells
Cells were grown overnight in VC medium at 30 °C, diluted 1/100 in VPG medium, and incubated for 4 h at 30 °C. Cells were washed once with V buffer. The cell suspension was passed through poly-L-lysine-coated tunnel slide with coverslip. To remove extra cells, the glass was washed once with V buffer. To label polar flagellum of the cells, they were incubated in V buffer containing the anti-polar flagellum antibody52 for 5 min and were washed twice in V buffer. Cells were incubated in V buffer containing an anti-rabbit IgG conjugated with rhodamine for 5 min. After washing the cells twice with V buffer, the immunolabeled cells were observed using a fluorescence microscope.
Detection of FliG protein
Cells were grown overnight in VC medium at 30 °C, diluted 1/100 in VPG medium, and incubated for 4 h at 30 °C. Cells were harvested and suspended in distilled water at a cell concentration equivalent to an optical density (OD) of 10 at 660 nm. Proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Immunoblotting was performed using an anti-FliG antibody (FliGB0164)53 as a primary antibody and goat anti-rabbit IgG-HRP as a secondary antibody; the proteins were detected using enhanced chemiluminescence (ECL) Western blotting reagents (GE Healthcare) as the substrate for chemiluminescence and a detector (LAS-3000 mini, Fujifilm).
MD simulation
The crystal structure of FliG bound to FliM from T. maritima (PDB code: 4FHR; Fig. 1A)20 for the MD simulations because the structure of FliG of Vibrio had not yet been experimentally determined. The region from D113 through M235 (named FliGMC1) of T. maritima FliG was included in the calculation. Since the region from 188 through 194 was disordered with respect to the structure, the atomic coordinates corresponding to the region were built, and point mutations (E126D, G196S, and G197A) were introduced by using Swiss Pdb Viewer software. Notably, the residues of E144, G214, and G215 in Vibrio corresponded to E126, G196, and G197 in T. maritima, respectively. After energy minimizations, the obtained structures were used as the initial structures. MD simulations were performed using Gromacs version 4.6.254. The topology was generated with standard amino-acid protonation states at pH 7.0. The force field of AMBER99SB-ILDN and the water model of TIP3P were used for the simulation. The starting structure was placed in a cubic box with 1.2 nm spaces around the solute, and the box was filled with water molecules. Na+ ions were added to a concentration of 150 mM. Finally, Cl− ions were added to make the total electrical charge neutral. Time was set at 2 fs. Energy minimization was performed by the steepest descent method. Following energy minimization, the system was equilibrated for 100 ps at 300 K under constant number of particles, volume, and temperature (NVT) and constant number of particles, pressure, and temperature (NPT) conditions. After equilibration, an all-atom MD simulation at 300 K under the NPT condition was performed at 100 ns (5 × 107 steps). Each protein was independently simulated three times. The principal component analysis (PCA) of the MD trajectory was performed by using ProDy55.
The model of C ring structure
The promoter of the full length FliG of V. alginolyticus was modeled based on the crystal structures of FliG from T. maritima and A. aeolicus (PDB code: 3HJL)12. The complex structure of FliG (containing FliGM and FliGC domains) and FliM was used as the template structure for the model of FliGM and FliM interaction. The model structure was fitted into the density map of the flagellar basal body from S. enterica subsp. (EMDB code: EMD-1887)56 to construct the C ring with 34-fold symmetry. The interaction between the FliGN domain and FliN was built according to the complex structure of the FliGN and the C-terminal domain of FliN from T. maritima (PDB code: 5TDY)18. The interaction between FliM and CheY was inferred on the basis of the complex structure of CheY and the N-terminal fragment of FliM from E. coli (PDB code: 1F4V)57.
Purification of Vibrio FliGMC2 fragment
Cells were grown overnight in 30 mL of M9 medium at 37 °C, inoculated in 1.5 L of M9 medium, and incubated at 37 °C. When the cell density at OD660nm was between 0.38 and 0.45, cells were incubated in ice for 1 h. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of 0.5 mM to induce overexpression of FliGMC fragment and inoculated for 1 day at 15 °C. The cells were harvested by centrifugation and stored at −80 °C to break the cell membrane. The frozen cells were suspended with T7.0-N150 buffer (50 mM Tris-HCl, pH 7.0, and 150 mM NaCl) and sonicated using a sonicator (Branson) set on duty cycle 50% and power 5 with proteinase inhibitor, complete EDTA free (Roche Life Science). Unbroken cells were removed by low-speed centrifugation. The samples were ultra-centrifuged at 118,000 × g for 30 min. The resultant supernatants were mixed with 5 mg of Talon Metal Affinity Resin (Takara) and incubated at least 10 min at room temperature in a polypropylene column by batch method. After eluting the supernatant in the column, 15 mL (3 fractions of the volume) of T7.0-N150 buffer was added to wash the column. To further wash the column, 5 mL (1 fraction volume) of I30 buffer (50 mM Tris-HCl, pH 7.0, 150 mM NaCl, and 30 mM imidazole) was added. To elute His-tag protein from the resin, 20 mL (4 fraction volume) of I120 buffer (50 mM Tris-HCl, pH 7.0, 150 mM NaCl, and 120 mM imidazole) was added and collected by 1 mL fractions.
The His-tag affinity-purified proteins were concentrated to 1 mL using 10 K Amicon device (Millipore). The samples were subjected to size exclusion chromatography using Superdex 200 Increase 10/300 column (GE Healthcare) in T7.0-N150 buffer with the flow rate at 0.75 mL per min. The peak fractions were collected, and the concentration of samples was measured using Direct Detect spectrometer (Millipore).
2D 1H-15N TROSY HSQC spectra of Vibrio FliGMC2 fragment
We used the fraction of the highest peak at OD280nm on 15N-labeled FliGMC2 fragments obtained by size exclusion chromatography whose concentrations were between 0.40 and 0.60 mM. The NMR sample buffer contained 50 mM Tris-HCl, 150 mM NaCl, 0.01% (w/v) sodium 2,2-dimethyl-2-silapentane-5-sulfonate (DSS), and 5% (w/v) D2O at pH 7.0. The slotted NMR tube was used, which provided higher signal-to-noise ratio and efficient use of sample mass compared to conventional NMR sample tubes58. NMR measurements were performed on an Avance-III HD 500 spectrometer (Bruker Biospin) equipped with a BBO cryogenic probe at 288 K.
In the experiments with 2D 1H-15N TROSY HSQC59 for observing backbone amide NH signals on 15N labeled FliGMC2 and its mutants, the data size and spectral width were 256 (t1) × 2048 (t2) and 1700 Hz (15N) × 7000 Hz (1H), respectively. The carrier frequencies of 15N and 1H were 119 and 4.7 ppm, respectively. The number of scans/FID was 16. All NMR spectra were processed by the Topspin 3.2 (Bruker Biospin).
References
Li, N., Kojima, S. & Homma, M. Sodium-driven motor of the polar flagellum in marine bacteria Vibrio. Genes Cells 16, 985–999 (2011).
Sato, K. & Homma, M. Multimeric structure of PomA, the Na+-driven polar flagellar motor component of Vibrio alginolyticus. J. Biol. Chem. 275, 20223–20228 (2000).
Blair, D. F. Flagellar movement driven by proton translocation. FEBS Lett. 545, 86–95 (2003).
Kojima, S. & Blair, D. F. The bacterial flagellar motor: structure and function of a complex molecular machine. Int. Rev. Cytol. 233, 93–134 (2004).
Terashima, H., Kojima, S. & Homma, M. Flagellar motility in bacteria structure and function of flagellar motor. Int. Rev. Cell Mol. Biol. 270, 39–85 (2008).
Kojima, S. et al. Stator assembly and activation mechanism of the flagellar motor by the periplasmic region of MotB. Mol Microbiol. 73, 710–718 (2009).
Zhu, S. et al. Conformational change in the periplamic region of the flagellar stator coupled with the assembly around the rotor. Proc. Natl Acad. Sci. USA 111, 13523–13528 (2014).
Kojima, S., Nonoyama, N., Takekawa, N., Fukuoka, H. & Homma, M. Mutations targeting the C-terminal domain of FliG can disrupt motor assembly in the Na+-driven flagella of Vibrio alginolyticus. J. Mol. Biol. 414, 62–74 (2011).
Francis, N. R., Sosinsky, G. E., Thomas, D. & DeRosier, D. J. Isolation, characterization and structure of bacterial flagellar motors containing the switch complex. J. Mol. Biol. 235, 1261–70 (1994).
McEvoy, M. M., Bren, A., Eisenbach, M. & Dahlquist, F. W. Identification of the binding interfaces on CheY for two of its targets, the phosphatase CheZ and the flagellar switch protein fliM. J. Mol. Biol. 289, 1423–1433 (1999).
Sarkar, M. K., Paul, K. & Blair, D. Chemotaxis signaling protein CheY binds to the rotor protein FliN to control the direction of flagellar rotation in Escherichia coli. Proc. Natl Acad. Sci. USA 107, 9370–9375 (2010).
Lee, L. K., Ginsburg, M. A., Crovace, C., Donohoe, M. & Stock, D. Structure of the torque ring of the flagellar motor and the molecular basis for rotational switching. Nature 466, 996–1000 (2010).
Minamino, T. et al. Structural insight into the rotational switching mechanism of the bacterial flagellar motor. PLoS Biol. 9, e1000616 (2011).
Brown, P. N., Hill, C. P. & Blair, D. F. Crystal structure of the middle and C-terminal domains of the flagellar rotor protein FliG. EMBO J. 21, 3225–3234 (2002).
Huber, A. H., Nelson, J. W. & Weis, W. I. Three-dimensional structure of the armadillo repeat region of β-catenin. Cell 90, 871–882 (1997).
Ogawa, R., Abe-Yoshizumi, R., Kishi, T., Homma, M. & Kojima, S. Interaction of the C-terminal tail of FliF with FliG from the Na+-driven flagellar motor of Vibrio alginolyticus. J. Bacteriol. 197, 63–72 (2015).
Xue, C. et al. Crystal structure of the FliF-FliG complex from Helicobacter pylori yields insight into the assembly of the motor MS-C ring in the bacterial flagellum. J. Biol. Chem. 293, 2066–2078 (2018).
Lynch, M. J. et al. Co-folding of a FliF-FliG split domain forms the basis of the MS:C ring interface within the bacterial flagellar motor. Structure 25, 317–328 (2017).
Paul, K., Brunstetter, D., Titen, S. & Blair, D. F. A molecular mechanism of direction switching in the flagellar motor of Escherichia coli. Proc. Natl Acad. Sci. USA 108, 17171–17176 (2011).
Vartanian, A. S., Paz, A., Fortgang, E. A., Abramson, J. & Dahlquist, F. W. Structure of flagellar motor proteins in complex allows for insights into motor structure and switching. J Biol Chem. 287, 35779–35783 (2012).
Lloyd, S. A. & Blair, D. F. Charged residues of the rotor protein FliG essential for torque generation in the flagellar motor of Escherichia coli. J. Mol. Biol. 266, 733–744 (1997).
Yakushi, T., Yang, J., Fukuoka, H., Homma, M. & Blair, D. F. Roles of charged residues of rotor and stator in flagellar rotation: comparative study using H+-driven and Na+-driven motors in Escherichia coli. J. Bacteriol. 188, 1466–1472 (2006).
Takekawa, N., Kojima, S. & Homma, M. Contribution of many charged residues at the stator-rotor interface of the Na+-driven flagellar motor to torque generation in Vibrio alginolyticus. J. Bacteriol. 196, 1377–1385 (2014).
Kinoshita, M. et al. Insight into adaptive remodeling of the rotor ring complex of the bacterial flagellar motor. Biochem. Biophys. Res. Commun. 496, 12–17 (2018).
Kinoshita, M., Namba, K. & Minamino, T. Effect of a clockwise-locked deletion in FliG on the FliG ring structure of the bacterial flagellar motor. Genes Cells. https://doi.org/10.1111/gtc.12565 (2018).
Onoue, Y., Kojima, S. & Homma, M. Effect of FliG three amino acids deletion in Vibrio polar-flagellar rotation and formation. J. Biochem. 158, 523–529 (2015).
Baker, M. A. et al. Domain-swap polymerization drives the self-assembly of the bacterial flagellar motor. Nat. Struct. Mol. Biol. 23, 197–203 (2016).
Nishikino, T. et al. Serine suppresses the motor function of a periplasmic PomB mutation in the Vibrio flagella stator. Genes Cells 21, 505–516 (2016).
Miyanoiri, Y. et al. Structural and functional analysis of the C-terminal region of FliG, an essential motor component of Vibrio Na+-driven flagella. Structure 10, 1540–1548 (2017).
Irikura, V. M., Kihara, M., Yamaguchi, S., Sockett, H. & Macnab, R. M. Salmonella typhimurium fliG and fliN mutations causing defects in assembly, rotation, and switching of the flagellar motor. J Bacteriol. 175, 802–810 (1993).
Lloyd, S. A., Tang, H., Wang, X., Billings, S. & Blair, D. F. Torque generation in the flagellar motor of Escherichia coli: Evidence of a direct role for FliG but not for FliM or FliN. J. Bacteriol. 178, 223–231 (1996).
Mashimo, T., Hashimoto, M., Yamaguchi, S. & Aizawa, S. Temperature-hypersensitive sites of the flagellar switch component FliG in Salmonella enterica serovar typhimurium. J. Bacteriol. 189, 5153–5160 (2007).
Homma, M., Oota, H., Kojima, S., Kawagishi, I. & Imae, Y. Chemotactic responses to an attractant and a repellent in the flagellar systems of Vibrio alginolyticus. Microbiology 142, 2777–2783 (1996).
Lam, K. H. et al. Structural basis of FliG-FliM interaction in Helicobacter pylori. Mol. Microbiol. 88, 798–812 (2013).
Van Way, S. M., Millas, S. G., Lee, A. H. & Manson, M. D. Rusty, jammed, and well-oiled hinges: Mutations affecting the interdomain region of FliG, a rotor element of the Escherichia coli flagellar motor. J. Bacteriol. 186, 3173–3181 (2004).
Brown, P. N., Terrazas, M., Paul, K. & Blair, D. F. Mutational analysis of the flagellar protein FliG: sites of interaction with FliM and implications for organization of the switch complex. J. Bacteriol. 189, 305–312 (2007).
Hashimoto et al. The hydrophobic core of FliG domain II is the stabilizer in the Salmonella flagellar motor. Microbiology 158, 2556–2567 (2012).
Zhu, S., Kojima, S. & Homma, M. Structure, gene regulation and environmental response of flagella in Vibrio. Front Microbiol 4, 410 (2013).
Parkinson, J. S., Hazelbauer, G. L. & Falke, J. J. Signaling and sensory adaptation in Escherichia coli chemoreceptors: 2015 update. Trends Microbiol 23, 257–266 (2015).
Pandini, A., Morcos, F. & Khan, S. The gearbox of the bacterial flagellar motor switch. Structure. 24, 1209–1220 (2016).
Okunishi, I., Kawagishi, I. & Homma, M. Cloning and characterization of motY, a gene coding for a component of the sodium-driven flagellar motor in Vibrio alginolyticus. J. Bacteriol. 178, 2409–2415 (1996).
Yorimitsu, T., Kojima, M., Yakushi, T. & Homma, M. Multimeric structure of the PomA/PomB channel complex in the Na+-driven flagellar motor of Vibrio alginolyticus. J. Biochem. (Tokyo) 135, 43–51 (2004).
Grant, S. G., Jessee, J., Bloom, F. R. & Hanahan, D. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl Acad. Sci. USA 87, 4645–4649 (1990).
Simon, R., Priefer, U. & Puhler, A. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in gram negative bacteria. Nat. Biotechnol. 1, 784–791 (1983).
Morales, B. M., Backman, A. & Bagdasarian, M. A series of wide-host-range low-copy-number vectors that allow direct screening for recombinants. Gene 97, 39–47 (1991).
Val, M. E., Skovgaard, O., Ducos-Galand, M., Bland, M. J. & Mazel, D. Genome engineering in Vibrio cholerae: a feasible approach to address biological issues. PLoS Genet. 8, e1002472 (2012).
Le Roux, F., Binesse, J., Saulnier, D. & Mazel, D. Construction of a Vibrio splendidus mutant lacking the metalloprotease gene vsm by use of a novel counterselectable suicide vector. Appl. Environ. Microbiol. 73, 777–784 (2007).
Kojima, S., Kuroda, M., Kawagishi, I. & Homma, M. Random mutagenesis of the pomA gene encoding the putative channel component of the Na+-driven polar flagellar motor of Vibrio alginolyticus. Microbiology 145, 1759–1767 (1999).
Onoue, Y. et al. Domain-based biophysical characterization of the structural and thermal stability of FliG, an essential rotor component of the Na+-driven flagellar motor. Biophys Physicobiol. 13, 227–233 (2016).
Bartolome, B., Jubete, Y., Martínez, E. & de la Cruz, F. Construction and properties of a family of pACYC184-derived cloning vectors compatible with pBR322 and its derivatives. Gene 102, 75–78 (1991).
Kawagishi, I., Okunishi, I., Homma, M. & Imae, Y. Removal of the periplasmic DNase before electroporation enhances efficiency of transformation in a marine bacterium Vibrio alginolyticus. Microbiology. 140, 2355–2361 (1994).
Yorimitsu, T., Mimaki, A., Yakushi, T. & Homma, M. The conserved charged residues of the C-terminal region of FliG, a rotor component of Na+-driven flagellar motor. J. Mol. Biol. 334, 567–583 (2003).
Koike, M., Terashima, H., Kojima, S. & Homma, M. Isolation of basal bodies with C-ring components from the Na+-driven flagellar motor of Vibrio alginolyticus. J Bacteriol. 192, 375–378 (2010).
Pronk, S. et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013).
Bakan, A., Meireles, L. M. & Bahar, I. ProDy: protein dynamics inferred from theory and experiments. Bioinformatics 27, 1575–1577 (2011).
Thomas, D. R., Francis, N. R., Xu, C. & DeRosier, D. J. The three-dimensional structure of the flagellar rotor from a clockwise-locked mutant of Salmonella enterica serovar Typhimurium. J. Bacteriol. 188, 7039–7048 (2006).
Lee, S. Y. et al. Crystal structure of an activated response regulator bound to its target. Nat. Struct. Biol. 8, 52–56 (2001).
Takeda, M. et al. Construction and performance of an NMR tube with a sample cavity formed within magnetic susceptibility-matched glass. J. Magn. Reson. 209, 167–173 (2011).
Pervushin, K., Riek, R., Wider, G. & Wuthrich, K. Attenuated T2 relaxation by mutual cancellation of dipole-dipole coupling and chemical shift anisotropy indicates an avenue to NMR structures of very large biological macromolecules in solution. Proc. Natl Acad. Sci. USA 94, 12366–12371 (1997).
Acknowledgements
This research was supported by Grants-in-aid for scientific research from the Ministry of Education, Science and Culture of Japan (24117004 and 23247024 to MH, and JP24657087 to S.K.), Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS) from AMED (17am0101111j0001 to A.H. and T.S.), and Grant-in-Aid for JSPS Research Fellow (17J11237 to T.N.). T.N. work was supported in part by the Integrative Graduate Education and Research program of Nagoya University. This work was performed in part using the NMR spectrometers with the ultra-high magnetic fields under the Cooperative Research Program of Institute for Protein Research, Osaka University, NMRCR-17-05.
Author information
Authors and Affiliations
Contributions
T.N., A.H., T.S. and M.H. designed the study; T.N., A.H. and Y.M. performed the research; A.H. and T.S. performed the M.D. simulations; T.N., A.H., Y.M., Y.O. and S.K. analyzed the data; T.N., A.H., Y.M. and M.H. wrote the paper.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nishikino, T., Hijikata, A., Miyanoiri, Y. et al. Rotational direction of flagellar motor from the conformation of FliG middle domain in marine Vibrio. Sci Rep 8, 17793 (2018). https://doi.org/10.1038/s41598-018-35902-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-35902-6
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
