
Abstract
Historical climatic oscillations and co-evolutionary dependencies were key evolutionary drivers shaping the current population structure of numerous organisms. Here, we present a genome-wide study on the biogeography of the bark beetle Pityogenes chalcographus, a common and widespread insect in Eurasia. Using Restriction Associated DNA Sequencing, we studied the population structure of this beetle across a wide part of its western Palaearctic range with the goal of elucidating the role of Pleistocene glacial-interglacial cycling and its close relationship to its main host plant Norway spruce. Genetic distance among geographic sites was generally low, but clustering analysis revealed three genetically distinct groups, that is, southern, central/south-eastern, and north-eastern locations. Thus, three key P. chalcographus glacial refugia were identified: in the Italian-Dinaric region, the Carpathians, and the Russian plain, shared with its main host. The current phylogeographic signal was affected by genetic divergence among geographically isolated refugia during glacial periods and postglacial re-establishment of genetic exchange through secondary contact, reflected by admixture among genetic groups. Additionally, certain life history traits, like the beetle’s dispersal and reproductive behaviour, considerably influenced its demographic history. Our results will help to understand the biogeography of other scolytine beetles, especially species with similar life history traits.
Similar content being viewed by others

Introduction
Historical events and processes have shaped the distribution and genetic structure of numerous organisms. Past climatic oscillations, like glacial and interglacial cycling during the Pleistocene, were major evolutionary drivers in many species1,2. During glacial periods major parts of the northern hemisphere were covered by a thick ice shield. In Europe, glaciation of northern regions and high elevation areas as well as permafrost made these habitats largely inhospitable for life. Glaciation contracted the ranges of many species, geographically restricting populations to glacial refugia3. Numerous European species endured glaciation events in the Mediterranean region, though Extra-Mediterranean refugia like the Carpathian Mountains have also been described3,4. In many cases, contraction of a species’ range to multiple, isolated refugia limited gene flow and caused genetic divergence among refugia2. Following the end of a glacial period, organisms expanded from refugia, recolonizing formerly inhospitable areas and re-establishing genetic exchange. Current population structure thus may reflect past divergence events followed by gene flow during secondary contact3,5.
Here, we present a genome-wide study on the biogeography of the bark beetle Pityogenes chalcographus (L.) (Coleoptera, Curculionidae, Scolytinae) that provides clear evidence for past genetic divergence in multiple glacial refugia. Pityogenes chalcographus has a wide geographic range that now covers major parts of Eurasia, from central Italy to northern Scandinavia and from western Europe to East Asia6. The distribution of western Palaearctic P. chalcographus is embedded in the range of its main host tree Norway spruce, Picea abies6, and the range patterns of these two species are likely impacted by climate-driven historical processes7,8,9,10.
Previous work using single molecular markers describes that European P. chalcographus is structured in three geographically distinct mitochondrial clades7,8. One clade mainly occurs in northern European regions, another in central Europe, and southern Europe harbours higher haplotype diversity7,8. The demography of P. chalcographus was likely affected by range changes during the Pleistocene, as these mitochondrial clades diverged at the beginning of the last ice ages about 100,000 years ago8. Pityogenes chalcographus might have survived the last ice ages in multiple, geographically separated refugia, concordant with those of Norway spruce7,8,9. These refugia were proposed to be located in the Apennines in Italy, in the Carpathians or the Bulgarian Mountains, and in the Russian plain8. Moreover, an additional refugium in the Dinaric Alps was suggested7.
Other evolutionary drivers resulting in population structure in P. chalcographus have been proposed, but with little supporting evidence. For example, heritable bacterial endosymbionts, like Wolbachia or Cardinium, can alter the reproduction of numerous arthropods, consequently affecting the population structure of their hosts11,12. Despite low-titre and low-prevalence endosymbiont infections, the reproductive outcome of P. chalcographus is unlikely affected by these bacteria13,14.
Pityogenes chalcographus is an oligophagous bark beetle, feeding in tree species of a few genera within the plant family Pinaceae6, and host specialization has also been proposed to drive population differentiation. Specialization to different host plants results in phenotypic and genetic differentiation among populations of many herbivorous insects15. Pityogenes chalcographus successfully uses various conifers as hosts in addition to Norway spruce6, but does not exhibit host-plant related lineage diversification16. Nevertheless, host preference and developmental performance in Norway spruce is highest17.
Instead, all available evidence suggests that Pleistocene ice ages were major drivers shaping the biogeography of P. chalcographus8. Nonetheless, due to the limited resolution of previously applied markers7,8 and the complex life history of this species, detailed knowledge on its demographic history, especially during glacial cycling, is scarce. In particular, in-depth information on the number and locality of Pleistocene refugia and impact of postglacial processes on the present genetic structure is lacking. The goal of this study was to make robust inferences about P. chalcographus phylogeography using a comprehensive set of genome-wide molecular markers. Studying large panels of single nucleotide polymorphisms (SNPs) provides a powerful approach to inferring phylogeographic history18,19. By examining SNP variation among 16 geographic sites spanning a wide part of the P. chalcographus range, we provide new insights into glacial-interglacial processes that have shaped the current population structure across geography. We identify key Pleistocene glacial refugia and infer postglacial secondary contact in the context of host plant availability and P. chalcographus life history traits. In addition, we discuss species-specific features of P. chalcographus and other bark beetles that may influence the inferred phylogeographic patterns. Finally, our study will help to understand demographic processes in other species of the weevil subfamily Scolytinae, especially species with similar life histories.
Materials and Methods
Study system
Pityogenes chalcographus is a Palaearctic species whose distribution tracks that of suitable host plants. It is oligophagous on various conifers with Norway spruce as the main host in Europe6. Pityogenes chalcographus preferentially breeds in thin-barked parts of trees, including branches or upper stem sections20 and can be a forest pest during environmentally favourable conditions21. Male beetles initiate host attacks and attract conspecifics using aggregation pheromones22. The species is polygynous; two to seven female beetles mate with one single male, and each female lays on average ten to 26 eggs23. After subcortical larval and pupal development, and maturation feeding of young beetles on phloem tissue, a new generation of adults disperses to attack fresh hosts. Pityogenes chalcographus can produce up to three generations per year20.
Insect collection, DNA extraction, ddRAD library preparation, and sequencing
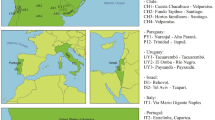
Pityogenes chalcographus was collected from 16 geographic sites between 2004 and 2009 (Fig. 1; Table S1). Living adults were sampled from breeding systems of standing trees or freshly cut logs of Norway spruce and stored in absolute ethanol at −20 °C. To avoid sampling of siblings, only one beetle per breeding system was collected.

Sample sites of Pityogenes chalcographus. Colours refer to assignment to a genetic cluster. Blue: Italian-Dinaric cluster. Red: north-eastern cluster. Black: central/south-eastern cluster. Black circles: key glacial refugia of Norway spruce, Picea abies, during the last ice ages. Yellow: present distribution of the primary host Norway spruce.
DNA was extracted from 192 whole beetles using the ‘GenElute Mammalian Genomic DNA miniprep kit’ (Sigma-Aldrich, St. Louis, MO) following the manufacturer’s instructions. DNA concentrations were estimated using a ‘NanoDrop 2000C’ and all samples were either concentrated or diluted the same concentration ±10% for downstream library preparation.
To study P. chalcographus on a multi-locus, genome-wide scale, we applied double digest Restriction Associated DNA Sequencing (ddRADSeq)24 using EcoRI and MseI restriction enzymes, as modified from25 (details on library preparation and sequencing see SI 2).
Sequence processing and data filtering
Raw reads were de-multiplexed and filtered as in18,26 (for details see SI 3). Due to the lack of a P. chalcographus reference genome only forward reads were used in this study. A de-novo ‘pseudo-reference’ genome was created using the ustacks algorithm in STACKS (v1.35)27,28 (parameters see SI 3). De-multiplexed and filtered reads were mapped to the ‘pseudo-reference’ genome using BWA-MEM (v0.7.12)29. SNP calling was performed using the Unified Genotyper in GATK (v3.3.0)30. We filtered out SNPs when i) the average base quality-phred score was <20, ii) the total depth per locus across all individuals was <10, iii) sites appeared to be over-assembled (when a chi-square test for the null hypothesis that each allele in a heterozygote has a representation of 0.5 is rejected at p < 0.05), and iv) the minor allele frequency across all individuals was <0.05. Moreover, only individuals with >10,000 retained SNPs were kept for downstream analyses. Location information of SNPs passing these filters was extracted and further analyses were performed in ANGSD (v0.913), using the GATK model30,31 (parameters used in ANGSD see SI 3). Individuals with more than 30% missing data were removed and loci that were present in less than 80% of samples were discarded.
Population statistics
Descriptive statistics and population genetic metrics were inferred from called genotypes calculated in ANGSD, using a final data set with 5,470 loci (requirements for passing filters see above and SI 3). SNPs were converted to a genind object in R via the package adegent (v2.0.1)32,33. Pairwise Nei’s FST values were calculated in the R package hierfstat34 and the G”ST value, as an additional, unbiased estimator35,36, in the R package mmod37. Additional population genetic statistics are reported in SI 4.
Isolation by distance
To assess whether the genetic structure of P. chalcographus follows an isolation-by-distance (IBD) pattern a Mantel test38, as implemented in the R package adegenet32,33, was performed. To estimate genetic distances among populations we calculated FST/(1 − FST)39 using pairwise Nei’s FST values as determined above. Geographical distances were inferred from the shortest distance between locations using the R package geosphere (v1.5-7)40 and were subsequently ln-transformed.
Relationship and structure among geographic sites
To assess the relationship among P. chalcographus geographic sites we used called genotypes as above, converted in a genpop object, that is, allele counts per population, in the R package adegenet32,33. Relationships were calculated applying a NJ-approach with 1,000 bootstrap replicates based on Nei’s distances in the R package poppr41,42. Results were visualized in FigTree (v1.4.3)43.
To study the admixture and population structure of individuals from different locations a structure analysis was performed. NGSadmix44 was run using genotype likelihoods, thus considering uncertainty of genotype calls45. Genotype likelihoods were calculated in ANGSD and loci were retained when the base quality phred-score was >20, the mapping quality phred-score >20, and the missing data rate was below 30%. Again, only loci present in at least 80% of individuals were retained. This resulted in a total number of 13,105 loci. The optimal number of groups (i.e., K) for clustering individuals was tested for K = 1 to K = 15, using changes in log-likelihood values between Ks46, running ten independent replicates with 10,000 iterations each. In addition to the optimal K-value, biologically meaningful Ks, i.e., K = 3 and K = 4, were tested (as previous studies proposed that P. chalcographus might have survived Pleistocene glaciation events in either three or four refugia7,8). Calculation of optimal K-values and visualization of admixture plots were done in R47.
Population structure was further studied by performing a Discriminant Analysis of Principal Components (DAPC), a multivariate statistical method identifying clusters based on linear combinations of genotypes produced by principal component analysis (PCA)48. DAPC was calculated in R using the package adegenet32,33,48 using called genotypes (5,470 loci) as described above. To achieve an optimal discrimination and avoid over-fitting of data, the optimum-α-score to estimate the optimal number of principal components was assessed.
Coalescent analysis of demography
To test different hypotheses of P. chalcographus demography, we performed an approximate Bayesian computation (ABC) analysis implemented in DIYABC (v2.1.0)49. We tested four evolutionary scenarios on a reduced data set (only loci with a missing data rate <3% were used, yielding 712 loci; to confirm that the reduced data set is representative a DAPC was performed, SI 5, Fig. S1). Scenario selection was based on results obtained from DAPC, the relationship among geographic sites, and NGSadmix. The following three recent groups (time = 0) were assumed to have originated from an unknown ancestor population (A): Italian-Dinaric (ITDI), north-eastern (NE), and central/south-eastern (CESE). In scenario 1 ITDI and CESE diverged from A in the past (t2) and NE branched off CESE later at t1. In scenario 2 ITDI and NE diverged from A at t2 and CESE branched off NE later at t1. In scenario 3 ITDI and NE originated from A at t2 and merged to CESE at t1. In scenario 4 ITDI, NE, and CESE diverged at the same time (t1) from A (details on DIYABC analysis see SI 5, Table S3).
Results
In total ~313 million reads were generated with an average of 1.6 million reads per individual. An individual barcode could be assigned to about 81% of raw reads yielding ~1.3 million retained reads per sample. 160 out of 192 individuals passed our filters and were included in the final analyses. Genotype calling yielded a data set with 5,470 SNPs, while 13,105 SNPs passed filters suitable for analysis of genotype likelihoods.
Population genetics metrics
Based on called genotypes, the average Nei’s pairwise FST value among geographic sites was 0.039 (details see SI 6, Table S2). The lowest FST value was observed between the French site FRDO and the Greek site GRDA (=0.021). The highest FST value was calculated between the Italian site ITTO and the eastern Carpathian site ROSA (=0.071). Further, an overall G”ST value of 0.021 was determined. For additional population statistics see SI 4, Table S2.
Isolation by distance
The genetic structure of P. chalcographus does not follow a strong IBD-pattern. The Mantel test did not reveal a significant positive correlation between genetic and geographic distances among locations (r Mantel = −0.011, p = 0.534, Fig. 2).

Mantel test for isolation by distance of Pityogenes chalcographus using Nei’s pairwise FST values and ln-transformed geographic distances (5,470 loci analysed).
Relationships among geographic sites
The NJ-tree based on Nei’s distances among geographic sites revealed a low level of genetic differentiation, reflected by relatively short branch lengths (Fig. 3). The average Nei’s distance was 0.020 with a maximum value of 0.033 between the Italian site ITTO and the Carpathian site ROSA. The minimum (=0.011) was calculated between the Greek location GRDA and the French site FRDO. Nonetheless, sites were largely grouped by geography with clusters consistent with proposed glacial refugia. Southern European P. chalcographus from Italian-Dinaric locations (CRSA, ITAB, ITAS, ITPA, ITTO; Fig. 3 blue) group together, supported by a bootstrap value of 100, and the Austrian site (ATRO) is sister to this Italian-Dinaric cluster (bootstrap value = 100). Within the Italian-Dinaric cluster, the two Apennine sites (ITAB, ITPA) are also supported by a bootstrap value of 100. Further, the north-eastern sites from Europe and Asian Russia (LIVI, POCH, RULU, RUSV, SWOV; Fig. 3 red) form one well-supported cluster (bootstrap value = 100). The remaining central/south-eastern European sites (FRDO, FRRA, GRDA, ROBI, ROSA; Fig. 3 black) did not form a distinct geographic cluster.

Neighbour Joining tree among Pityogenes chalcographus geographic sites based on Nei’s distances using 1,000 bootstrap replicates, using called genotypes (5,470 loci analysed; abbreviations of geographic sites see Fig. 1). Blue: Italian-Dinaric cluster. Red: north-eastern cluster. Black: central/south-eastern cluster. • bootstrap value = 100.
Admixture and structure among individuals
NGSadmix analysis resulted in an optimal K = 2 (SI 7, Fig. S2), also suggesting two distinct geographic clusters. One clear cluster covered individuals from north-eastern sites (LIVI, POCH, RULU, RUSV, SWOV; Fig. 4a red). Individuals sampled from these locations showed relatively low amounts of admixture. The second distinct cluster comprised samples from the Italian-Dinaric region (CRSA, ITAB, ITAS, ITPA, ITTO; Fig. 4a blue). Within the Italian-Dinaric cluster (blue), the two northern Italian sites (southern slopes of the Alps; ITAS, ITTO) and the Dinaric location (CRSA) had higher amounts of admixture than the southernmost Italian sites from the Apennines (ITAB, ITPA). Samples belonging to the central/south-eastern European sites had a higher amount of admixture and could not clearly be assigned to one of the two clusters. In contrast to the other central/south-eastern sites (FRDO, FRRA, GRDA, ROBI, ROSA), the highly admixed Austrian location (ATRO) had a greater proportion of the Italian-Dinaric (blue) cluster than of the north-eastern (red) cluster.

Admixture plot (NGSadmix) for K = 2 (a) and K = 3 (b) for Pityogenes chalcographus, using genotype likelihoods (13,105 loci analysed; abbreviations of geographic sites see Fig. 1).
NGSadmix analysis with K = 3, which would be consistent with hypothesized three key glacial refugia of P. chalcographus8, again supported the same two, relatively cohesive, geographic clusters in the north-eastern (LIVI, POCH, RULU, RUSV, SWOV; Fig. 4b red) and the Italian-Dinaric regions (CRSA, ITAB, ITAS, ITPA, ITTO; Fig. 4b blue). Again, the Austrian site (ATRO) was highly admixed but its major cluster is from the Italian-Dinaric region (blue). The third cluster (black) was relatively diffuse across geography; the remaining central/south-eastern populations (FRDO, FRRA, GRDA, ROBI, ROSA) did not form a clear cluster as was the case for K = 2 suggesting high levels of admixture. In the north-eastern cluster, the three locations furthest northeast (RULU, RUSV, SWOV) demonstrated relatively low admixture, as did the two southernmost from the Apennines (ITAB, ITPA). Admixture in other north-eastern and remaining Italian-Dinaric sites was more extensive.
Structure analysis with K = 4, consistent with an alternative hypothesis of four glacial refugia for P. chalcographus7, yielded qualitatively similar results to the K = 3 analysis (SI 8, Fig. S3).
Grouping of individuals across geography
DAPC results provide support for three distinct, geographic P. chalcographus groups (Fig. 5). Again, one group comprised north-eastern sites (LIVI, POCH, RULU, RUSV, SWOV; Fig. 5 red). Individuals from this group were clearly separated from the other locations. A second group included the Italian-Dinaric sites and the Austrian location (ATRO, CRSA, ITAB, ITAS, ITPA, ITTO; Fig. 5 blue). Individuals from this group did not overlap with the north-eastern group and had a very limited overlap with the remaining central/south-eastern group (Fig. 5 black). Individuals from the Italian-Dinaric sites (CRSA, ITAB, ITAS, ITPA, ITTO) had no overlap with the central/south-eastern group, only the Austrian location (ATRO) had a slight overlap with the central site in France (FRDO). Although being highly admixed, the remaining central/south-eastern sites (FRDO, FRRA, GRDA, ROBI, ROSA) were found to form an additional group. Apart from the French location mentioned above, they did not overlap with other groups (Fig. 5).

Grouping of Pityogenes chalcographus individuals across geography, using a Discriminant Analysis of Principal Components (DAPC) and called genotypes (5,470 loci analysed; abbreviations of geographic sites see Fig. 1). Blue: Italian-Dinaric group. Red: north-eastern group. Black: central/south-eastern group.
Additionally, distances on the DAPC axes reflect finer-scale geographic information: For example, the Italian-Dinaric sites, including the Austrian location, are closer to the remaining central/south-eastern sites than to the north-eastern sites. Within the central/south-eastern group (black), the Greek site GRDA is the south-eastern-most and closest to the two Carpathian sites (ROSA, ROBI). The Austrian location, for example, is close to the northern Italian sites (ITAS, ITTO), and to the two central sites in France (FRDO, FRRA). The Apennine sites ITAB and ITPA are the southernmost and therefore on the edge of the Italian-Dinaric (blue) group. Within the north-eastern group (red), the Polish location POCH is closest to the central/south-eastern group (Fig. 5).
Demographic history
The best-supported scenario describing the demographic history of P. chalcographus was scenario 4 (Fig. 6). The posterior probability (PP) of scenario 4 was 0.9847 (95% CI 0.9821–0.9873) and was higher than the posterior probabilities of the other three scenarios (PP scenario 1 = 0.0076, 95% CI 0.0060–0.0092; PP scenario 2 = 0.0066, 95% CI 0.0052–0.0079; PP scenario 3 = 0.0012, 95% CI 0.0009–0.0014). This suggests that the Italian-Dinaric (ITDI), north-eastern (NE), and central/south-eastern (CESE) groups most probably diverged from an unknown ancestral population (A) at the same time point. The type I error for scenario 4 was 0.058 (i.e., probability that another scenario is chosen although scenario 4 is correct), the type II error for this scenario was 0.089 (i.e., probability that this scenario is chosen although it is not the correct one).

Evolutionary scenarios to describe the demographic history of Pityogenes chalcographus using approximate Bayesian computation (ABC) analysis on a reduced data set (712 loci). A (ancestral population), CESE (central/south-eastern sites), NE (north-eastern sites), ITDI (Italian-Dinaric sites). Scenario 4 was best supported (red frame). Posterior probability (PP) scenario 1 = 0.0076, 95% CI 0.0060–0.0092; PP scenario 2 = 0.0066, 95% CI 0.0052–0.0079; PP scenario 3 = 0.0012, 95% CI 0.0009–0.0014, PP scenario 4 = 0.9847, 95% CI 0.9821–0.9873.
Discussion
We present a genome-wide, biogeographic analysis of the bark beetle P. chalcographus with emphasis on the influence of Pleistocene climatic oscillations on its present population structure. Analyses of clustering, relationships of geographic sites as well as a Bayesian approach testing population ancestry provide evidence for past contraction into Pleistocene glacial refugia and NGSadmix shows subsequent population admixture. Our data suggest a grouping of current P. chalcographus populations into three main, geographic clusters: one cluster comprising north-eastern sites, a second cluster covering Italian-Dinaric locations and a central European site from Austria, and a third cluster consisting of other central/south-eastern European sites. The genetic structure of P. chalcographus is characterized by high levels of admixture and low levels of differentiation among geographic sites. Pleistocene climatic events and certain species-specific traits were likely the main drivers that shaped the genetic architecture of P. chalcographus. Furthermore, the combination of our phylogeographic data and that of Norway spruce9 allows for the re-evaluation of the locality and number of glacial refugia and to assess the importance of secondary contact during postglacial expansion in this important bark beetle.
Past climatic fluctuations resulted in range changes of many organisms, shaping much of their present day genetic structure3. Demographic processes of certain insect groups were additionally affected by co-evolutionary dependencies. Bark beetles belong to the species-rich family Curculionidae, a group with long-standing plant-associations50. As most bark beetle life-cycle events occur in plant tissues, the geographic range of these insects is tightly associated with the geographic range of their main hosts8,18,51,52,53. Pityogenes chalcographus is oligophagous on conifers6. Although this beetle shows no host-related lineage diversification16, Norway spruce is favoured over other tree species17. Further, present range overlaps of European P. chalcographus are most consistent with Norway spruce6. Thus, we hypothesize that this insect-plant relationship resulted in range contraction of both, the host tree and the beetle, during glaciation events in the Pleistocene.
Cold conditions during Pleistocene glaciation forced Norway spruce to retreat to multiple refugia. Supported by pollen fossils and molecular data, these key refugia were located in the Russian plain, the Carpathians/Bulgarian Mountains, the Apennines, and the Dinaric Alps9,10,54,55. The refugium in the Apennines, however, is contentious56. In addition, numerous small refugia in Europe have been described9.
Judging by single molecular markers, early events (about 100,000 years BP) during the last ice ages caused divergence of mitochondrial clades8 and up to four P. chalcographus glacial refugia were hypothesized, consistent with those of Norway spruce7,8. Our analyses provide comprehensive insights in Pleistocene contraction-expansion processes of P. chalcographus to infer the number and locality of refugia, and assess subsequent postglacial admixture. DAPC results and the NJ-tree suggest two distinct genetic clusters and one diffuse genetic cluster, and the ABC analysis clearly supports a scenario with three clusters originating from a single, ancestral population. Moreover, these results suggest that current P. chalcographus populations clearly cluster by geography. Taking these results together and considering data on the Pleistocene history of Norway spruce, we suggest three P. chalcographus glacial refugia in the western Palaearctic.
Clustering of P. chalcographus from Russian, Swedish, Polish, and Lithuanian sites (north-eastern group) indicates that these individuals survived periods of increased glaciation in the Russian plain. Grouping of beetles from the French, Romanian, and Greek sites (central/south-eastern group) suggests a refugium in the Carpathians (or Bulgarian Mountains)9,10. Italian-Dinaric P. chalcographus, including beetles from the Austrian site, might have survived glaciation events in southern Europe, where major Norway spruce refugia in the Apennines and the Dinaric Alps were found10,55. These two mountain ranges are geographically relatively close and several small refugia between them have been reported57. Because P. chalcographus is a long-range disperser58, we propose one Italian-Dinaric P. chalcographus refugium.
In addition to these refugia, P. chalcographus might have survived Pleistocene ice ages also in the more eastern part of its range. Due to its oligophagous feeding behaviour, it can utilize Asian conifers, like Picea obovata, Picea jezoensis, or Pinus koraiensis, as hosts6. Several refugia of these tree species have been described in Asia, for example in Siberia (P. obovata)59 or in the Far East (P. koraiensis60, P. jezoensis61). Thus, P. chalcographus might have endured unfavourable conditions during the Pleistocene even in these regions, shared with a respective host. However, detailed knowledge on the genetic population structure of P. chalcographus from the eastern Palaearctic and its influence on European populations are not available and future research should focus on extensive sampling of this region to get a more complete picture of the beetle’s biogeography.
After the end of the last ice ages about 10,000 years ago, P. chalcographus expanded its range to previously inhospitable habitats. As for glacial periods, we assume that recolonization of the beetle and its main host also occurred parallel. For example, present Norway spruce in Fennoscandia was recolonized from a single refugium in the Russian plain9,62,63. Clustering of P. chalcographus from north-eastern locations suggests that these beetles also originated predominately from this Russian refugium, consistent with a tight insect-host relationship. Assuming three P. chalcographus refugia, NGSadmix results suggest secondary contact, for example, in Poland where north-eastern and central/south-eastern European beetles admixed (Fig. 4). We cannot rule out the possibility of shared polymorphisms among refugia contributing to the genetic clustering results. However, the geographic areas with the greatest inferred admixture for the beetles were also a secondary contact zones for Norway spruce62, again supporting a shared contraction and expansion history of bark beetle and host tree. Pityogenes chalcographus from central/south-eastern European sites show high amounts of admixture. These beetles might predominantly have expanded from a refugium in the Carpathians with individuals from sites in relative close proximity to this mountain range (ROBI, ROSA, GRDA) having lower amounts of admixture. Moreover, NGSadmix results suggest that P. chalcographus from this central European refugium also contributed to the genetic architecture of north-eastern European and northern Italian locations, reflecting additional secondary contact, for example, in the Southern Alps and Austria.
Recolonization from the Italian-Dinaric refugium mainly accounted for the genetic structure of southern sites. The southernmost populations in this area (ITAB, ITPA, CRSA) show low amounts of admixture compared to the northern Italian sites and the Austrian location. Furthermore, NGSadmix results suggest that P. chalcographus from the Italian-Dinaric refugium may also have contributed to the genetic structure of central/south-eastern sites and only limited to north-eastern sites.
Similar biogeographic patterns, that is, climate-driven range contractions and expansions and a tight host plant association, have also been reported from other insect species. For example, numerous butterflies (Lepidoptera) are characterized by their close relationship to plants, resulting in mutual adaptations to each other and overlapping distribution areas of insect and host64. Furthermore, various European lepidopteran species show demographic patterns affected by climatic fluctuations during the Pleistocene similar to P. chalcographus: range contractions to geographically isolated refugia in Mediterranean and Extra-Mediterranean regions resulted in divergence among these refugia, followed by postglacial secondary contact and re-establishment of genetic exchange2,65,66,67. In addition, diversification of the butterfly family Nymphalidae was found as a result of angiosperm radiation during the Cretaceous era68, a comparable co-evolutionary history as reported for weevils50. Taking this knowledge together underlines our hypothesis that glacial-interglacial cycling and the association to its main host plant were major evolutionary drivers in P. chalcographus.
In addition to the influence of past climatic oscillations, specific life history traits likely contributed to the current genetic structure of P. chalcographus. One important trait that affects the genetic structure of bark beetles is their dispersal behaviour18. IBD describes an increase of genetic differentiation among sites with increasing geographic distance69, presumably because of limited dispersal70. Pityogenes chalcographus can spread over long distances, more than 80 km aided by wind58, facilitating exchange of genetic material among sites. This feature might explain the absence of an IBD pattern in P. chalcographus, as long-range dispersing insects are generally characterized by a weak relationship between genetic and geographic distance71. In addition to the beetle’s intrinsic, species-specific traits, anthropogenic influence might also explain the lack of IBD. Various insects, like the bark beetle Tomicus piniperda72, the fire ant Solenopsis invicta73, or the leafhopper Scaphoideus titanus74 exhibit an absence of this pattern, for example, due to human trade. As humans have been trading across Eurasia for more than 2000 years75,76, we therefore cannot rule out a human influence on the genetic structure of P. chalcographus.
Further, the main host of P. chalcographus, Norway spruce, has a wide, current range10 and was also very common before the last ice ages77,78. The absence of insuperable physical barriers in the ranges of P. chalcographus and its main host9 probably results the low level of population differentiation and high amounts of admixture found in this study. This is corroborated by the relatively close relationship of P. chalcographus from Italian-Dinaric and the Austrian sites, suggesting that the Alps were not a strong barrier for dispersal of these two species. In contrast, physical barriers were found to affect the genetic architecture of other bark beetles, for example the mountain pine beetle Dendroctonus ponderosae in North America, where deserts and mountain ranges hampered gene flow among populations18,52,53.
Another factor that might have influenced the genetic architecture of P. chalcographus is its reproductive behaviour. It releases aggregation pheromones, a complex system of semiochemicals22, to attract male and female conspecifics. It is polygynous where one male can mate with several females, has a high fecundity, and can produce up to three generations per year20,23. Pityogenes chalcographus might have established thousands of generations of different origin since the end of the last glaciation events, likely reflected in high levels of admixture and low levels of differentiation.
In conclusion, the phylogeography of P. chalcographus was shaped by past climatic oscillations during the Pleistocene and co-evolutionary processes with its main host plant. Limited exchange of genetic material among different refugia during glaciation events is still reflected in the phylogeographic signal observed today; however, extensive postglacial admixture as a result of certain intrinsic traits has resulted in low levels of differentiation.
Data Availability
Sequence data will be provided upon request.
References
Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 405, 907–913, https://doi.org/10.1038/35016000 (2000).
Schmitt, T. Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front. Zool. 4, 1110.1186/1742-9994-4-11 (2007).
Hewitt, G. M. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 58, 247–276, https://doi.org/10.1111/j.1095-8312.1996.tb01434.x (1996).
Taberlet, P., Fumagalli, L., Wust-Saucy, A. G. & Cosson, J. F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 7, 453–464, https://doi.org/10.1046/j.1365-294x.1998.00289.x (1998).
Hewitt, G. M. Genetic consequences of climatic oscillations in the Quaternary. Philos. T. Roy. Soc. B 359, 183–195, https://doi.org/10.1098/rstb.2003.1388 (2004).
Pfeffer, A. Zentral- und westpaläarktische Borken- und Kernkäfer (Coleoptera, Scolytidae, Platypodidae). (Naturhistorisches Museum Basel, 1995).
Avtzis, D. N., Arthofer, W. & Stauffer, C. Sympatric occurrence of diverged mtDNA lineages of Pityogenes chalcographus (Coleoptera, Scolytinae) in Europe. Biol. J. Linn. Soc. 94, 331–340, https://doi.org/10.1111/j.1095-8312.2008.01004.x (2008).
Bertheau, C. et al. Divergent evolutionary histories of two sympatric spruce bark beetle species. Mol. Ecol. 22, 3318–3332, https://doi.org/10.1111/mec.12296 (2013).
Tollefsrud, M. M. et al. Genetic consequences of glacial survival and postglacial colonization in Norway spruce: combined analysis of mitochondrial DNA and fossil pollen. Mol. Ecol. 17, 4134–4150, https://doi.org/10.1111/j.1365-294X.2008.03893.x (2008).
Schmidt-Vogt, H. Die Fichte. (Verlag Paul Parey, 1977).
Engelstadter, J. & Hurst, G. D. D. The Ecology and evolution of microbes that manipulate host reproduction. Annu. Rev. Ecol. Evol. Syst. 40, 127–149, https://doi.org/10.1146/annurev.ecolsys.110308.120206 (2009).
Hurst, G. D. D. & Jiggins, F. M. Problems with mitochondrial DNA as a marker in population, phylogeographic and phylogenetic studies: the effects of inherited symbionts. Proc. Biol. Sci. 272, 1525–1534, https://doi.org/10.1098/rspb.2005.3056 (2005).
Schebeck, M. et al. Reproductive manipulators in the bark beetle Pityogenes chalcographus (Coleoptera: Curculionidae)-the role of Cardinium, Rickettsia, Spiroplasma, and Wolbachia. J. Insect Sci. 18, 4, https://doi.org/10.1093/jisesa/iey044 (2018).
Arthofer, W., Riegler, M., Avtzis, D. N. & Stauffer, C. Evidence for low-titre infections in insect symbiosis: Wolbachia in the bark beetle Pityogenes chalcographus (Coleoptera, Scolytinae). Environ. Microbiol. 11, 1923–1933, https://doi.org/10.1111/j.1462-2920.2009.01914.x (2009).
Dres, M. & Mallet, J. Host races in plant-feeding insects and their importance in sympatric speciation. Philos. Trans. R Soc. Lond. B Biol. Sci. 357, 471–492, https://doi.org/10.1098/rstb.2002.1059 (2002).
Bertheau, C., Bankhead-Dronnet, S., Martin, C., Lieutier, F. & Roux-Morabito, G. Lack of genetic differentiation after host range extension argues for the generalist nature of Pityogenes chalcographus (Curculionidae: Scolytinae). Ann. Forest Sci. 69, 313–323, https://doi.org/10.1007/s13595-011-0161-4 (2012).
Bertheau, C. et al. Preference-performance relationship and influence of plant relatedness on host use by Pityogenes chalcographus L. Agr. Forest Entomol. 11, 389–396, https://doi.org/10.1111/j.1461-9563.2009.00442.x (2009).
Dowle, E. J. et al. Reproductive isolation and environmental adaptation shape the phylogeography of mountain pine beetle (Dendroctonus ponderosae). Mol. Ecol. 26, 6071–6084, https://doi.org/10.1111/mec.14342 (2017).
Bagley, R. K., Sousa, V. C., Niemiller, M. L. & Linnen, C. R. History, geography and host use shape genomewide patterns of genetic variation in the redheaded pine sawfly (Neodiprion lecontei). Mol. Ecol. 26, 1022–1044, https://doi.org/10.1111/mec.13972 (2017).
Postner, M. In Die Forstschädlinge Europas Vol. 2 (ed. Schwenke, W.) 334–482 (Verlag Paul Parey, 1974).
Grégoire, J.-C. & Evans, H. F. In Bark and wood boring insects in living trees in Europe, a synthesis. (eds Lieutier, F. et al.) 19–37 (Kluwer Academic Publishers, 2004).
Francke, W., Heemann, V., Gerken, B., Renwick, J. A. A. & Vite, J. P. 2-Ethyl-1,6-Dioxaspiro[4.4]Nonane, principal aggregation pheromone of Pityogenes chalcographus (L). Naturwissenschaften 64, 590–591, https://doi.org/10.1007/Bf00450651 (1977).
Schwerdtfeger, F. E. B. zur Fortpflanzungsbiologie des Borkenkäfers Pityogenes chalcographus L. J. Appl. Entomol. 15, 335–427 (1929).
Parchman, T. L. et al. Genome-wide association genetics of an adaptive trait in lodgepole pine. Mol. Ecol. 21, 2991–3005, https://doi.org/10.1111/j.1365-294X.2012.05513.x (2012).
Egan, S. P. et al. Experimental evidence of genome-wide impact of ecological selection during early stages of speciation-with-gene-flow. Ecol. Lett. 18, 817–825, https://doi.org/10.1111/ele.12460 (2015).
Assour, L. A., LaRosa, N. & Emrich, S. J. Hot RAD: a tool for analysis of next-gen RAD tag data. arXiv, 1511.06754 (2015).
Catchen, J., Hohenlohe, P. A., Bassham, S., Amores, A. & Cresko, W. A. Stacks: an analysis tool set for population genomics. Mol. Ecol. 22, 3124–3140, https://doi.org/10.1111/mec.12354 (2013).
Catchen, J. M., Amores, A., Hohenlohe, P., Cresko, W. & Postlethwait, J. H. Stacks: building and genotyping loci de novo from short-read sequences. G3-Genes Genom. Genet. 1, 171–182, https://doi.org/10.1534/g3.111.000240 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760, https://doi.org/10.1093/bioinformatics/btp324 (2009).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303, https://doi.org/10.1101/gr.107524.110 (2010).
Korneliussen, T. S., Albrechtsen, A. & Nielsen, R. ANGSD: analysis of next generation sequencing data. Bmc Bioinformatics 15:35610.1186/s12859-014-0356-4 (2014).
Jombart, T. adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24, 1403–1405, https://doi.org/10.1093/bioinformatics/btn129 (2008).
Jombart, T. & Ahmed, I. adegenet 1.3-1: new tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071, https://doi.org/10.1093/bioinformatics/btr521 (2011).
Goudet, J. H. I. E. R. F. S. T. A. T. a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 5, 184–186, https://doi.org/10.1111/j.1471-8278.2004.00828.x (2005).
Hedrick, P. W. A standardized genetic differentiation measure. Evolution 59, 1633–1638, https://doi.org/10.1111/j.0014-3820.2005.tb01814.x (2005).
Meirmans, P. G. & Hedrick, P. W. Assessing population structure: F-ST and related measures. Mol. Ecol. Resour. 11, 5–18, https://doi.org/10.1111/j.1755-0998.2010.02927.x (2011).
Winter, D. J. MMOD: an R library for the calculation of population differentiation statistics. Mol. Ecol. Resour. 12, 1158–1160, https://doi.org/10.1111/j.1755-0998.2012.03174.x (2012).
Mantel, N. Detection of disease clustering and a generalized regression approach. Cancer Res. 27, 209–220 (1967).
Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145, 1219–1228 (1997).
Geosphere: Spherical Trigonometry v. 1.5-7 (2017).
Kamvar, Z. N., Brooks, J. C. & Grunwald, N. J. Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet. 6, https://doi.org/10.3389/fgene.2015.00208 (2015).
Kamvar, Z. N., Tabima, J. F. & Grunwald, N. J. Poppr: an R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. Peerj 2, https://doi.org/10.7717/peerj.281 (2014).
FIGTREE v. 1. 4.3 (http://tree.bio.ed.ac.uk/software/figtree/, 2009).
Skotte, L., Korneliussen, T. S. & Albrechtsen, A. Estimating individual admixture proportions from next generation sequencing data. Genetics 195, 693–702, https://doi.org/10.1534/genetics.113.154138 (2013).
Nielsen, R., Paul, J. S., Albrechtsen, A. & Song, Y. S. Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 12, 443–451, https://doi.org/10.1038/nrg2986 (2011).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620, https://doi.org/10.1111/j.1365-294X.2005.02553.x (2005).
R: A language and environment for statistical computing. (R Foundation for Statistical Computing, Vienna, Austria, 2013).
Jombart, T., Devillard, S. & Balloux, F. Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. Bmc Genet. 11, https://doi.org/10.1186/1471-2156-11-94 (2010).
Cornuet, J. M. et al. DIYABC v2.0: a software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 30, 1187–1189, https://doi.org/10.1093/bioinformatics/btt763 (2014).
McKenna, D. D., Sequeira, A. S., Marvaldi, A. E. & Farrell, B. D. Temporal lags and overlap in the diversification of weevils and flowering plants. P. Natl. Acad. Sci. USA 106, 7083–7088, https://doi.org/10.1073/pnas.0810618106 (2009).
Cognato, A. I., Harlin, A. D. & Fisher, M. L. Genetic structure among pinyon pine beetle populations (Scolytinae: Ips confusus). Environ. Entomol. 32, 1262–1270, https://doi.org/10.1603/0046-225x-32.5.1262 (2003).
Mock, K. E. et al. Landscape-scale genetic variation in a forest outbreak species, the mountain pine beetle (Dendroctonus ponderosae). Mol. Ecol. 16, 553–568, https://doi.org/10.1111/j.1365-294X.2006.03158.x (2007).
Batista, P. D., Janes, J. K., Boone, C. K., Murray, B. W. & Sperling, F. A. H. Adaptive and neutral markers both show continent-wide population structure of mountain pine beetle (Dendroctonus ponderosae). Ecol. Evol. 6, 6292–6300, https://doi.org/10.1002/ece3.2367 (2016).
Vendramin, G. G., Anzidei, M., Madaghiele, A., Sperisen, C. & Bucci, G. Chloroplast microsatellite analysis reveals the presence of population subdivision in Norway spruce (Picea abies K.). Genome 43, 68–78, https://doi.org/10.1139/gen-43-1-68 (2000).
Giannini, R., Morgante, M. & Vendramin, G. G. Allozyme variation in Italian populations of Picea abies (L) Karst. Silvae Genet. 40, 160–166 (1991).
Scotti, I. et al. Postglacial recolonization routes for Picea abies K. in Italy as suggested by the analysis of sequence-characterized amplified region (SCAR) markers. Mol. Ecol. 9, 699–708, https://doi.org/10.1046/j.1365-294x.2000.00911.x (2000).
Latalowa, M. & van der Knaap, W. O. Late Quaternary expansion of Norway spruce Picea abies (L.) Karst. in Europe according to pollen data. Quaternary Sci. Rev. 25, 2780–2805, https://doi.org/10.1016/j.quascirev.2006.06.007 (2006).
Nilssen, A. C. Long-range aerial dispersal of bark beetles and bark weevils (Coleoptera, Scolytidae and Curculionidae) in northern Finland. Ann. Entomol. Fenn. 50, 37–42 (1984).
Tollefsrud, M. M., Latalowa, M., van der Knaap, W. O., Brochmann, C. & Sperisen, C. Late Quaternary history of North Eurasian Norway spruce (Picea abies) and Siberian spruce (Picea obovata) inferred from macrofossils, pollen and cytoplasmic DNA variation. J. Biogeogr. 42, 1431–1442, https://doi.org/10.1111/jbi.12484 (2015).
Aizawa, M., Kim, Z. S. & Yoshimaru, H. Phylogeography of the Korean pine (Pinus koraiensis) in northeast Asia: inferences from organelle gene sequences. J. Plant Res. 125, 713–723, https://doi.org/10.1007/s10265-012-0488-4 (2012).
Aizawa, M. et al. Phylogeography of a northeast Asian spruce, Picea jezoensis, inferred from genetic variation observed in organelle DNA markers. Mol. Ecol. 16, 3393–3405, https://doi.org/10.1111/j.1365-294X.2007.03391.x (2007).
Tollefsrud, M. M. et al. Combined analysis of nuclear and mitochondrial markers provide new insight into the genetic structure of North European Picea abies. Heredity 102, 549–562, https://doi.org/10.1038/hdy.2009.16 (2009).
Giesecke, T. & Bennett, K. D. The Holocene spread of Picea abies (L.) Karst. in Fennoscandia and adjacent areas. J. Biogeogr. 31, 1523–1548, https://doi.org/10.1111/j.1365-2699.2004.01095.x (2004).
Ehrlich, P. R. & Raven, P. H. Butterflies and plants - a study in coevolution. Evolution 18, 586–608, https://doi.org/10.2307/2406212 (1964).
Habel, J. C., Schmitt, T. & Muller, P. The fourth paradigm pattern of post-glacial range expansion of European terrestrial species: the phylogeography of the Marbled White butterfly (Satyrinae, Lepidoptera). J. Biogeogr. 32, 1489–1497, https://doi.org/10.1111/j.1365-2699.2005.01273.x (2005).
Schmitt, T., Rakosy, L., Abadjiev, S. & Muller, P. Multiple differentiation centres of a non-Mediterranean butterfly species in south-eastern Europe. J. Biogeogr. 34, 939–950, https://doi.org/10.1111/j.1365-2699.2006.01684.x (2007).
Schmitt, T. & Seitz, A. Intraspecific allozymatic differentiation reveals the glacial refugia and the postglacial expansions of European Erebia medusa (Lepidoptera: Nymphalidae). Biol. J. Linn. Soc. 74, 429–458, https://doi.org/10.1006/bijl.2001.0584 (2001).
Wahlberg, N. et al. Nymphalid butterflies diversify following near demise at the Cretaceous/Tertiary boundary. P. Roy. Soc. B Biol. Sci. 276, 4295–4302, https://doi.org/10.1098/rspb.2009.1303 (2009).
Wright, S. Isolation by Distance. Genetics 28, 114–138 (1943).
Orsini, L., Vanoverbeke, J., Swillen, I., Mergeay, J. & De Meester, L. Drivers of population genetic differentiation in the wild: isolation by dispersal limitation, isolation by adaptation and isolation by colonization. Mol. Ecol. 22, 5983–5999, https://doi.org/10.1111/mec.12561 (2013).
Peterson, M. A. & Denno, R. F. The influence of dispersal and diet breadth on patterns of genetic isolation by distance in phytophagous insects. Am. Nat. 152, 428–446, https://doi.org/10.1086/286180 (1998).
Kerdelhue, C., Magnoux, E., Lieutier, F., Roques, A. & Rousselet, J. Comparative population genetic study of two oligophagous insects associated with the same hosts. Heredity 97, 38–45, https://doi.org/10.1038/sj.hdy.6800836 (2006).
Yang, C. C., Shoemaker, D. D., Wu, W. J. & Shih, C. J. Population genetic structure of the red imported fire ant. Solenopsis invicta, in Taiwan. Insect Soc. 55, 54–65, https://doi.org/10.1007/s00040-007-0969-y (2008).
Bertin, S. et al. Diffusion of the Nearctic leafhopper Scaphoideus titanus Ball in Europe: a consequence of human trading activity. Genetica 131, 275–285, https://doi.org/10.1007/s10709-006-9137-y (2007).
Hulme, P. E. T. transport and trouble: managing invasive species pathways in an era of globalization. J. Appl. Ecol. 46, 10–18, https://doi.org/10.1111/j.1365-2664.2008.01600.x (2009).
Preston, C. D., Pearman, D. A. & Hall, A. R. Archaeophytes in Britain. Bot. J. Linn. Soc. 145, 257–294, https://doi.org/10.1111/j.1095-8339.2004.00284.x (2004).
Frenzel, B. Pleistocene vegetation of Northern Eurasia - recent vegetation of Northern Eurasia Resulted from a relentless contest between steppe and forest. Science 161, 637–649, https://doi.org/10.1126/science.161.3842.637 (1968).
Ravazzi, C. Late Quaternary history of spruce in southernEurope. Rev. Palaeobot. Palyno. 120, 131–177, https://doi.org/10.1016/S0034-6667(01)00149-X (2002).
Acknowledgements
We thank the Austrian Science Fund FWF for financial support of this study (project number P26749-B25 to CS and J3527-B22 to HS). Data were processed using the Vienna Scientific Cluster, Austria, and the Beocat Computing Facilities at Kansas State University, Manhattan, KS. We thank Josef Pennerstorfer (BOKU Vienna) for assistance with GIS and the ‘BOKU Vienna Open Access Publishing Fund’ for covering the open access charges.
Author information
Authors and Affiliations
Contributions
M.S., H.S., and C.S. designed the study. M.S., D.N.A., and C.B. conducted lab work before library preparation. H.S. and J.L.F. prepared the ddRAD libraries. M.S., E.J.D., G.J.R. analysed the data. M.S. wrote the first draft of the manuscript and all authors contributed to the final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schebeck, M., Dowle, E.J., Schuler, H. et al. Pleistocene climate cycling and host plant association shaped the demographic history of the bark beetle Pityogenes chalcographus. Sci Rep 8, 14207 (2018). https://doi.org/10.1038/s41598-018-32617-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-32617-6
Keywords
This article is cited by
-
Insect adaptation: unveiling the physiology of digestion in challenging environments
Chemical and Biological Technologies in Agriculture (2024)
-
Climatic oscillations in Quaternary have shaped the co-evolutionary patterns between the Norway spruce and its host-associated herbivore
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
