
Abstract
Plutonium and radiocaesium are hazardous contaminants released by the Fukushima Daiichi nuclear power plant (FDNPP) disaster and their distribution in the environment requires careful characterisation using isotopic information. Comprehensive spatial survey of 134Cs and 137Cs has been conducted on a regular basis since the accident, but the dataset for 135Cs/137Cs atom ratios and trace isotopic analysis of Pu remains limited because of analytical challenges. We have developed a combined chemical procedure to separate Pu and Cs for isotopic analysis of environmental samples from contaminated catchments. Ultra-trace analyses reveal a FDNPP Pu signature in environmental samples, some from further afield than previously reported. For two samples, we attribute the dominant source of Pu to Reactor Unit 3. We review the mechanisms responsible for an emergent spatial pattern in 134,135Cs/137Cs in areas northwest (high 134Cs/137Cs, low 135Cs/137Cs) and southwest (low 134Cs/137Cs, high 135Cs/137Cs) of FDNPP. Several samples exhibit consistent 134,135Cs/137Cs values that are significantly different from those deposited on plant specimens collected in previous works. A complex spatial pattern of Pu and Cs isotopic signature is apparent. To confidently attribute the sources of mixed fallout material, future studies must focus on analysis of individual FDNPP-derived particles.
Similar content being viewed by others

Introduction
The release of considerable quantities of radioactive material from the Fukushima Daiichi Nuclear Power Plant (FDNPP) accident in March 2011 contaminated large areas of the surrounding terrestrial environment. The dispersion and deposition of radioactive material were strongly influenced by the prevailing weather systems and a complex pattern of intermittent emission from multiple reactor units1. The meteorological conditions during emission periods are well defined, but there remains considerable uncertainty about the isotopic composition and release rates of radioactive contaminants from the various potential sources at FDNPP2.
Determining the spatial distribution of radionuclides can be used to infer the quantity of material released, its chemical composition and the environmental pathways. Additionally, subsequent temporal variation provides insight into the mechanisms that transfer or retain radionuclides of interest. Monitoring and understanding post-depositional remobilisation of potentially harmful radionuclides in the environment allows the risk they present to be managed more effectively. This information is highly valued, not only for decision-making processes regarding public health, but also the longer-term remediation efforts across the region and any future nuclear incidents that may occur.
Radiocaesium and Pu are recognised as two of the most significant percieved concerns from the disaster. The release of Pu to terrestrial areas of Japan remained unconfirmed for up to one year following the Fukushima disaster3. Since then, several studies have provided evidence of its release to the land surface north, northwest and south of FDNPP, up to distances of 120 km from the site3,4,5,6,7. It is estimated that only a fraction (~0.002%) of Pu from the total reactor core inventory was introduced to the environment3. For the data available, only trace quantities of FDNPP-derived Pu have been measured in environmental materials and mixing of the FDNPP Pu with trace amounts of Pu derived from atmospheric nuclear weapons testing complicates interpretation of the observed isotopic signature5,8. Heterogeneity in both isotopic ratio and specific activity of Pu in the vicinity of FDNPP has raised questions about the source and composition of contamination4,9. The low volatility of Pu and the incidence of hydrogen explosions suggests Pu may have been distributed in a particulate form. The introduction of particulate Pu to the environment raises concern because of the risk such material poses to public health10,11.
In contrast to Pu, Cs has high volatility. Consequently, an extremely large quantity of radiocaesium was released from FDNPP (9–37 PBq 137Cs) and is considered to be the principal source of radiation dose to the public2,12,13,14. Following the FDNPP accident, an intensive programme of 134Cs and 137Cs measurement was conducted to determine the spatial distribution of these isotopes15. Recently, 134Cs/137Cs data obtained since the accident has been combined with atmospheric transport models to identify the reactor units responsible for the highest dose rate areas16. The determination of 134Cs/137Cs activity ratio has been a useful tool within decontamination efforts, risk assessment and source term attribution following the FDNPP accident, but it is recognised that the additional measurement of 135Cs/137Cs atom ratio enables much better discrimination of the source terms6,17,18,19,20,21. While variations in 134Cs/137Cs activity ratio are observed due to changes in fuel burnup, 135Cs/137Cs atom ratios vary as a function of neutron capture by the fission product 135Xe, the parent nuclide of 135Cs. Concentrations of 135Xe change considerably according to reactor conditions22. Adding 135Cs to the suite of isotopic analyses is necessary because all three reactors were boiling-water reactors with similar fuels (Reactor Units 1 & 2 - UO2; Reactor Unit 3 - UO2/MOX) and operating conditions23. Also, mass-spectrometric determination of 135Cs/137Cs can achieve greater relative precisions20 and this signal will remain useful in environmental samples for much longer (134Cs t1/2 = 2.07 a, 135Cs t1/2 = 2.3 Ma, 137Cs t1/2 = 30.08 a).
Here, we explore the spatial pattern of the Pu and Cs isotopes in a variety of environmental samples to evaluate the source term, potential environmental pathways and influence of source mixing between FDNPP contamination and atmospheric fallout9,19,24,25. Critical, was the development of a new simultaneous procedure to separate both Cs and Pu fractions from the same environmental samples. Isotopic analyses were performed by thermal ionization mass spectrometry (TIMS) and multicollector inductively coupled plasma mass spectrometry (MC-ICPMS). We present 239+240Pu activities, 240Pu/239Pu atom ratios, 137Cs activity, 134Cs/137Cs activity ratios and 135Cs/137Cs atom ratios for samples collected from locations primarily in the Fukushima prefecture.
Results
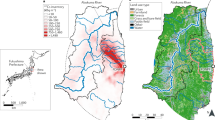
Isotopic analysis of Pu and Cs has been performed on range of environmental samples (soil, sediment and various types of vegetation), collected from several locations in the Fukushima prefecture, two from the Abukuma River and one sample from Chiba, near Tokyo, during May 2014 and October 2015 (Fig. 1; Tables S1 and S2). All environmental samples represent, to varying degrees, a combination of background signal, contamination during the FDNPP accident and also post-depositional migration. These factors complicate the interpretation of the spatial distributions of Pu and Cs. We recognise that the affinity of Pu and Cs may vary considerably between even the most similar types of environmental sample as a function of their respective physico-chemical properties. We broadly categorise sample types into vegetation and sediment for Fig. 1, but recognise that the two populations will exhibit overlap. Reported values of Pu and Cs activity (symbol size) and activity/atom ratios (symbol colour) are for the strong acid leach (8 M HNO3) fraction of ~1 g sub-samples. Therefore, these data are representative of the leachable fraction only and are not necessarily indicative of local radioactivity levels. We expect results from vegetation samples (moss, litter etc) to be biased to the more recent deposition of FDNPP contamination, whereas sediment and soil samples are expected to contain a greater proportion of background signal from the intensive period of atmospheric weapons testing during the 1950s and 1960s. All results presented are decay corrected to 11/03/2011.

Isoscapes of 134, 135, 137Cs and 239, 240Pu for part of the Fukushima prefecture surrounding FDNPP. The green marker is used to highlight an anomalous 240Pu/239Pu atom ratio of 0.64. R1, R2 and R3 correspond to ORIGEN estimated isotope ratio values for Reactor Units 1, 2 and 3, respectively27. SW indicates the mean value for the Cs isotope ratios measured to the southwest of FDNPP by Snow et al.19. 240Pu/239Pu atom ratio for Northern Hemisphere integrated global fallout is denoted by NHF28.
Specific activity of 239+240Pu ranges from 0.012 ± 0.005 Bq kg−1 in coarse sediment from Minamisoma to 0.761 ± 0.182 Bq kg−1 for a soil sample collected from Katsurao. A sample from Iitate Village exhibited a high 239+240Pu activity of 0.757 ± 0.079 Bq kg−1, while the remaining samples were ≤0.2 Bq kg−1. These 239+240Pu specific activities are similar to baseline values (0.004–1.46 Bq kg−1) obtained from samples collected across Japan prior to the Fukushima disaster26. All 240Pu/239Pu atom ratios lie between the integrated Northern Hemisphere global fallout value (0.180 ± 0.014) and the ORIGEN estimated value for Reactor Unit 3 (0.356)27,28, except one anomaly (Iitate), 0.645 ± 0.024.
Except for two samples collected from Date City, the data obtained for 135Cs/137Cs atom ratios and 134Cs/137Cs activity ratios fall within the range of ORIGEN calculated values for reactor cores of Units 1–3. The 137Cs activities in these samples range from 1.2–1040 Bq g−1.
Discussion
Of 22 samples analysed in this study, 15 exhibit a 240Pu/239Pu atom ratio greater than the upper limit of the values reported for pre-FDNPP soils collected from central-eastern Japan in the 1970s26. While this is a higher fraction than observed in other recent studies3,4,25,29 direct comparisons should avoided because of differences in experimental protocol, i.e. sample type, locations and chemical treatment (leachable fraction). Some of our data illustrates that FDNPP-derived Pu is found in environmental samples collected considerably further afield than previously reported4,7: Two of the furthest study sites from FDNPP, Date and Minamiosawa (>50 km direct distance), exhibit FDNPP-derived 240Pu/239Pu atom ratios of 0.323 ± 0.021 and 0.324 ± 0.029, respectively. Previous efforts to assess the presence of FDNPP-derived Pu in environmental samples >45 km have been compromised by either limits of detection or by material being derived of Pu predominately sourced from weapons testing fallout7. While the 239+240Pu activity for all of the samples remains similar or lower than typical global fallout measurements for the region (0.5–1.5 Bq kg−1)30, such a heterogeneous spatial distribution of 240Pu/239Pu indicates that particulates have been transported further afield from FDNPP than was previously reported.
There is no apparent relationship between 239+240Pu activity and 240Pu/239Pu atom ratio in Fig. 1. Indeed, the two samples with highest 239+240Pu activities exhibit end-member values of 240Pu/239Pu atom ratios representing weapons testing fallout and FDNNP reactor cores; 240Pu/239Pu = 0.165 ± 0.003 and 240Pu/239Pu = 0.359 ± 0.013, respectively. We also find a poor relationship between distance from the FDNPP site and Pu isotopic data. One might expect to observe higher 239+240Pu activities and 240Pu/239Pu atom ratios similar to the ORIGEN estimated values (0.320–0.356) at sites closer to FDNPP, but this is not the case. The non-uniform spatial distribution of Pu isotope data is in agreement with previous work3,4,25 and may be a product of one or a combination of two possibilities: (1) contamination during FDNPP hydrogen explosions4, which may have ejected fuel particles with an uneven size distribution and variable dispersion pathway to the surrounding environment31; (2) non-uniform pattern of background, pre-FDNPP, Pu. For the latter case, because FDNPP Pu concentations are similar to background levels, the observed spatial distribution of Pu isotopes may be caused by different degrees of mixing with a non-uniform pre-FDNPP Pu concentration and the range of Pu-affinity for different sample types studied here and elsewhere4,9,25,29. The heterogeneous signal observed in bulk leach analysis of environmental samples demands further attention, with more focus on sophisticated particle characterization techniques.
Due to the limitations associated with bulk analysis, it is challenging to identify potential source terms that are end members of mixing combinations. For FDNPP-derived Pu in environmental materials, according to the ORIGEN estimated reactor values, the end-members of the 240Pu/239Pu mixing spectrum are global fallout (240Pu/239Pu atom ratio = 0.180 ± 0.014) and Reactor Unit 3 (240Pu/239Pu atom ratio = 0.356). It is not possible to identify Pu sourced from Reactor Units 1 (240Pu/239Pu atom ratio = 0.344) or 2 (240Pu/239Pu atom ratio = 0.320) because any 240Pu/239Pu atom ratios that correspond to these values would appear as a mixture of Pu from Reactor Unit 3 and global fallout27,28,32. Two moss samples collected from Iitate Village and Okuma exhibit 240Pu/239Pu atom ratios corresponding to Pu sourced from Reactor Unit 3 (Fig. 2) and are distinguishable from the average ORIGEN value of Reactor Unit 127. This is the first report of Pu isotope ratios for environmental samples that correspond to a specific FDNPP reactor unit, but it is not possible to statistically evaluate this observation because no uncertainties are reported for the ORIGEN estimations27. There also remains some possibility that the 240Pu/239Pu atom ratios associated with these mosses may be a mixture of higher 240Pu/239Pu atom ratios from spent fuel ponds (SFPs) that have mixed with lower 240Pu/239Pu atom ratios such as global fallout. However, this scenario is unlikely considering SFP-derived Pu has not been observed in this work or in any other studies of FDNPP-derived Pu in the environment. Furthermore, it should be highlighted that both samples are mosses and highly unlikely to retain background Pu sourced from atmospheric fallout4.

240Pu/239Pu atom ratios for moss samples from Okuma and Iitate village (2SE), FDNPP Reactor Units27 and Northern Hemisphere integrated global fallout (NHF).
A moss sample from Iitate Village exhibited the only 240Pu/239Pu atom ratio that did not align with either global fallout or the estimated ORIGEN model simulations (0.645 ± 0.045). A similar mean value was obtained for a vegetation sample collected from Odaka, Minamisoma (0.64 ± 0.37 (1σ)) by Schneider et al.4, but the relatively large uncertainty associated with this value indicates that it is not statistically different from the ORIGEN estimations. If material with this high 240Pu/239Pu atom ratio was sourced from FDNPP, it implies a higher fuel burn-up than otherwise estimated. A pressure-tube boiling-water reactor (RBMK) is the only reactor type reported in the literature that yields a 240Pu/239Pu atom ratio (0.67) corresponding to this anomalous value33,34. Extremely large quantities of radionuclides were released to the environment from a RBMK reactor during the Chernobyl accident in 1986. A recent study indicates that the more volatile Chernobyl-derived radionuclides contribute a significant propotion of background radiation levels in Japan24. However, 240Pu/239Pu atom ratios measured in soil samples collected after the Chernobyl accident, but before the FDNPP accident, were not indicative of Chernobyl-derived Pu26. Furthermore, 240Pu/239Pu atom ratios measured in environmental samples contaminated by the Chernobyl accident were found to be <0.6035,36,37,38. These observations infer that the anomalously high 240Pu/239Pu atom ratio measured here is unlikely to have been sourced from the Chernobyl disaster. Considering the complexity of the reactor conditions during the accident at FDNPP, and potential heterogeneity in 240Pu/239Pu atom ratios through partially-spent fuel, it is feasible that such conditions may have yielded a higher fuel burn-up. It should also be highlighted that the 241Pu/239Pu atom ratios observed elsewhere also point towards a high fuel burn-up3,29,39. Information regarding fuel burn-up can be related to accident mechanisms and is particularly valuable where high radiation levels prevent investigation of damaged reactors.
In order to discriminate potential source terms for the volatile Cs contamination, we supplement existing 134,135Cs/137Cs values and 137Cs specific activities for environmental samples from the region using TIMS and SF-ICPMS. Key to any interpretation of the Cs isoscape revealed in Fig. 1 is consideration of the competing influence of background signal prior to the accident and the extent of the mixing between this and one or more sources of Cs since the accident. Activity and isotopic composition before and after the event are required to deconvolve the source term and vector of transport. In addition to the isoscapes, we take advantage of a bivariate plot (Fig. 3) where 134,135Cs/137Cs data from this study are compared with data from Snow et al.19, which comprises 11 measurements of 5 environmental samples (1 soil, 4 vegetation) collected 100 to 250 km southwest of FDNPP19.

135Cs/137Cs atom ratios and 134Cs/137Cs activity ratios obtained in this study (samples collected northwest of FDNPP, red shading) and by Snow et al.19 (samples collected southwest of FDNPP, blue shading). Frequency distributions for 135Cs/137Cs atom ratios and 134Cs/137Cs activity ratios, respectively also illustrated. The larger diamond symbols correspond to the ORIGEN estimated reactor values27. Uncertainties are estimated to 2SE.
For sediments and vegetation samples obtained to the northwest of FDNPP, values for both 134Cs/137Cs and 135Cs/137Cs were obtained: 1.03–1.08 and 0.31–0.36, respectively. These data differ markedly from those established for sample material obtained from the southwest of FDNPP: 0.98–1.00 (134Cs/137Cs) and 0.37–0.39 (135Cs/137Cs) (Fig. 3)19. As mentioned previously, this could be a function of relative differences in global fallout contribution and/or mixing between one or more FDNPP source terms (fuel burn up and reactor conditions).
First, we consider the potential contribution of atmospheric fallout radiocaesium to the spatial pattern post-FDNPP. Atmospheric nuclear weapons tests conducted during the 1950s and 1960s and the Chernobyl disaster in 1986 have left a legacy of background radiocaesium throughout the Japanese terrestrial environment24. Remaining 134Cs (t1/2 = 2.07 a) from these releases is now negliglble, while 137Cs (t1/2 = 30.08 a) has decayed by a factor of <0.5 (Chernobyl) or <0.25 (atmospheric weapons testing) of the original concentrations. With a half-life of 2.3 × 106 a, 135Cs totals remain effectively the same. In comparison to the releases from Reactor Units at FDNPP, these older releases had higher 135Cs/137Cs atom ratios (0.48–0.59 for Chernobyl fallout, 3.3–4.2 for atmospheric weapons testing; values decay corrected to 11/03/2011)40. A greater relative abundance of background radiocaesium will therefore increase 135Cs/137Cs atom ratios and decrease 134Cs/137Cs activity ratios, while larger fractions of FDNPP radiocaesium will have the opposite effect. The areas northwest of FDNPP received considerably more 137Cs contamination in comparison to the southwest15. The higher 135Cs/137Cs and lower 134Cs/137Cs in the southwest relative to the northwest may therefore be an indication of a relatively greater contribution from global fallout material.
Recently, Yang et al.24 analysed heavily-contaminated leaves to obtain isotope ratios for FDNPP radiocaesium that were expected to be unaffected by fallout. A mean 134Cs/137Cs activity ratio (1.033 ± 0.006) and 135Cs/137Cs atom ratio (0.334 ± 0.005) was recommended for all contamination from FDNPP. Subsequently, these values were used to estimate 135Cs activity in environmental samples prior to the FDNPP disaster24. For the data obtained in this study, the 135Cs/137Cs atom ratio for the highest activity vegetation sample (Katsurao moss = 1041 Bq g−1) differs from the Yang et al.24 recommended value by 6%. To attribute this variation solely to residual fallout material would require an inconceivable quantity of background radiocaesium. While we accept the quantity of fallout material may vary between different samples, it should be recognized that this may not be the only factor responsible for variation in 134, 135Cs/137Cs.
High activity samples from Katsurao and Minamisoma exhibit 135Cs/137Cs and 134Cs/137Cs are statistically indistinguishable (Table 1), as are samples from Okuma and Minamisoma. While it is possible that lower activity samples may have been shifted to equivalent residual fallout values, as discussed, it would require an inconceivable quantity of fallout 135Cs to do the same for higher activity samples. It is also highly unlikely that appropriate quantities of global fallout material have been retained in these environmental samples to coincidently yield equivalent 135Cs/137Cs and 134Cs/137Cs. We therefore conclude that these values are representative of FDNPP-derived radiocaesium and have not been significantly perturbed by global fallout material. Notably, these isotope ratios are in agreement with the ORIGEN estimated value for Reactor Unit 3. However, the radiocaesium isotope ratio values for Reactor Unit 3 fall in-between those of Reactor Units 1 and 2. Therefore, it is not possible to rule out a scenario where mixing between Reactor Unit 1 and 2 generated these isotope ratios. Furthermore, as is the case for Pu, the Cs isotope ORIGEN values should be used with some caution given that they correspond to a mean value with no reported uncertainty. It is also interesting to note that the difference between the signature proposed here and that presented in Yang et al.24 occurs even though the samples from the respective studies were collected from locations within relatively close proximity to one another: the highest activity samples measured in Yang et al.24 were collected from Namie Town and Minamisoma, while the highest activity samples in this work were collected from Minamisoma and Katsurao. This indicates variability in the isotopic composition of radiocaesium in a more localized manner than the proposed northwest-southwest trend. Further work is required to explore this further, ideally on material that does not contain fallout material such as isolated FDNPP particles.
The issue of global fallout inhibiting interpretation of isotopic ratios in environmental samples highlights the requirement for more sophisticated particle characterisation techniques. For radiocaesium, we recommend that isotopic analysis should be performed on a suite of FDNPP fallout material ranging from sub-millimeter heterogenous agglomerations41 to smaller micron-sized spherical Cs-rich particles42,43. Analysis of material collected on atmospheric sampling filters is also expected to provide Cs isotope ratios that are not influenced by global fallout material.
In addition to the effects of global fallout material, mixing between FDNPP source terms is also expected to be a cause of spatial trends in the isotopic composition of radiocaesium. The contributing factors for this are a function of several variables:
-
Distinctive 134Cs/137Cs and 135Cs/137Cs associated with each of the FDNPP Reactor Units 1–327,32. In addition, 135Cs/137Cs atom ratios are expected to vary through reactor cores, therefore individual reactor cores may have emitted inconsistent 135Cs/137Cs44.
-
The Reactor Units released material in variable quantities between 12/03/2011 and 23/03/20111: For total 137Cs release by FDNPP, reported contributions for Reactor Units 1, 2 and 3 are 3.18%, 91.5% and 4.64%, respectively24.
-
Upon release, transportation of volatile radionuclides such as radiocaesium was defined by wind direction, while the predominant cause of deposition was precipitation1.
It is the interaction between these processes that governed spatial distribution of radiocaesium isotopes in the environment: The location of radiocaesium contamination in the environment with a specific isotopic signature will reflect the proportional amounts of material emitted from the respective reactors, their corresponding isotopic composition, the wind direction that coincides with the time of release and the location of the plume when rainout occurred. The location of rainout is reported to be the cause of highest radiocaesium deposition1,45,46. There were two plumes released from FDNPP that were affected by precipitation:
-
1)
An explosion in Reactor Unit 2 on the morning of 15/03/2011 released a plume of contamination that was subsequently transported north-northwest by prevailing wind. Rainout occurred during the afternoon of 15/03/2011 and continued until the morning of 16/03/20111,23. These events are acknowledged to be the cause of the high 137Cs activities monitored to the northwest of FDNPP19.
-
2)
On 20/03/2011 a release from Reactor Unit 1 was transported south with a plume previously emitted from Reactor Unit 1 (Plume 1) on 12/03/2011. Prevailing winds carried the contamination southwards to the Tokyo Metropolitan Area when precipitation occurred1.
The contamination associated with these deposition events is a mixture of radiocaesium sourced from Reactor Units 1, 2 and 3. However, according to information obtained from air pollution monitoring stations it is apparent that the material in the northwest is predominantly sourced from Reactor Unit 2, while the radiocaesium deposited in the south is reported to consist of a comparatively larger fraction of material from Reactor Unit 11. We see that this is reflected in the radiocaesium isotope ratio data: samples collected northwest of FDNPP 134Cs/137Cs and 135Cs/137Cs are weighted towards Reactor Units 2 and 3 and samples from south of FDNPP appear to contain more radiocaesium from Reactor Unit 1 (Fig. 3). Contrary to these trends however, are environmental samples from Tsukuba (135Cs/137Cs atom ratio = 0.341 ± 0.016) and Kamagaya (135Cs/137Cs atom ratio = 0.355 ± 0.0020), located 140 km and 230 km southwest of FDNPP respectively47. These values are in closer agreement with the samples collected northwest of FDNPP in this study than those from the southwest19. Considering Reactor Units 1, 2 and 3 all contributed to multiple plumes that may have undergone multiple release events1, one might expect to observe this wider spectrum of Cs isotope ratio values. Such observations again highlight the requirement for a larger data set with respect to 134, 135Cs/137Cs particularly for southwest of FDNPP and provide yet another example of the complexity of the FDNPP Cs isoscape.
It is evident that both global fallout and the interplay between reactor release chronology and meteorological conditions have contributed to the observed spatial trends in isotopic signature of radiocaesium. The effect of global fallout has previously been confirmed and is certainly responsible for some of the variation in 134,135Cs/137Cs24. However, the work conducted here, combined with data and insights from elsewhere, indicates that the influence of reactor emission chronology in combination with prevailing meteorological conditions is the more dominant factor responsible for the lower 135Cs/137Cs and higher 134Cs/137Cs in the northwest relative to southwest FDNPP6,17,18,19,24. Isotopic analyses of isolated radiocaesium-rich particulates in environmental materials and air filters that captured FDNPP contamination during different periods is required to improve our understanding of these processes. Constraining the spatial distribution of the isotopic signature of radiocaesium facilitates the tracing of post-depositional migration of these radionuclides through the environment, which is key for remediation efforts and public health. The use of 134Cs/137Cs activity ratios in combination with the more sensitive 135Cs/137Cs atom ratios demonstrates how these techniques may be used to explore the transport and behavior of radiocaesium through the environment.
Methods
Sampling
A range of environmental samples (soil, sediment and various types of vegetation), were collected from several locations in the Fukushima prefecture, the mouth of the Abukuma River and one sample from the Chiba prefecture during 2014 and 2015. A joint sampling effort was conducted by the University of Bristol and Kyoto University. Sample descriptions are available in supplementary information (Table S1).
Analytical procedures
A portion of dried sample (~1 g) was dry-ashed at 450 °C for 12 hours before being transferred to a polytetrafluoroethylene vial and spiked with ~1 mBq 242Pu (IRMM-085, Certified Spike Isotopic Reference Material, Institute of Reference Materials and Methods, Brussels, Belgium). Samples were refluxed overnight at 165 °C using 8 M HNO3 (all reagents used were analytical grade or higher purity). Particulates were separated from the leachate by centrifugation at 4000 rpm for 10 minutes. Supernatant was dried down and re-dissolved in 10 ml 8 M HNO3 − 0.2 M NaNO2 and heated at 90 °C for 20 minutes. BioRad AG1-X8 Cl− form anion exchange resin (~1 ml) was placed into a column with dimensions of 30 × 80 mm and pre-conditioned with 10 ml 8 M HNO3. Sample was loaded onto the column in two 5 ml aliquots, the two aliquots, containing species with no affinity for the resin, were collected and kept for Cs separation. The columns were subsequently rinsed with 10 ml 3 M HNO3, 10 ml 9 M HCl and eluted with 12 ml 9 M HCl-0.1 M NH4I. The eluent was gently dried down and the precipitate was oxidised by refluxing with 1 ml concentrated (69% v/v) HNO3 and 1 ml concentrated H2O2 (30% v/v) overnight at 165 °C. The sample was adjusted to 2 ml 0.6 M HNO3 for analysis by MC-ICPMS.
Plutonium isotopic measurements were conducted using a Neptune multi-collector inductively coupled plasma mass spectrometer (MC-ICPMS) (Thermo Fisher Scientific Inc., MA, USA), at the Bristol Isotope Group, University of Bristol. An Aridus sample introduction system (CETAC Technologies Inc., NE, USA) was connected to a microflow PFA-50 nebuliser for sample uptake. The instrument was tuned daily using a 30 ppb natural U standard (CRM U112a). All analyses were performed in low-resolution mode (m/Δm ≈ 400). Plutonium isotopes (239, 240, 242Pu) were measured via peak-jumping mode using the axial secondary electron multiplier (SEM). 238U+ was simultaneously monitored by Faraday cups L1, L2, L3 (Table S3). SEM dark noise remained at <3 cpm. The SEM sits behind a retarding potential quadrupole lens (RPQ), which is a highly selective filter that removes ions with a disturbed energy or trajectory improving abundance sensitivity. In the case of Pu, the RPQ is particularly advantageous for reducing tailing effects from large 238U+ beams on 239Pu.
Uranium decontamination factors for the chemical separation procedure for certified reference material IAEA-385 (estuarine sediment) were 1.5 × 103 (n = 5). The procedure gave a chemical yield of approximately 80%. For Pu measurements by MC-ICPMS, residual 238U may form a hydride (238UH+) in the interface region of the instrument and interfere at m/z 239. Tailing effects from 238U+ may also interfere at m/z 239. To account for this, a solution of CRM U112a yielding 8 V (≈5 × 108 cps) was monitored periodically during each measurement sequence. (238UH+ + Utail)/238U+ was typically 1 × 10−6 and the 238U+ intensity for the FDNPP Pu samples ranged from 100–300 mV. Repeat measurements of CRM U112a were also performed throughout the measurement sequence to correct for instrumental mass bias.
Repeat measurements of certified reference materials IAEA-367, IAEA-384 and IAEA-385 validate the accuracy and precision of this methodology for the determination of 239+240Pu activity and 240Pu/239Pu atom ratios in complex environmental materials (Tables S4–S7). Sub-samples of IAEA-385 were processed alongside FDNPP samples to ensure analytical proficiency was upheld.
Caesium was successfully separated from the retained solution using a double cation exchange method with ammonium molybdophosphate-polyacrylonitrile (AMP-PAN). Purified Cs was loaded onto a single standard-purity Re filament with glucose activator for analysis by TIMS. This method is described in Dunne et al.20.
To determine 137Cs activity of the leachate, 137Cs/133Cs atom ratios measured by TIMS were combined with 133Cs concentration measurements of a fraction of the initial leachate. The 133Cs concentration was determined using a Thermo Element 2XR sector field-inductively coupled plasma mass spectrometer (SF-ICPMS). All samples were introduced in 2 ml 0.6 M HNO3 using a PFA nebuliser (ESI Scientific, Omaha, NE) and a quartz spray chamber introduction system. Measurements were performed in low resolution mode as there are no significant interferences at m/z 133. Analytical grade 133Cs standards (1 g ml−1) were diluted accordingly and used as concentration standards (Sigma-Aldrich, Steinheim, Germany). Concentrations are derived from calibration curves (n = 5), spanning the range of beam intensities produced by the respective samples (Table S8).
Data Availability
All data generated in this study are included in Supplementary Information.
References
Tsuruta, H., Oura, Y., Ebihara, M., Ohara, T. & Nakajima, T. First retrieval of hourly atmospheric radionuclides just after the Fukushima accident by analyzing filter-tapes of operational air pollution monitoring stations. Sci. Rep. 4, 6717 (2014).
United Nations Scientific Committee on the effects of atomic radiation (UNSCEAR). Sources, effects and risks of ionizing radiation: United Nations Scientific Committee on the effects of atomic radiation 2013 Report, Vol 1 (available from, http://www.unscear.org/docs/reports/2013/13-85418_Report_2013_Annex_A.pdf, last access Jan 2018) (2013).
Zheng, J. et al. Isotopic evidence of plutonium release into the environment from the Fukushima DNPP accident. Sci. Rep. 2, 304 (2012).
Schneider, S. et al. Plutonium release from Fukushima Daiichi fosters the need for more detailed investigations. Sci. Rep. 3, 2988 (2013).
Shinonaga, T., Steier, P., Lagos, M. & Ohkura, T. Airborne plutonium and non-natural uranium from the Fukushima DNPP found at 120 km distance a few days after reactor hydrogen explosions. Environ. Sci. Technol. 48, 3808–3814 (2014).
Shibahara, Y. et al. Analysis of cesium isotope compositions in environmental samples by thermal ionization mass spectrometry – 3. Measurement of isotopic ratios in soil samples obtained in Fukushima prefecture. J. Nucl. Sci. Technol. 51, 575–579 (2016).
Yamamoto, M. et al. Isotopic Pu, Am and Cm signatures in environmental samples contaminated by the Fukushima Dai-ichi Nuclear Power Plant accident. J. Environ. Radioact. 132, 31–46 (2014).
Yang, G., Tazoe, H., Hayano, K., Okayama, K. & Yamada, M. Isotopic compositions of 236U, 239Pu, and 240Pu in soil contaminated by the Fukushima Daiichi Nuclear Power Plant accident. Sci. Rep. 7, 13619 (2017).
Zheng, J., Tagami, K. & Uchida, S. Release of plutonium isotopes into the environment from the fukushima daiichi nuclear power plant accident: what is known and what needs to be known. Environ. Sci. Technol. 47, 9584–95 (2013).
IAEA. The Fukushima Daiichi accident, Technical Volume 1/5, description and context of the accident (available from, http://www-pub.iaea.org/MTCD/Publications/PDF/AdditionalVolumes/P1710/Pub1710-TV1-Web.pdf, last access Jan 2018) (2015).
Eriksson, M., Lindahl, P., Roos, P., Dahlgaard, H. & Holm, E. U, Pu, and Am nuclear signatures of the Thule hydrogen bomb debris. Environ. Sci. Technol. 42, 4717–22 (2008).
Stohl, A. et al. Xenon-133 and caesium-137 releases into the atmosphere from the Fukushima Dai-ichi nuclear power plant: Determination of the source term, atmospheric dispersion, and deposition. Atmos. Chem. Phys. 12, 2313–2343 (2012).
Chino, M. et al. Preliminary estimation of release amounts of 131I and 137Cs accidentally discharged from the Fukushima Daiichi Nuclear Power Plant into the atmosphere. J. Nucl. Sci. Technol. 48, 1129–1134 (2011).
Morino, Y., Ohara, T. & Nishizawa, M. Atmospheric behavior, deposition, and budget of radioactive materials from the Fukushima Daiichi nuclear power plant in March 2011. Geophys. Res. Lett. 38, 1–7 (2011).
Ministry of Education Culture, Sports, Science and Technology (MEXT). Preparation of distribution map of radiation doses, etc. (Map of Radioactive Cesium Concentration in Soil) by MEXT (available from: http://radioactivity.nsr.go.jp/en/contents/5000S/4165/24/1750_083014.pdf, last access Jan 2018) (2011).
Chino, M. et al. Utilization of 134Cs/137Cs in the environment to identify the reactor units that caused atmospheric releases during the Fukushima Daiichi accident. Sci. Rep. 6, 31376 (2016).
Zheng, J. et al. 135Cs/137Cs isotopic ratio as a new tracer of radiocesium released from the Fukushima nuclear accident. Environ. Sci. Technol. 48, 5433–8 (2014).
Shibahara, Y. et al. Analysis of cesium isotope compositions in environmental samples by thermal ionization mass spectrometry – 1. A preliminary study for source analysis of radioactive contamination in Fukushima prefecture. J. Nucl. Sci. Technol. 51, 575–579 (2014).
Snow, M. S., Snyder, D. C. & Delmore, J. E. Fukushima Daiichi reactor source term attribution using cesium isotope ratios from contaminated environmental samples. Rapid Commun. Mass Spectrom. 30, 523–532 (2016).
Dunne, J. A., Richards, D. A. & Chen, H. W. Procedures for precise measurements of 135Cs/137Cs atom ratios in environmental samples at extreme dynamic ranges and ultra-trace levels by thermal ionization mass spectrometry. Talanta 174, 347–356 (2017).
Ohno, T. & Muramatsu, Y. Determination of radioactive cesium isotope ratios by triple quadrupole ICP-MS and its application to rainwater following the Fukushima Daiichi Nuclear Power Plant accident. J. Anal. At. Spectrom. 29, 347–351 (2014).
Hayes, A. C. & Jungman, G. Determining reactor flux from xenon-136 and cesium-135 in spent fuel. Nucl. Instruments Methods Phys. Res. Sect. A Accel. Spectrometers, Detect. Assoc. Equip. 690, 68–74 (2012).
Tokyo Electric Power Company Inc. Fukushima nuclear accidents investigation report: Main body (summary) (available from: http://www.tepco.co.jp/en/press/corp-com/release/betu12_e/images/120620e0102.pdf, last access Jan 2018) (2012).
Yang, G., Tazoe, H. & Yamada, M. 135Cs activity and 135Cs/137Cs atom ratio in environmental samples before and after the Fukushima Daiichi Nuclear Power Plant accident. Sci. Rep. 6, (2016).
Schneider, S. et al. Radionuclide pollution inside the Fukushima Daiichi exclusion zone, part 2: Forensic search for the ‘Forgotten’ contaminants Uranium-236 and plutonium. Appl. Geochemistry (2017).
Yang, G., Zheng, J., Tagami, K. & Uchida, S. Plutonium concentration and isotopic ratio in soil samples from central-eastern Japan collected around the 1970s. Sci. Rep. 5, 9636 (2015).
Nishihara, K., Iwamoto, H. & Suyama, K. Estimation of fuel compositions in Fukushima-Daiichi Nuclear Power Plant. JAEA-Data/ Code 2012-018, 65–117 (2012).
Kelley, J. M., Bond, L. A. & Beasley, T. M. Global distribution of Pu isotopes and 237Np. Sci. Total Environ. 237-238, 483–500 (1999).
Shibahara, Y. et al. Determination of isotopic ratios of plutonium and uranium in soil samples by thermal ionization mass spectrometry. J. Radioanal. Nucl. Chem. 307, 2281–2287 (2016).
Hardy, E. P., Krey, P. W. & Volchok, H. L. Global inventory and distribution on fallout plutonium. Nature 241, 444–445 (1973).
Kirchner, G., Bossew, P. & De Cort, M. Radioactivity from Fukushima Dai-ichi in air over Europe; part 2: What can it tell us about the accident? J. Environ. Radioact. 114, 35–40 (2012).
Nishihara, K. et al. Radionuclide release to stagnant water in the Fukushima-1 nuclear power plant 1. J. Nucl. Sci. Technol. 52, 1–7 (2014).
Warneke, T., Croudace, I. W., Warwick, P. E. & Taylor, R. N. A new ground-level fallout record of uranium and plutonium isotopes for northern temperate latitudes. Earth Planet. Sci. Lett. 203, 1047–1057 (2002).
Carlson, J., Bardsley, J., Bragin, V. & Hill, J. Plutonium isotopics – non proliferation and safeguards issues, Australian Safeguards Office, Report IAEA-SM-351/64 (1988).
Salminen-Paatero, S., Nygren, U. & Paatero, J. 240Pu/239Pu mass ratio in environmental samples in Finland. J. Environ. Radioact. 113, 163–70 (2012).
Jakopič, R., Richter, S., Kühn, H. & Aregbe, Y. Determination of 240Pu/239Pu, 241Pu/239Pu and 242Pu/239Pu isotope ratios in environmental reference materials and samples from Chernobyl by thermal ionization mass spectrometry (TIMS) and filament carburization. J. Anal. At. Spectrom. 25, 815–821 (2010).
Boulyga, S. F. & Becker, J. S. Isotopic analysis of uranium and plutonium using ICP-MS and estimation of burn-up of spent uranium in contaminated environmental samples. J. Anal. At. Spectrom. 17, 1143–1147 (2002).
Ketterer, M. E., Hafer, K. M. & Mietelski, J. W. Resolving Chernobyl vs. global fallout contributions in soils from Poland using plutonium atom ratios measured by inductively coupled plasma mass spectrometry. J. Environ. Radioact. 73, 183–201 (2004).
Evrard, O. et al. Novel insights into Fukushima nuclear accident from isotopic evidence of plutonium spread along coastal rivers. Environ. Sci. Technol. 48, 9334–9340 (2014).
Chen, H., Lee, T., Ku, T. & Das, J. P. Production ratio of nuclear fallout 137Cs/135Cs. Chinese J. Phys. 46, 560–569 (2008).
Martin, P. G. et al. In-situ removal and characterisation of uranium-containing particles from sediments surrounding the Fukushima Daiichi Nuclear Power Plant. Spectrochim. Acta - Part B At. Spectrosc. 117, 1–7 (2016).
Adachi, K., Kajino, M., Zaizen, Y. & Igarashi, Y. Emission of spherical cesium-bearing particles from an early stage of the Fukushima nuclear accident. Sci. Rep. 3, 12–15 (2013).
Imoto, J. et al. Isotopic signature and nano-texture of cesium-rich micro-particles: Release of uranium and fission products from the Fukushima Daiichi Nuclear Power Plant. Sci. Rep. 7, 5409 (2017).
Galperin, A., Goldfield, A. & Karni, A. A simple model for 135Xe-level evaluation in neutronic analysis of a PWR core. Ann. Nucl. energy 12, 259–261 (1985).
Gonze, M. et al. Assessment of dry and wet atmospheric deposits of radioactive aerosols: application to Fukushima radiocaesium fallout. Environ. Sci. Technol. 48, 11268–76 (2014).
Morino, Y., Ohara, T., Watanabe, M., Hayashi, S. & Nishizawa, M. Episode analysis of deposition of radiocesium from the Fukushima Daiichi Nuclear Power Plant accident. Environ. Sci. Technol. 47, 2314–22 (2013).
Zheng, J. et al. Determination of 135Cs and 135Cs/137Cs atomic ratio in environmental samples by combining ammonium molybdophosphate (AMP)-selective Cs adsorption and ion exchange chromatographic separation to triple-quadrupole inductively coupled plasma-mass spectrometry. Anal. Chem. 86, 7103–10 (2014).
Acknowledgements
This research was funded by a Natural Environment Research Council (NERC) doctoral training grant (NE/K500823/1). Funding from the Cabot Institute, University of Bristol, enabled sample collection. We thank Yoichi Ozawa, F1 monitoring project, Tomio Kano, citizen of Kawamata Town, Koichi Oyama, member of the city council of Minamisoma City, and Hachiro Sato, member of the village council of Iitate Village, for their assistance with sample collection. We also acknowledge the collaboration with Professor Hideo Yamashita and Dr. Masanobu Ishida, former Kindai University, who provided some of the samples for this work.
Author information
Authors and Affiliations
Contributions
J.A.D. and D.A.R. developed strategy for isotopic determination of Pu and Cs. J.A.D. conducted all mass-spectrometric analyses. I.A.X.Y. performed gamma spectrometry. D.A.R., T.B.S., P.J.M. and Y.Y. collected samples and provided logistical support. J.A.D. and D.A.R. wrote the article.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dunne, J.A., Martin, P.G., Yamashiki, Y. et al. Spatial pattern of plutonium and radiocaesium contamination released during the Fukushima Daiichi nuclear power plant disaster. Sci Rep 8, 16799 (2018). https://doi.org/10.1038/s41598-018-34302-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-34302-0
Keywords
This article is cited by
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
