
Abstract
Marine infrastructure can favor the spread of non-indigenous marine biofouling species by providing a suitable habitat for them to proliferate. Cryptic organisms or those in early life stages can be difficult to distinguish by conventional morphological taxonomy. Molecular tools, such as metabarcoding, may improve their detection. In this study, the ability of morpho-taxonomy and metabarcoding (18S rRNA and COI) using three reference databases (PR2, BOLD and NCBI) to characterize biodiversity and detect non-indigenous species (NIS) in biofouling was compared on 60 passive samplers deployed over summer and winter in a New Zealand marina. Highest resolution of metazoan taxa was identified using 18S rRNA assigned to PR2. There were higher assignment rates to NCBI reference sequences, but poorer taxonomic identification. Using all methods, 48 potential NIS were identified. Metabarcoding detected the largest proportion of those NIS: 77% via 18S rRNA/PR2 and NCBI and 35% via COI/BOLD and NCBI. Morpho-taxonomy detected an additional 14% of all identified NIS comprising mainly of bryozoan taxa. The data highlight several on-going challenges, including: differential marker resolution, primer biases, incomplete sequence reference databases, and variations in bioinformatic pipelines. Combining morpho-taxonomy and molecular analysis methods will likely enhance the detection of NIS from complex biofouling.
Similar content being viewed by others


Introduction
Biological invasions of non-indigenous species (NIS) can cause severe economic and environmental impact contributing to biodiversity loss1,2. One of the major vectors responsible for the transfer of marine NIS is global shipping via ballast water3 and hull fouling4. The ports and marinas where these vessels berth often act as hubs for the spread of NIS5,6. Artificial substrata such as wharf piles and pontoons, which might be less attractive for native taxa, provide opportunistic NIS with vacant niches where they can settle and establish thriving populations6,7.
Early detection of NIS is a critical factor to inform timely implementation of measures and allow the greatest chance of successful management1. Current marine surveillance programmes largely rely on traditional morphological identification of NIS during visual surveys by divers and from biological samples collected by a variety of methods, including grabs, benthic sleds, trawls, and passive sampling devices such as settlement plates8,9,10. Morpho-taxonomy is particularly well suited for conspicuous organisms, such as macrofauna or macroalgae that can be readily identified. However, the identification of cryptic or small juvenile life stages in situ or within complex samples remains challenging11,12.
Recent advances in molecular technologies, in particular the emergence of high-throughput sequencing (HTS), are rapidly changing the way biomonitoring programmes are undertaken13. For example, metabarcoding is increasingly being used for biodiversity assessments from a range of aquatic ecosystems14,15,16,17,18,19,20. The potential for high sensitivity, accuracy and standardizing and automating these methods make metabarcoding a particularly well-suited approach for use in marine biosecurity surveillance programmes21,22,23,24,25.
Nonetheless, metabarcoding approaches have limitations3,14,26,27. For example, databases of reference DNA sequences are incomplete28 and are often tailored for certain genetic markers or taxonomic groups (e.g. Protist Ribosomal Reference [PR2] database for the nuclear small ribosomal subunit 18S rRNA)29. GenBank30 contains reference sequences from many different genetic markers and includes all domains of life but is prone to erroneously identified sequences31. Another limitation of metabarcoding is a lack of ‘universal’ markers across phyla5, therefore not all taxa can be equally detected due to primer selectivity and resulting amplification biases32. Recent studies have highlighted the advantage of using multiple barcode regions, including mitochondrial Cytochrome oxidase c subunit 1 (COI) for discriminating between many metazoan species, and 18S rRNA that enables the detection of a much broader range of taxa5,33,34.
Appropriate bioinformatic pipelines can improve the robustness of molecular biodiversity assessment, and derived taxonomic assignments may vary based on the parameters applied35. One of the critical steps in a pipeline is the grouping of similar sequences into Operational Taxonomic Units (OTUs), to count for intraspecific variation within taxa and sequencing errors. Although a 97% similarity threshold is commonly applied for general biodiversity assessments, a higher threshold may increase rare taxa detection, which may be particularly well suited when targeting NIS36.
Combining the use of conventional morpho-taxonomic and metabarcoding approaches may enhance detection sensitivity37 and lead to the minimization of Type-I errors (false positives) and Type-II errors (false negatives) in marine NIS surveillance programmes2. In this study, an integrated morpho-taxonomic and metabarcoding approach was used to characterize biodiversity on settlement plates, with a focus on the detection of NIS within the samples. The overall goal was to investigate the benefits of integrating molecular methods such as metabarcoding into conventional marine NIS surveillance programmes to collectively enhance the likelihood of early detections. The metabarcoding approach comprised two molecular marker regions, the V4 region of 18S rRNA and a small region of COI. Taxonomy was assigned against three sequence databases (The Protist Ribosomal Reference database [PR2], The Barcode of Life Database [BOLD] and the nucleotide collection of the National Centre for Biotechnology Information [NCBI]). The following hypotheses were tested: that biofouling diversity and NIS detection would differ significantly (1) between morphological and molecular approaches due to varying efficiency in identifying micro-, meio- and macrofaunal communities, and (2) between molecular markers (18S rRNA and COI) due to differences in the taxonomic resolution and coverage of sequence reference databases.
Material and Methods
Experimental design
This research was implemented as a companion study to a project that used morpho-taxonomic identification to investigate the optimal design of settlement arrays for sampling non-indigenous biofouling species10,38. Five settlement arrays each consisting of twelve polyvinyl chloride (PVC) settlement plates (14.5 cm × 14.5 cm) attached to PVC pipes were deployed at randomly chosen pontoons in Westhaven Marina, Auckland, New Zealand. Separate deployments were made in winter (June to October 2015) and summer (November 2015 to February 2016) months. Each settlement array was placed horizontally at two meters depth and comprised of three different treatments in a crossed experimental design. The design included assessing the effect of different antifouling coatings, light intensities and surface textures on NIS detection as previously described in von Ammon, et al.39 and Tait, et al.38. The present study does not address the effect of these treatments but considers combined data from all five arrays. Each plate was considered as an individual sample while most downstream analyses (apart from rarefaction curves) were processed on combined data of all 60 samples for summer and winter each.
Morpho-taxonomic approach
All plates were retrieved after three-month deployment periods and immediately transported to the laboratory on ice and in individual bags filled with seawater. Visible taxa were identified and species richness was determined under a light microscope at 10x magnification as in Tait, et al.38. Abundant species from the upper layer of biofouling were partially removed to identify biofouling organisms in secondary cover. Voucher specimens of unknown species were preserved in ethanol and sent to taxonomic specialists for identification. During this handling, care was taken to ensure that a portion of each morphologically identified taxon remained on the plate for subsequent metabarcoding analyses. All specimens were identified to the lowest possible taxonomic level, and categorized as either indigenous to New Zealand, non-indigenous, cryptogenic (undetermined geographic origin), or unresolved. Unresolved individuals accounted for less than 10% of taxa and were excluded from downstream NIS analyses, as their biosecurity status could not be determined.
Metabarcoding approach
Following the morpho-taxonomic screening, the five identical arrays (n = 60 plates) were sampled for metabarcoding analysis. All biofouling material was removed from the plate surface using sterile stainless steel surgical blades (Swann-MortonR, Sheffield, UK) and transferred into a 10 mL sterile tube (Merck KGaA, Darmstadt, Germany) in February (summer) or sterilized sponges (Whirl-pak™, Speci-sponges™, Nasco, WI, USA) stored in individual sterile plastic bags in October (winter). All samples were stored immediately at −70 °C until further processing. The sampling technique varied between summer and winter samples due to the high amounts of calcifying organisms on the summer plates which could not be removed with the sponge method used for winter samples.
The summer samples were centrifuged (4000 × g, 15 min) and the supernatant discarded. The winter samples (sponges with biofilm samples) were macerated using a stomacher (Colworth 400; AJ Seward, London, UK) for 2 min at maximum speed, then squeezed to remove excess liquid. The resulting biofouling suspensions were pelleted by centrifugation (4000 × g, 15 min) and the supernatant discarded. DNA was extracted from the resulting pellets using the PowerMax® Soil DNA Isolation Kit (QIAGEN, CA, USA) following the manufacturer’s protocol. The quantity and quality of extracted DNA were measured using a NanoPhotometer (Implen, Munich, Germany).
For the characterization of eukaryotic communities, a segment (approximately 400 base pairs [bp]) of the V4 region of the 18S rRNA gene and an approximately 300 bp fragment of the mitochondrial COI gene were amplified by Polymerase Chain Reaction (PCR). For the 18S rRNA gene, the eukaryotic-specific primers were Uni18SF: 5′-AGG GCA AKY CTG GTG CCA GC-3′ and Uni18SR: 5′-GRC GGT ATC TRA TCG YCT T-3′40 modified to include IlluminaTM overhang adaptors following von Ammon, et al.39. Thermocycling PCR conditions were: 94 °C for 3 min followed by 35 cycles of 94 °C for 30 s, 52 °C for 30 s, 72 °C for 1 min, with a final extension step at 72 °C for 7 min. For the COI gene, the eukaryotic-specific primers mlCOIintF: 5′-GGW ACW GGW TGA ACW GTW TAY CCY CC-3′ and jgHCO2198: 5′-TAI ACY TCI GGR TGI CCR AAR AAY CA-3′ were used41. PCR amplification was undertaken in a total volume of 50 μL using 25 μL of MyTaq™ Red Mix (Bioline, USA), 2 μL of each primer, 16 μL of DNA-free water, 3 μL of BSA (0.2 mg/mL) and 2 μL of template DNA. Thermocycling conditions were: 95 °C for 3 min, followed by 40 cycles of 95 °C for 30 s, 50 °C for 30 s, 72 °C for 90 s, and a final extension of 72 °C for 10 min. Two samples of 20 μL of ddH2O were used as negative controls following the same protocol as described above.
Purification and quantification of amplicons were performed following the Agencourt® AMPure® XP protocol (Beckman Coulter, USA), using magnetic beads and a Qubit® 2.0 Fluorometer (Invitrogen). Purified amplicons were diluted to 3 ng μL−1 and libraries sent to New Zealand Genomics Limited at the University of Auckland for sequencing. Paired-end sequences (2 × 250) were generated on a MiSeq instrument using the TruSeqTM SBS kit v3 (IlluminaTM).
Bioinformatics analyses
The raw sequence files were demultiplexed using fastq-multx (version 1.3.1) and bi-directional reads were paired using SolexaQA++42. By running the following pipeline on QIIME 143, the reads were truncated on the 3′ end from the first base where the Phred score dropped below 3 and which explains, along with differences in sequence length among taxa, the variation in the overlap size and when paired-end reads were assembled. For quality filtering, merging and dereplication, the software package VSEARCH was used44. Merged reads with more than five bp expected error were discarded. Sequence chimeras were detected using VSEARCH Uchime de novo method by mapping unique 18S rRNA sequences against the PR2 database29 and COI sequences against the combined MIDORI45 and Barcode Of Life Database (BOLD; Ratnasingham and Hebert46) that were trimmed using the COI primers of this study. Sequence reads were clustered into OTUs at 99% similarity to retain maximum sensitivity for NIS detection. OTUs found in negative controls were discarded across all samples. Singletons and all low read OTUs were kept. Taxonomy was assigned using the QIIME package43 and the default UCLUST classifier with 0.9 minimum sequence identity47, with the PR2 and BOLD databases for 18S rRNA and COI clusters, respectively.
For cross-validation, both 18S rRNA and COI sequence datasets were also aligned against NCBI’s nucleotide collection (nr/nt) database (NCBI) using Megablast48, searching for a maximum of 10 best-matching sequences (Megablast+, default e-value of 0.001, word size 28). Using hits with the lowest e-value, query sequences were assigned at species level if similarity of the hit was greater or equal to 97%. Otherwise, if sequences did not fulfill the 97% or above threshold, the naïve Last Common Ancestor (LCA) and default parameters in MEGAN v.549 among the best hits was used for assignment to higher taxonomic ranks. This process separates reads that align specifically to a single taxon and assigns these together and less specific reads that were aligned to different taxonomies by the 10 best matching hits in Megablast. In the latter case, if less than 75% of the hits share the same taxonomy, no assignment is made at this specific taxonomic rank and the algorithm repeats the process to the next level.
Sequences for the COI region have been deposited in the NCBI’s Sequence Read Archive under BioProject ID PRJNA478269, sample accession SAMN09508561–679 and for the 18S rRNA region in the European Nucleotide Archive (ENA) under BioProject PRJEB25036, sample accession SAMEA104599381-499.
Statistical analyses
The phyloseq R package and associated tools50 were used for the following diversity analyses. Read abundance of metabarcoding data was rarefied to 5,000 reads. Ten samples were below this threshold and were discarded for the diversity analysis. The datasets were then filtered for Eukaryotes and merged at a genus level to be consistent with the morpho-taxonomic data and presence/absence transformed. Overall taxonomic richness (genus level) of samples from each dataset was visualized using boxplots.
Unrarefied OTU tables were filtered for Metazoa, Chlorophyta, Ochrophyta and Rhodophyta (taxonomic groups commonly identified in biofouling by morpho-taxonomic analysis). The abundance data was merged at the genus level. Bar plots for each approach were computed using standardized data from most abundant taxa.
A general list of NIS for New Zealand was assembled by consulting the Marine Invasive Taxonomic Service51 and through comparison with the New Zealand Organisms Register52 and the Marine Biosecurity Porthole51. This list was used to filter non-indigenous taxa on genus and species levels from the metabarcoding and morpho-taxonomic datasets. Euler diagrams were constructed on the filtered data using the online Venn diagram creator at http://bioinformatics.psb.ugent.be/webtools/Venn/ to quantify overlaps in NIS detected using the different approaches and proportionally visualized using the R package ‘eulerr’53. For assessing performance of NIS detections, rarefaction curves were computed on the filtered datasets of all approaches combining the information of NIS from PR2/NCBI for 18S rRNA and BOLD/NCBI for COI and their combinations using the iNEXT R package54,55.
Results
Morpho-taxonomy
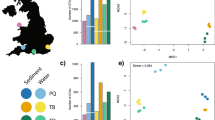
The morpho-taxonomic assessment resulted in the total observation of 39 taxa in summer (average 6.4 per sample plate) and 31 taxa in winter (4.2 per sample) of which 35 and 27, respectively, could be identified down to at least genus level, and 33 and 24 to species level (Fig. 1). The main phyla present were Bryozoa, Chordata, Mollusca and Annelida. For winter samples, Annelida were less abundant while Cnidaria increased in abundance (Fig. 2).

Boxplots displaying number of eukaryotic genera (left) and species (right) detected per summer and winter sample plates, using 18S ribosomal RNA (18S rRNA) and Cytochrome c oxidase Subunit I (COI) metabarcoding assigned to the three databases PR2 (18S rRNA), BOLD (COI) and NCBI (both 18S rRNA and COI) and morpho-taxonomy (morph). Molecular datasets were rarefied to 5,000 reads, with all genus-level unassigned sequences removed.

OTU proportions of the most abundant metazoan and algal taxa, standardized for all datasets ignoring the unassigned fraction: 18S ribosomal RNA (18S rRNA) and Cytochrome c oxidase Subunit I (COI) metabarcoding (PR2, BOLD and NCBI database assignments) and morphological taxonomy (morph) for summer and winter samples.
Metabarcoding
The total paired-end, quality filtered and non-chimeric and de novo non-chimeric sequences obtained from combined summer and winter plates for 18S rRNA and COI PCR amplicons were 2,831,859 reads (95,751 unique OTUs) and 3,084,751 reads (211,164 unique OTUs), respectively (Table 1). Negative controls contained 41 reads assigned to 19 OTUs for 18S rRNA and COI, indicating insignificant background contamination, from which no new potential NIS were identified.
The comparative analysis of three databases and related taxonomic assignment methods showed that assigning sequence data to the NCBI reference database resulted in fewer unassigned sequences for both genes. When only metazoan sequences are assessed, the use of the PR2 database resulted in 1,766 (i.e. 7%) less 18S rRNA sequences assigned to genus compared to NCBI, while the BOLD database enabled assignment of 18,579 (i.e. 34%) of COI NCBI assigned sequences. A similar ratio was also observed for sequences assigned to species (Table 1).
Comparative taxonomic richness and diversity among metabarcoding data
Metabarcoding data yielded up to 76 and 50 (18S rRNA; PR2 database; winter season) taxa per sample plate identified to genus and species level, respectively (Fig. 1).
Notable differences in the number of identified genera or species were observed between markers and seasons, and amongst the three databases (Fig. 1). For example, the number of identified genera or species from 18S rRNA data was higher in winter samples regardless of the database used, whereas an opposite trend was observed in COI (BOLD and NCBI data). Using BOLD, a large number of sequences remained unassigned (Table 1), resulting in fewer than 11 genera and species per sample being identified in summer and winter season. Using COI assigned to NCBI, up to 58 genera could be identified on average while the average number of COI NCBI assigned species was with 25 considerably lower (Fig. 1).
Assessment of the taxonomic diversity among all datasets showed that 80–90% of eukaryote OTUs were assigned to metazoan taxa, with the exception of the COI NCBI dataset (35%; Table 1). The remainder of the latter data were unassigned eukaryotes (51,787 taxa) or belonged to algal groups (65,379). The 18S rRNA data showed a very similar diversity across PR2 and NCBI assignments (Fig. 2), and was mainly comprised of Chordata (more abundant in summer), Nematoda (more abundant in winter) and Bryozoa (consistent across seasons). Cnidaria and Mollusca OTUs were present predominantly in winter. There were more OTUs assigned to Metazoa in the COI NCBI data than in COI BOLD (Table 1). This is reflected in the differences between NCBI and BOLD COI databases in the proportions of the major taxa (Fig. 2). Only NCBI identified the algal groups (Chlorophyta and Ochrophyta), and appeared to identify more Bryozoa and Mollusca. The taxa found using the COI marker gene were more consistent between summer and winter seasons compared to the 18S rRNA marker. Overall, the metabarcoding analyses using COI assigned to NCBI appeared to identify highest diversity of all major taxa compared to the morphological taxonomy and other metabarcoding approaches.
Detection of putative non-indigenous taxa
In total, there were 51 NIS genera and 48 NIS species identified across all approaches and assignment methods. The morpho-taxonomic screening for NIS identified three putative non-indigenous genera and seven species (5%/14%, respectively) that were not detected by either molecular markers or the database assignments (Fig. 3; Table S1). These predominantly consisted of bryozoan taxa, e.g. Bugulina (flabellate), Schizoporella japonica, Celleporaria umbonatoidea and Tricellaria (inopinata). Overall, the dataset using 18S rRNA and PR2 yielded the highest number of unique NIS detections with just two genera but 14 species (3%/29%, respectively). Three genera and three species were uniquely detected using 18S rRNA NCBI such as Styela clava. The dataset using COI and BOLD resulted in the detection of one unique species (Figs 3A and 4C), while the dataset using COI and NCBI identified two unique algal NIS (Striaria and Pzeudo-nitzschia; Fig. 3B; Table S1). A complete list of the potential non-indigenous genera and species detected using morphological and metabarcoding approaches and the different databases is given in Table S1.

Euler diagrams of non-indigenous taxa identified using the morpho-taxonomy (morph) and metabarcoding approaches, 18S ribosomal RNA (18S rRNA) and Cytochrome c oxidase Subunit I (COI). Comparisons at; (A) genus level using the PR2 or BOLD databases for taxonomic assignment, (B) genus level using NCBI for taxonomic assignment, (C) species level using the PR2 or BOLD for taxonomic assignment, and (D) species level using NCBI for taxonomic assignment. The size of circles is proportional to the number of genera/species identified by each method.

Rarefaction curves of NIS diversity calculated from all detected non-indigenous taxa identified during summer at (A) genus and (B) species levels and during winter at (C) genus and (D) species levels. Detected using morpho-taxonomy and metabarcoding (18S ribosomal RNA [18S rRNA] and Cytochrome c oxidase Subunit I [COI]) approaches, individually and in combination.
A total of 21 genera (e.g. Arcuatula) and 11 species (e.g. Ciona savignyi, Arcuatula senhousia, and Hydroides elegans) were detected by at least two of the metabarcoding approaches and database assignments but not by morpho-taxonomy. The combination of morpho-taxonomy with at least one metabarcoding approach led to the detection of 22 non-indigenous genera (e.g. Bugula, Ciona or Ectopleura) and 12 species (e.g. Ascidiella aspersa, Styela plicata or Sabella spallanzanii). Only nine genera (e.g. Botryllus, Styela and Hydroides) and two species (Botryllus schlosseri and Styela plicata) were detected by all approaches and database assignment methods combined.
Sample-based rarefaction curves for the detection of non-indigenous taxa (Fig. 4) showed that, overall, morpho-taxonomy followed by COI, especially at species level, individually and in combination returned the poorest average per sample diversity, although COI required fewer samples to detect the complete suite of taxa detectable with that method. The 18S rRNA approach was individually the most successful method at detecting diversity with the fewest samples. The different combinations of approaches displayed higher richness of detected NIS than the individual methods. The increase in richness of each combination was approximately equal to the sum of the individual component curves, indicative of the low overlap in detections between all approaches.
In winter, all approaches required more sampling effort than in summer to detect all potential NIS present (Fig. 4C,D). Overall, the combination of all approaches (morph + 18S rRNA + COI) resulted in highest detected richness with the fewest samples, although for summer the combination of 18S rRNA with COI at genus level and 18S rRNA and morpho-taxonomy nearly reached the same efficiency.
Discussion
The results of the present study add to the growing body of research advocating for the use of parallel morpho-taxonomy and metabarcoding to enhance biodiversity studies and for the sensitive detection of non-indigenous species5,14,37,56. This study reinforced the finding that the effectiveness of metabarcoding is strongly influenced by the genetic marker targeted and by the completeness of the sequence databases used for taxonomic assignment57. Nevertheless, it is also acknowledged that the use of different parameters or algorithms in the bioinformatic workflow may also influence the final results as observed in e.g. Hatzenbuhler, et al.58. We assessed the efficiency of two markers (18S rRNA and COI) and three reference databases for taxonomic assignment; PR2 for 18S rRNA, BOLD for COI and NCBI for both characterizing the biodiversity and detecting NIS in marine biofilms/biofouling. The results reveal clear benefits using some approaches, with the greatest success being derived from a combination of approaches.
The bioinformatics pipeline used in this study clustered sequences into OTUs at a 99% similarity threshold and maintained all sequences including singletons. While the voluminous data produced from this approach is resource intensive to analyze, it enhances the likelihood of keeping rare, but potentially important sequences. The removal of singletons is usually employed to eliminate false positives35. However, in the context of marine biosecurity surveillance, a false negative could be costly, e.g. the NIS is not detected prior to, or during the early incursion stage when management intervention might be possible. In contrast, a positive result (even if false) can be used to trigger a series of actions to confirm the presence of a NIS including; additional sampling/surveying, further analysis of technical replicates and the use of alternative species-specific molecular approaches such as real-time PCR59,60,61. Nonetheless, the high number of potential false positives observed here and which were likely related to the available bioinformatics workflow, may have serious implications if interpreted in isolation as they might also trigger the deployment of unnecessary and costly investigations. The process of quality control over the interpretation of sequence reads is important in determining its utility. Recent bioinformatics algorithms such as UNOISE62 or DADA2 (Divisive Amplicon Denoising Algorithm)36, which uses amplicon sequence variants or unique sequences instead of clustering sequences in order to retain as much information as possible, are becoming extremely efficient at denoising data (i.e. removing erroneous sequences). This approach represents an interesting research avenue for analyzing HTS data aimed at detecting rare taxa.
Recent metabarcoding studies have highlighted the advantages of using sequences from several genetic regions to characterize biological communities. This may overcome issues such as limitations in the universality of primers, and extend the different taxonomic levels resolved63. In the present study, the COI analysis yielded over twice the number of OTUs compared to the 18S rRNA. A similar result was also reported by Borrell, et al.64 who investigated the applicability of these two markers to detect NIS in estuaries. The most likely explanation for this result is that the rate of evolution in the COI gene is much faster than in the 18S rRNA and therefore there is greater sequence variability between species65. The use of the 99% similarity clustering (described above) contributed to the preservation of this effect28,35.
Taxonomy was assigned to OTUs using the PR2 database for 18S rRNA, and BOLD for COI. Additionally, all unique sequences were blasted directly against the NCBI database but using MEGAN’s LCA algorithm for taxonomy assignment when species hits were below 97%. This resulted in marked differences in the number of taxa detected. For the COI data, almost all OTUs remained unassigned when using the BOLD database, however this value was less than 2% when blasted against NCBI. This was largely attributed to the high abundance of bacteria, protists and micro algal taxa in the samples that are not well represented in the BOLD database, which focuses on invertebrates and vertebrates5,66. When the COI data was interrogated at genus level, the number of assigned OTUs was also markedly different between the BOLD or NCBI database. The higher Megablast taxonomic assignment rates at genus and species level were reached using 97% species hits and the LCA algorithm for the remaining assignments. The reason was to reach conservative results and to avoid similar false positive detections from both, the UCLUST and Megablast algorithms. However, the NCBI database contains a high number of non-curated data entries many of which come from environmental studies57. BLAST searches often return these as the closest match and prevent the algorithm searching for reference sequences which are present in lower abundance but have more definitive taxonomic identity67. Customized databases can largely avoid this problem as they contain exclusively curated data and therefore increasing taxon-specific resolution29,65. However, the 97% similarity threshold and the LCA algorithm of lower assigned hits was applied to aim for the most conserved taxonomic level in case of several Blast hits of the same similarity threshold but different taxonomy67.
A similar pattern was observed for 18S rRNA data. About 8% of OTUs were unassigned using the PR2 database whereas this reduced to about 2% when using the NCBI database. When only Eukaryotes and Metazoans were considered, the assignment using either database was comparable31. When taxonomy was assigned at the genus level there were up to six times more uniquely assigned taxa in NCBI compared to PR2 while the overall genus assignments showed very little difference (7%); this again is likely related to the high abundance of non-curated data-entries or not further determined ‘sp.’ species in NCBI and the Megablast search on the highly conserved 18S region that can reveal several hits of the same similarity threshold.
The morpho-taxonomic approach employed in the present study resulted in the lowest rate of identified taxa, a result which has been shown in many other studies23,57,68. A plethora of factors likely contribute to the low detection rate of taxa including; an inability to detect micro-organisms, cryptic taxa, larval stages, and the presence of extracellular DNA which is only detected using molecular techniques39. Morpho-taxonomy, however, is still the technique that results in the fewest false positive errors. Additionally, in the present study a number of taxa, most notably Bryozoan taxa, were detected using morphology but not metabarcoding. A search of the customized databases (PR2 and BOLD) used in the present study revealed that there were no representative sequences for most bryozoans detected by the morphological approach. However, NCBI has several entries for all these taxa (18S rRNA and COI regions). This suggests that amplification of bryozoan taxa failed possibly due to incompatible primer binding sites, primer affinity or degraded DNA within the bulk samples. Lejzerowicz, et al.15 targeted the same V4 18S rRNA region and also failed to identify bryozoans that were morphologically detected. Berry, et al.57 identified a similar issue with the COI mini-barcode, failing to detect most mollusc taxa. In the present study, the non-detection of some species using metabarcoding could also be due to the highly diverse communities found in the biofouling samples, as sensitivity and accuracy of metabarcoding has been shown to be influenced by taxon composition and abundance58,60. Collectively, the present results and those from similar studies indicate that the greatest and most complete biodiversity inventories will be obtained using a combined metabarcoding and morphological approach, which has the additional benefit of enabling cross-verification of specific detections, such as rare or invasive species of interest58.
In addition to exploring biodiversity, this study aimed to evaluate each approach for its ability to identify potential NIS. Considering only the molecular approaches, the lowest NIS detection was obtained when taxonomic assignment was undertaken using NCBI and the LCA algorithm alone. To avoid the low assignment rates using the LCA algorithm, we combined the taxonomical assignment for potential NIS retrieving direct best hits of at least 97% identity. With the exception of five algal genera uniquely identified using NCBI for 18S rRNA and COI, all other NIS were also identified when assignment was undertaken using the PR2 or BOLD databases (Table S1). It is usually anticipated that taxonomic resolution will be low when targeting the 18S rRNA compared to COI65. However, in this study the greatest detection of potential non-indigenous species (n = 14) occurred when using the 18S rRNA and PR2 database. These results should be treated with some caution due to the highly conserved nature of 18S that might result in assignment of an OTU to a closely related genus or species using less stringent algorithms such as UCLUST. However, a less stringent approach is still valid for flagging potential NIS. We recommend interrogating the flagged data more closely to assess the validity of the positive results prior to initiating further actions. For example, Asterias forbesi was identified only with 18S rRNA. A manual blast assigned it to the closely related species Asterias amurensis, the highly undesirable seastar, which is blacklisted in the World Register of Introduced Marine Species (WRIMS), and is not yet known to occur in New Zealand. However, these uncertain observations likely represent a false positive detection. The sequence was only present in low abundance (8 reads in 20 samples) and when using the NCBI database and the LCA algorithm for the taxonomy assignment, these sequences were assigned taxonomically only to the family level, i.e. ‘Asteroidea’. It is most likely that this detection is not A. forbesi, but that of a closely related taxon whose sequence is absent from current databases. Even in this case, this result is valuable in the marine surveillance context as it might be effectively used for triggering the application of a species-specific A. amurensis diagnostic test (e.g. real-time PCR) possibly confirming the presence of this unwanted organism in these samples. A further example of the challenges associated with NIS detection using metabarcoding is the invasive marine fanworm Sabella spallanzanii, which was detected by three approaches (18S rRNA [PR2], COI [BOLD] and morpho-taxonomic data) but not when taxonomy was assigned using NCBI. When the NCBI database was used, taxonomy could not be assigned below the genus level i.e. ‘Sabella’. Overall, these examples highlight the on-going need for the development of regionally specific reference databases, and the requirement for additional markers that provide accurate species-specific resolution. By contrast, both molecular markers identified the presence of the invasive taxa Arcuatula senhousia, Ciona savignyi and Amathia gracilis. However, they were not identified using morpho-taxonomy. As noted above this is most likely because they were in their larval stage, the metabarcoding detected extracellular DNA, or a closely related species was present and taxonomy was incorrectly assigned (e.g. Ciona intestinalis).
Rarefaction curves were used to explore how including additional approaches enhanced the detection of NIS. When the sequencing depth was kept similar among molecular data, the 18S rRNA gene resulted in the highest NIS detection, while metabarcoding of the COI and morpho-taxonomy performed equally. In the present study, 25% of NIS taxa identified at species level were present in both morphological and metabarcoding datasets, a result which is similar to the 20% shared by both methods in Lejzerowicz, et al.15. Eighty percent of NIS detections did not overlap between morphological and metabarcoding approaches, highlighting how using these approaches in parallel may enhance the detection of NIS. Interestingly, the number of NIS detected using metabarcoding did not reach diversity saturation in the winter samples. There is the possibility that some of the variation observed in this study between winter and summer datasets resulted from using different sampling techniques (swabbing versus scraping). It is also likely that the lower number of dominating macro-organisms in the winter settlement plates favored the colonization of a more diverse microbial assemblage among biofouling meta-communities39.
Conclusions
Bioinformatic pipelines are continually evolving and careful consideration must be given to the methodology applied, which should be based on the specific aims of the study. In the present investigation, we employed stringent clustering (99%) without denoising steps. Although this likely maintains some sequencing errors which may result in inflation of biodiversity values, and potentially the false positive detection of NIS, it increases the likelihood of NIS detection and the initiation of more stringent detection procedures. Further important considerations are the target gene and reference database used for taxonomic assignment. In this study, the taxonomy of the communities and levels of resolution varied markedly according to the gene and database used. Our results highlight the benefits of including at least two molecular markers when attempting to obtain a detailed overview of the diversity of highly complex marine biofouling communities. The results also highlight biases in each of the different databases and identification algorithms. Each method showed both strengths and weaknesses, suggesting that the most accurate results come from the use of a combination of methods. This allows for cross-validation and maximizes the coverage of NIS. While new markers are continually being developed and databases improved, caution should be applied when interpreting metabarcoding data due to primer bias and incomplete or incorrectly annotated references databases. This was evidenced in this study by the absence of most Bryozoa from the metabarcoding data, despite being highly abundant in morphological data, and the likely false detection such as the differing Asterias assignments. We therefore advocate for the use of metabarcoding as a screening method when aiming to detect NIS, with positive detections triggering the application of more targeted molecular methods or in-depth morphological analysis. The inclusion of data from the metabarcoding of two markers and morpho-taxonomy resulted in the highest number of potential NIS detections, suggesting that combining these methods will enhance marine biosecurity surveillance.
References
Ricciardi, A. et al. Invasion Science: A Horizon Scan of Emerging Challenges and Opportunities. Trends in Ecology & Evolution 32, 464–474, https://doi.org/10.1016/j.tree.2017.03.007 (2017).
Trebitz, A. S. et al. Early detection monitoring for aquatic non-indigenous species: Optimizing surveillance, incorporating advanced technologies, and identifying research needs. Journal of Environmental Management 202, 299–310, https://doi.org/10.1016/j.jenvman.2017.07.045 (2017).
Zaiko, A. et al. Metabarcoding approach for the ballast water surveillance-an advantageous solution or an awkward challenge? Marine Pollution Bulletin 92, 25–34, https://doi.org/10.1016/j.marpolbul.2015.01.008 (2015).
Seebens, H., Gastner, M. T. & Blasius, B. The risk of marine bioinvasion caused by global shipping. Ecology Letters 16, 782–790, https://doi.org/10.1111/ele.12111 (2013).
Borrell, Y. J., Miralles, L., Do Huu, H., Mohammed-Geba, K. & Garcia-Vazquez, E. DNA in a bottle-Rapid metabarcoding survey for early alerts of invasive species in ports. PLoS One 12, e0183347, https://doi.org/10.1371/journal.pone.0183347 (2017).
Olenin, S. et al. New arrivals: An indicator for non-indigenous species introductions at different geographical scales. Frontiers in Marine Science 3, https://doi.org/10.3389/fmars.2016.00208 (2016).
Dafforn, K. A. et al. Marine urbanization: an ecological framework for designing multifunctional artificial structures. Front. Ecol. Environ. 13, 82–90, https://doi.org/10.1890/140050 (2015).
Campbell, M. L., Gould, B. & Hewitt, C. L. Survey evaluations to assess marine bioinvasions. Marine Pollution Bulletin 55, 360–378, https://doi.org/10.1016/j.marpolbul.2007.01.015 (2007).
Chan, F. T., MacIsaac, H. J. & Bailey, S. A. Survival of ship biofouling assemblages during and after voyages to the Canadian Arctic. Marine Biology 163, https://doi.org/10.1007/s00227-016-3029-1 (2016).
Tait, L., Inglis, G., Seaward, K., Spong, K. & Wilkens, S. L. Optimising settlement arrays for surveillance of non-indigenous biofouling species. Results and recommendations following settlement array field trials. MPI Technical –Paper 70, 978-1-77665-425-3 (2016).
Abad, D. et al. Is metabarcoding suitable for estuarine plankton monitoring? A comparative study with microscopy. Marine Biology 163, https://doi.org/10.1007/s00227-016-2920-0 (2016).
Harvey, J. B. J., Hoy, M. S. & Rodriguez, R. J. Molecular detection of native and invasive marine invertebrate larvae present in ballast and open water environmental samples collected in Puget Sound. J Exp Mar Biol Ecol 369, 93–99, https://doi.org/10.1016/j.jembe.2008.10.030 (2009).
Baird, D. J. & Hajibabaei, M. Biomonitoring 2.0: a new paradigm in ecosystem assessment made possible by next-generation DNA sequencing. Mol. Ecol. 21, 2039–2044, https://doi.org/10.1111/j.1365-294X.2012.05519.x (2012).
Cowart, D. A. et al. Metabarcoding is powerful yet still blind: a comparative analysis of morphological and molecular surveys of seagrass communities. PLoS One 10, e0117562, https://doi.org/10.1371/journal.pone.0117562 (2015).
Lejzerowicz, F. et al. High-throughput sequencing and morphology perform equally well for benthic monitoring of marine ecosystems. Scientific Reports 5, https://doi.org/10.1038/srep13932 (2015).
Laroche, O. et al. First evaluation of foraminiferal metabarcoding for monitoring environmental impact from an offshore oil drilling site. Marine Environmental Research 120, 225–235, https://doi.org/10.1016/j.marenvres.2016.08.009 (2016).
Darling, J. A. & Piraino, S. MOLTOOLS: a workshop on “Molecular tools for monitoring marine invasive species”. Biological Invasions, 809–813, https://doi.org/10.1007/s10530-015-0855-x (2015).
Darling, J. A. & Mahon, A. R. From molecules to management: adopting DNA-based methods for monitoring biological invasions in aquatic environments. Environmental Research 111, 978–988, https://doi.org/10.1016/j.envres.2011.02.001 (2011).
Dowle, E. J., Pochon, X. J. C. B., Shearer, K. & Wood, S. A. Targeted gene enrichment and high-throughput sequencing for environmental biomonitoring: a case study using freshwater macroinvertebrates. Molecular Ecology Resources 16, 1240–1254, https://doi.org/10.1111/1755-0998.12488 (2016).
Pawlowski, J., Lejzerowicz, F., Apotheloz-Perret-Gentil, L., Visco, J. & Esling, P. Protist metabarcoding and environmental biomonitoring: Time for change. European Journal of Protistology 55, 12–25, https://doi.org/10.1016/j.ejop.2016.02.003 (2016).
Zhang, J., Kapli, P., Pavlidis, P. & Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29, 2869–2876, https://doi.org/10.1093/bioinformatics/btt499 (2013).
Pochon, X., Zaiko, A., Hopkins, G. A., Banks, J. C. & Wood, S. A. Early detection of eukaryotic communities from marine biofilm using high-throughput sequencing: an assessment of different sampling devices. Biofouling 31, 241–251, https://doi.org/10.1080/08927014.2015.1028923 (2015).
Zaiko, A. et al. Metabarcoding improves detection of eukaryotes from early biofouling communities: implications for pest monitoring and pathway management. Biofouling 32, 671–684, https://doi.org/10.1080/08927014.2016.1186165 (2016).
Darling, J. A. et al. Recommendations for developing and applying genetic tools to assess and manage biological invasions in marine ecosystems. Marine Policy 85, 54–64, https://doi.org/10.1016/j.marpol.2017.08.014 (2017).
Comtet, T., Sandionigi, A., Viard, F. & Casiraghi, M. DNA (meta)barcoding of biological invasions: a powerful tool to elucidate invasion processes and help managing aliens. Biological Invasions 17, 905–922, https://doi.org/10.1007/s10530-015-0854-y (2015).
Ficetola, G. F. et al. Replication levels, false presences and the estimation of the presence/absence from eDNA metabarcoding data. Molecular Ecology Resources 15, 543–556, https://doi.org/10.1111/1755-0998.12338 (2015).
Groendahl, S., Kahlert, M. & Fink, P. The best of both worlds: A combined approach for analyzing microalgal diversity via metabarcoding and morphology-based methods. PLoS One 12, e0172808, https://doi.org/10.1371/journal.pone.0172808 (2017).
Brown, E. A. et al. Early detection of aquatic invaders using metabarcoding reveals a high number of non-indigenous species in Canadian ports. Diversity and Distributions 22, 1045–1059, https://doi.org/10.1111/ddi.12465 (2016).
Guillou, L. et al. The Protist Ribosomal Reference database (PR2): a catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucleic Acids Research 41, D597–604, https://doi.org/10.1093/nar/gks1160 (2013).
Coordinators, N. R. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res 45, D12–D17, https://doi.org/10.1093/nar/gkw1071 (2017).
Lopez-Escardo, D. et al. Metabarcoding analysis on European coastal samples reveals new molecular metazoan diversity. Sci Rep 8, 9106, https://doi.org/10.1038/s41598-018-27509-8 (2018).
Xiong, W., Li, H. & Zhan, A. Early detection of invasive species in marine ecosystems using high-throughput sequencing: technical challenges and possible solutions. Marine Biology 163, https://doi.org/10.1007/s00227-016-2911-1 (2016).
Chesters, D., Zheng, W., Zhu, C. D. & Yu, D. W. A DNA Barcoding system integrating multigene sequence data. Methods in Ecology and Evolution 6, 930–937, https://doi.org/10.1111/2041-210x.12366 (2015).
Wangensteen, O. S., Palacín, C., Guardiola, M. & Turon, X. DNA metabarcoding of littoral hard-bottom communities: high diversity and database gaps revealed by two molecular markers. PeerJ 6, e4705, https://doi.org/10.7717/peerj.4705 (2018).
Scott, R. et al. Optimization and performance testing of a sequence processing pipeline applied to detection of nonindigenous species. Evolutionary Applications 11, 891–905, https://doi.org/10.1111/eva.12604 (2018).
Callahan, B. J., McMurdie, P. J. & Holmes, S. P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. The Multidisciplinary Journal of Microbial Ecology 11, 2639–2643, https://doi.org/10.1038/ismej.2017.119 (2017).
Deiner, K. et al. Environmental DNA metabarcoding: transforming how we survey animal and plant communities. Molecular Ecology 26, 5872–5895, https://doi.org/10.1111/mec.14350 (2017).
Tait, L. & Inglis, G. & Seaward, K. Enhancing passive sampling tools for detecting marine bioinvasions. Marine Pollution Bulletin 128, 41–50, https://doi.org/10.1016/j.marpolbul.2018.01.015 (2018).
von Ammon, U. et al. The impact of artificial surfaces on marine bacterial and eukaryotic biofouling assemblages: A high-throughput sequencing analysis. Marine Environmental Research 133, 57–66, https://doi.org/10.1016/j.marenvres.2017.12.003 (2018).
Zhan, A. et al. High sensitivity of 454 pyrosequencing for detection of rare species in aquatic communities. Methods in Ecology and Evolution 4, 558–565, https://doi.org/10.1111/2041-210x.12037 (2013).
Leray, M. et al. A new versatile primer set targeting a short fragment of the mitochondrial COI region for metabarcoding metazoan diversity: application for characterizing coral reef fish gut contents. Front. Zool. 10, 14, https://doi.org/10.1186/1742-9994-10-34 (2013).
Cox, M. P., Peterson, D. A. & Biggs, P. J. SolexaQA: At-a-glance quality assessment of Illumina second-generation sequencing data. BMC Bioinformatics 11, 485, https://doi.org/10.1186/1471-2105-11-485 (2010).
Caporaso, J. G. et al. QIIME allows analysis of highthroughput community sequencing data. Nature Methods 7, 335, https://doi.org/10.1038/nmeth0510-335 (2010).
Rognes, T., Flouri, T., Nichols, B., Quince, C. & Mahé, F. VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584, https://doi.org/10.7717/peerj.2584 (2016).
Machida, R. J., Leray, M., Ho, S. L. & Knowlton, N. Metazoan mitochondrial gene sequence reference datasets for taxonomic assignment of environmental samples. Sci Data 4, 170027, https://doi.org/10.1038/sdata.2017.27 (2017).
Ratnasingham, S. & Hebert, P. D. N. BOLD: The Barcode of Life Data System (www.barcodinglife.org). Molecular Ecology Notes 7, 355–364, https://doi.org/10.1111/j.1471-8286.2006.01678.x (2007).
Edgar, R. C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461, https://doi.org/10.1093/bioinformatics/btq461 (2010).
Morgulis, A. et al. Database indexing for production MegaBLAST searches. Bioinformatics 24, 1757–1764, https://doi.org/10.1093/bioinformatics/btn322 (2008).
Huson, D. H. et al. MEGAN community edition - Interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Computational Biology 12, e1004957, https://doi.org/10.1371/journal.pcbi.1004957 (2016).
McMurdie, P. J. & Holmes, S. phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8, e61217, https://doi.org/10.1371/journal.pone.0061217 (2013).
Seaward, K. et al. The Marine Biosecurity Porthole – a web-based information system on non-indigenous marine species in New Zealand. Management of Biological Invasions 6, 177–184, https://doi.org/10.3391/mbi.2015.6.2.08 (2015).
NZOR. The New Zealand Organisms Register (NZOR), http://nzor.org.nz (2011).
Larsson, J. Eulerr: Area-Proportional Euler and Venn Diagrams with Circles or Ellipses R package version 3.0.1 URL (2017).
Hsieh, T. C., Ma, K. H. & Chao, A. iNEXT: iNterpolation and EXTrapolation for species diversity. R package version 2.0.12 (2016).
Chao, A. et al. Rarefaction and extrapolation of phylogenetic diversity. Methods in Ecology and Evolution 6, 380–388, https://doi.org/10.1111/2041-210x.12247 (2015).
Zimmermann, J., Glockner, G., Jahn, R., Enke, N. & Gemeinholzer, B. Metabarcoding vs. morphological identification to assess diatom diversity in environmental studies. Molecular Ecology Resources 15, 526–542, https://doi.org/10.1111/1755-0998.12336 (2015).
Berry, O. et al. Comparison of morphological and DNA metabarcoding analyses of diets in exploited marine fishes. Marine Ecology Progress Series 540, 167–181, https://doi.org/10.3354/meps11524 (2015).
Hatzenbuhler, C., Kelly, J. R., Martinson, J., Okum, S. & Pilgrim, E. Sensitivity and accuracy of high-throughput metabarcoding methods for early detection of invasive fish species. Scientific Reports 7, 46393, https://doi.org/10.1038/srep46393 (2017).
Alberdi, A., Aizpurua, O., Gilbert, M. T. P., Bohmann, K. & Mahon, A. Scrutinizing key steps for reliable metabarcoding of environmental samples. Methods in Ecology and Evolution 9, 134–147, https://doi.org/10.1111/2041-210x.12849 (2018).
Leray, M. & Knowlton, N. Random sampling causes the low reproducibility of rare eukaryotic OTUs in Illumina COI metabarcoding. PeerJ 5, e3006, https://doi.org/10.7717/peerj.3006 (2017).
Wood, S. A., Zaiko, A., Richter, I., Inglis, G. J. & Pochon, X. Development of a real-time polymerase chain reaction assay for the detection of the invasive Mediterranean fanworm, Sabella spallanzanii, in environmental samples. Environmental Science and Pollution Research International 24, 17373–17382, https://doi.org/10.1007/s11356-017-9357-y (2017).
Edgar, R. C. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing, https://doi.org/10.1101/081257 (2016).
De Barba, M. et al. DNA metabarcoding multiplexing and validation of data accuracy for diet assessment: application to omnivorous diet. Molecular Ecology Resources 14, 306–323, https://doi.org/10.1111/1755-0998.12188 (2014).
Borrell, Y. J. et al. Metabarcoding and post-sampling strategies to discover non-indigenous species: A case study in the estuaries of the central south Bay of Biscay. Journal for Nature Conservation 42, 67–74, https://doi.org/10.1016/j.jnc.2017.07.002 (2018).
Deagle, B. E., Jarman, S. N., Coissac, E., Pompanon, F. & Taberlet, P. DNA metabarcoding and the cytochrome c oxidase subunit I marker: not a perfect match. Biology Letters 10, https://doi.org/10.1098/rsbl.2014.0562 (2014).
Bucklin, A., Steinke, D. & Blanco-Bercial, L. DNA Barcoding of Marine Metazoa. Annual Review of Marine Science 3, 471–508, https://doi.org/10.1146/annurev-marine-120308-080950 (2011).
Huson, D. H., Auch, A. F., Qi, J. & Schuster, S. C. MEGAN analysis of metagenomic data. Genome Research 17, 377–386, https://doi.org/10.1101/gr.5969107 (2007).
Fletcher, L. M. et al. Bilge water as a vector for the spread of marine pests: a morphological, metabarcoding and experimental assessment. Biological Invasions 19, 2851–2867, https://doi.org/10.1007/s10530-017-1489-y (2017).
Acknowledgements
We thank staff from Ministry for Primary Industries and here especially Eugene Georgiades for their valuable input during the preparation of this manuscript. We also thank the taxonomists Dennis Gordon, Mike Page, Geoff Read, Kimberley Seaward and Serena Cox for morphological identification of biofouling species. This work was funded by the New Zealand Ministry for Business, Innovation and Employment Strategic Science Investment Fund (SSIF) under NIWA Coasts and Oceans Research Programme 6, Marine Biosecurity (2017/18 SCI) and Ministry for Primary Industries’ Operational Research Project RFP16696 – Settlement Arrays.
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: U.V.A., S.W., A.Z., L.T., G.I. and X.P. Performed the experiments: U.V.A. and L.T. Analysed the data: U.V.A. and O.L. Contributed reagents/materials/analysis tools: U.V.A., S.W., O.L., A.Z., L.T., G.I. and X.P. Wrote the paper: U.V.A., S.W. and X.P. Reviewed manuscript drafts: U.V.A., S.W., O.L., A.Z., L.T., S.L., G.I. and X.P.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ammon, U.v., Wood, S.A., Laroche, O. et al. Combining morpho-taxonomy and metabarcoding enhances the detection of non-indigenous marine pests in biofouling communities. Sci Rep 8, 16290 (2018). https://doi.org/10.1038/s41598-018-34541-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-34541-1
Keywords
This article is cited by
-
VESPA: an optimized protocol for accurate metabarcoding-based characterization of vertebrate eukaryotic endosymbiont and parasite assemblages
Nature Communications (2024)
-
Harnessing environmental DNA to reveal biogeographical patterns of non-indigenous species for improved co-governance of the marine environment in Aotearoa New Zealand
Scientific Reports (2023)
-
Microbial community shift on artificial biological reef structures (ABRs) deployed in the South China Sea
Scientific Reports (2023)
-
eDNA metabarcoding of small plankton samples to detect fish larvae and their preys from Atlantic and Pacific waters
Scientific Reports (2021)
-
Monitoring of biofouling communities in a Portuguese port using a combined morphological and metabarcoding approach
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
