
Abstract
Leptospira is a phylogenetically unique group of bacteria, and includes the causative agents of leptospirosis, the most globally prevalent zoonosis. Bacteriophages in Leptospira are largely unexplored. To date, a genomic sequence is available for only one temperate leptophage called LE1. Here, we sequenced and analysed the first genomes of the lytic phages LE3 and LE4 that can infect the saprophyte Leptospira biflexa using the lipopolysaccharide O-antigen as receptor. Bioinformatics analysis showed that the 48-kb LE3 and LE4 genomes are similar and contain 62% genes whose function cannot be predicted. Mass spectrometry led to the identification of 21 and 23 phage proteins in LE3 and LE4, respectively. However we did not identify significant similarities with other phage genomes. A search for prophages close to LE4 in the Leptospira genomes allowed for the identification of a related plasmid in L. interrogans and a prophage-like region in the draft genome of a clinical isolate of L. mayottensis. Long-read whole genome sequencing of the L. mayottensis revealed that the genome contained a LE4 phage-like circular plasmid. Further isolation and genomic comparison of leptophages should reveal their role in the genetic evolution of Leptospira.
Similar content being viewed by others
Introduction
Recently, there has been a renewed interest in bacteriophages for their potential use as alternatives to conventional antibiotics1, and also in understanding their contribution in evolution of bacteria2,3. In addition, phages could be used to develop new genetic tools such as replicative vectors from different compatibility groups and phage-delivery systems. Unfortunately, little is known about the diversity of phages among the genus Leptospira. Leptospira are ubiquitous organisms that are found as free-living saprophytes in environmental water and soil, or as pathogens that can cause acute or chronic infections in animals. A third group that is composed of intermediate species (in regards to their pathogenesis) of Leptospira, is phylogenetically closely related to the pathogenic species and can cause mild infections4. Leptospirosis is an emerging waterborne zoonosis which results in more than one million human cases a year with a fatality rate frequently exceeding 10%5.
To the best of our knowledge, the only phages that have been isolated, purified, and phenotypically characterized in the genus Leptospira are: vB_LbiM_LE1 (renamed6, and abbreviated LE1), vB_LbiM_LE3 (LE3), and vB_LbiM_LE4 (LE4)7. These tailed phages have been isolated from urban sewage and infect the saprophyte L. biflexa. The LE1 temperate phage genome was previously sequenced8 and an Escherichia coli-L. biflexa shuttle vector was generated by cloning the replication origin of LE19. Until now, the virulent phages LE3 and LE4 have not been further characterized at the genomic and proteomic levels. In addition to these three phages, phage-like particles were also observed following mitomycin C induction of a pathogenic strain carrying a circular plasmid with phage-related genes, but these phage-like particles were not purified10.
Recently, comparative analyses of genome sequences have suggested the existence of prophages and genomic islands within the genus Leptospira11,12. Putative prophages are found in infectious Leptospira species, including pathogenic and intermediate species, and absent in saprophytic Leptospira species, suggesting that phages may have had a major role in the emergence of the pathogens and/or in the acquisition of virulence factors. Analysis of the complete genomes of 20 Leptospira species led to the description of several predicted prophage regions11, including LE1-like and Mu-like prophages. In addition, the existence of two other groups of prophages have been proposed: a 22-kb region which was initially described in L. interrogans serovar Lai but that is present in most pathogenic species and the intermediate species L. licerasiae11,12,13 and a 65-kb region, observed in L. interrogans serovar Manilae, which can excise from the chromosome and form a circular replicon14,15 that is closely related to a 70-kb circular plasmid, pLIMLP116.
Characterizing leptophages—phages that infect Leptospira—sequencing their genomes, identifying major proteins, understanding their genetic relationships, and studying their interactions with various host strains are all essential steps in the understanding of Leptospira evolution. In addition, most replicative vectors are based on the replication systems derived from phage or prophage-like elements17. These vectors are used for genetic complementation, heterologous expression and controlled gene expression. However, pathogenesis research is slowed down by the lack of practical tools for routine genetic manipulation of pathogenic Leptospira strains. For all these reasons, we report herein the characterization of the first virulent leptophages at the phenotypic, genomic, and proteomic levels. We also demonstrate that these leptophages use lipopolysaccharides (LPS) as a receptor on bacterial cells. A general analysis of LE3 and LE4 genomes did not reveal significant similarities with other viral genomes, suggesting they may constitute a new group of phages. We also searched for related prophages in Leptospira genomes available in public databases, leading to the identification of novel circular plasmids in a clinical isolate.
Results and Discussion
Morphology
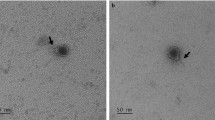
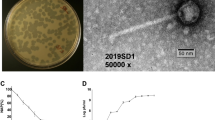
Three tailed phages, designated LE1, LE3, and LE4, were previously isolated from urban sewage by direct plating of water samples using L. biflexa as a host7. Cultures of L. biflexa were inoculated with LE3 and LE4 at a multiplicity of infection (MOI) of 0.001, showing a moderate lysis—up to 50%—after one month. Purified viruses observed by electron microscopy share the same common tailed-phage morphology consistent with their affiliation with the Myoviridae family (from the order Caudovirales) which is one of the most represented and characterized in bacteriophages18. Myoviridae cannonically possess tails that mechanically contract during infection. LE1 has a hexahedral capsid of 80 nm in diameter, and a contractile tail of 75 to 115 nm in length (Fig. 1A, n = 15 phages measured). LE3 and LE4 have a hexahedral capsid of 65 nm in diameter, and a contractile tail of 65 to 80 nm in length (Fig. 1B–D n = 12 and n = 20 phages measured). Most of the LE3 and LE4 particles were found to be associated with membrane-derived vesicles. It is actually unclear if these vesicles are artifacts due to the lysis or if they are produced by the bacterial cells to serve as decoys, as previously reported for E. coli19.

Transmission electron micrographs of leptophages. Morphology of leptophages (A) LE1, (B) LE3, and (C) LE4. Scale bar represents 100 nm. (D) Magnification of L. biflexa strain Patoc 1 (cell length of 10 µm) six hours after infection with phage LE4 (MOI=10). Arrowheads indicate representative phages. Scale bar represents 200 nm.
To better characterize the lytic phage infectious cycle, L. biflexa was challenged with LE4 at an MOI of 10 and the cells were observed by electron microscopy at different times (Supplementary Fig. S1). At two hours post-infection, approximately two phages are attached per cell. This small number of cell-associated phage particles may be due to poor adsorption of phages, vesicles and remnants of cells adsorbing phages, or loss of phages during the grid preparation. Six hours post-infection, cells start to release LE4 virions. An electron micrograph shows the release of at least 25 phage particles along the length (10 µm) of the infected cell (Fig. 1D). This number is most likely underestimated, as additional particles are probably being assembled and matured within the host cell or not visible in the two-dimension micrographs. The phage particles seem to exit through the cell envelop without lysing the whole infected cell, at least during the early stage of the burst. At 24 hours, infected cultures present phage particles, cell remnants and rare intact cells (Supplementary Fig. S1).
Phage absorption and one-step growth kinetics
The adsorption behaviour of LE4 to L. biflexa strain Patoc 1 was studied at a MOI of 0.0001. Approximately 20% of the phages were adsorbed in 30 min and the adsorbed phages reached a maximum of 70% after 3 h of incubation (Supplementary Fig. S2).
One-step growth studies were also conducted to investigate the different phases of the phage infection. We then determined the latent period and burst size of LE4 on L. biflexa, which are relatively slow growing bacteria, with a doubling time of about 24 hours. According to the one-step growth experiments, the latent period of LE4 on L. biflexa was approximately 6 hours and the rise period was 2 hours. The burst size corresponds to ten phages per infected cell (Fig. 2).

One-step growth curve of LE4 infecting L. biflexa. Data from six experiments; error bars indicate standard deviation.
Host range of lytic phages LE3 and LE4
For the determination of the LE3/LE4 host range, 25 strains belonging to 10 Leptospira species (L. biflexa, L. interrogans, L. borgpetersenii, L. kirschneri, L. weilii, L. noguchii, L. meyeri, L. yanagawae, L. fainei, and L. licerasiae) were tested by spot assays. Lysis activity was limited to the L. biflexa strain Patoc 1, and the L. meyeri strain 201601301, supporting the hypothesis that these phages have a narrow host range7, even though the host strains do not belong to the same species and have been isolated in different geographical regions (Europe and Indian Ocean).
As expected for lytic (virulent) phages, plaques of LE3 and LE4 on lawns of L. biflexa formed clear and non-turbid zones of heterogenous sizes ranging from 0.3 mm to 3 mm, with no colonies growing inside the zone of clearance even after prolonged incubation (Supplementary Fig. S3). Moreover, isolated bacterial clone resistant to LE4 were further tested for the presence of LE4 by PCR (see below). We were unable to detect LE4 DNA via colony PCR, further suggesting that the phages LE3 and LE4 are lytic phages and not lysogenic.
Selection, identification, and characterization of phage resistant mutants
Phages infect bacteria by attaching to surface-exposed receptors and injecting their DNA into the cells. In Gram-negative bacteria, outer membrane proteins, pili, flagella, oligosaccharides, and LPS have all been characterized as phage receptors20,21. To better understand the genetic factors influencing bacterial susceptibility to phage infection, we selected for phage-resistant strains. Consequently, we isolated and sequenced the genome of an L. biflexa spontaneous mutant clone that was resistant to LE4 (Fig. 3A). This clone, called RLE4, was later screened by spot assays, which confirmed it as a true phage-resistant clone not only for LE4 but also for LE1 and LE3 (Fig. 3B). However, at a high concentration (105 phage), LE4 is able to induce some cell lysis (Supplementary Fig. S4) probably due to the emergence of phages overcoming this resistance.

LPS as a receptor of the leptophages LE1, LE3, and LE4. (A) Growth curves of L. biflexa infected by LE3 and LE4. (B) Plaque assay of LE1 LE3 and LE4 (103 PFU) on lawn of L. biflexa strain Patoc 1 and L. biflexa strain RLE4. (C) Genetic map of the LPS locus. The location of SNP is noted on the RLE4 map as an arrow. (D) Schematic representation of targeted mutagenesis of LEPBIa2019 in WT L. biflexa.
RLE4, as well as the parent susceptible strain, were sequenced on an Illumina platform. Mutations that were detected in sequenced strains were mapped against the L. biflexa reference genome (Fig. 3C). A single-nucleotide polymorphism (C->T at position 2,100,832) was identified. This mutation causes a premature stop codon in the gene LEPBIa2019 which encodes a probable acetyltransferase. This nonsense mutation leads to a truncated protein of 30 kDa instead of 37 kDa for the native protein. Attempts to complement RLE4 with wild-type LEPBIa2019 were unsuccessful (data not shown). Interestingly, LEPBIa2019 is located in the rfb/O-antigen locus of the LPS that could be part of a large operon.
To confirm this initial observation, we generated a deletion mutant of LEPBIa2019 by allelic exchange in L. biflexa (Fig. 3D) and tested for phage sensitivity. Spot assays demonstrated that the LEPBIa2019 mutant was resistant to LE4 similarly to RLE4 (Supplementary Fig. S4), thereby validating that LE4 infectious cycle is O-antigen dependent as previously reported for other phages22. However, the adsorption of LE4 was not significantly reduced in the LEPBIa2019 mutant (Supplementary Fig. S2), suggesting LEPBIa2019 gene product is not essential for viral adsorption under our experimental conditions, but may play a role in the entry of viral DNA into the bacterial cell. As shown in other phages, receptor requirements are complex and other cell surface components may play a role in adsorption and/or penetration of the phage22.
The host selectivity is probably determined by the specific interaction of the tail fibers of the phage with LPS of the host cell. An investigation of the O-antigen locus in 21 Leptospira species revealed that the susceptible strain L. meyeri 201601301 has an homologous region with L. biflexa, especially for the gene LEPBIa2019 that shares an unusually high percentage (90.3%) of similarities between both species that is higher than percentages of similarities share with all the other species (Supplementary Fig. S5).
Leptophage genome analysis
To date, the 73,622-bp genome sequence of the temperate phage LE1 is the only genome of a phage infecting Leptospira to be available in public databases8. Here, we present the first complete genome sequences of lytic leptophages. The genomes of LE3 and LE4 are composed of 48,288 bp and 47,866 bp of double-stranded DNA, respectively, and have a GC content of 37.3%; similar to that of L. biflexa (38.6%) and LE1 (38.5%). In total, the genome of LE3 and LE4 had respectively 83 and 81 ORF, with all the ORF on the same strand (Fig. 4). Whole-genome sequence alignments reveal that the phages LE3 and LE4 are closely related (95% identity at the nucleotide level and 78 shared ORF), but are genetically distinct from LE1 (Fig. 4). In terms of gene repertoire, LE3 has only four genes encoding hypothetical proteins that do not have clear homologues in LE4 while three genes, also encoding for hypothetical proteins, are predicted to be unique to LE4. Given the high identity between LE3 and LE4, we decided to further focus our analysis on LE4. Of the 81 ORF of LE4, only 31 (38%) had a predicted function assigned according to BLASTp, PFAM or COG (Table 1). Accordingly, the majority of genes encode for proteins of unknown functions. Following the genome annotations, it was apparent that the phage genomes displayed a modular organisation which is common for the majority of tailed bacteriophage genomes such as LE18. There are distinguishable gene modules whose products are predicted to be involved in DNA replication, structure/morphogenesis, and DNA packaging (Table 1, Fig. 4). It was not possible to identify genes encoding integrase, capsid proteins, or proteins involved in the process of host cell lysis (holins and lysins), further confirming that the majority of the phages’ genes remain uncharacterized.

Linear genomic maps and protein identity comparisons between the leptophages. Based on nucleotide sequences of the phage genomes and predicted open reading frames. The direction of arrows represents the direction of transcription. Predicted functions and percent identity at the protein level are indicated.
Proteomic analysis of LE3 and LE4
To further characterize LE3 and LE4 and identify major structure proteins, we decided to perform bottom-up proteomics experiments on purified phages prepared in biological replicates. A homemade protein sequence database composed of (i) the 83 and 81 different putative protein sequences deduced from LE3 and LE4 genomics data; (ii) the L. biflexa Uniprot database (UP000001847, 3723 entries); and (iii) usual proteomics contaminants was used for the search. We processed 2 to 3 replicates per phage and analyzed all 5 samples. The liquid chromatography/tandem mass spectrometry (LC-MS/MS) analysis reproducibly detected 8 and 11 proteins in LE3 and LE4, respectively (Supplementary Table 1). All the proteins found in LE3 were also present in LE4 confirming that the two phages are closely related. Among a total of 23 proteins found in LE4, the most abundant proteins, as expected, are structural proteins such as portal (gp19), head to tail connector (gp25), tail sheath (gp29 and gp33), tail collar (gp41), and baseplate (gp39) proteins. Other abundant proteins of unknown functions encoded by genes in the same locus, such as gp19 and gp22, may also belong to the same morphogenesis module. Surprisingly, no capsid protein was identified, suggesting that the capsid protein(s) in leptophages are highly divergent from other known capsid proteins. Three non-structural phage proteins, including proteins involved in DNA replication (gp12) and recombination (gp8, gp58), were also identified as part of the phage particle by mass spectrometry analysis, suggesting a possible contamination of the samples during the phage purification process. Alternatively, high-sensitivity LC-MS/MS may have lowered detection limits, making it possible to analyze not only structural proteins but also encapsidated enzymes.
Taken together, our data show that, even if bacteriophages are hugely abundant and crucial for various ecosystems23, their genomic sequences are underrepresented in public databases, comparatively to bacteria. This is particularly true for leptophages.
Search of LE4-like prophages in Leptospira genomes
The phages LE3 and LE4 revealed strong DNA sequence identity, whereas no homologies to previously-known phage sequences were uncovered—even at the protein level. Comparison of the sequences of known or putative phages and prophages in Leptospira genomes suggested their partition into three main groups: the LE1-like (similar to the circular plasmid Laicp14), the Mu-like prophages11, and the LE4-like prophages. This latter group contains LE3, LE4, and two prophage-like regions in the pathogens L. mayottensis and L. interrogans (Figs 5 and 6). The LE4-like region in the pathogenic L. mayottensis strain 200901116 shares a strong synteny with the LE4 morphogenesis module and contains a phage-like replication origin (Fig. 4) that has been previously used to generate a replicative vector for pathogenic Leptospira strains24. A draft genome sequence of this strain with 84 contigs had been obtained previously from Illumina sequencing25. In an attempt to define all of the replicons present in L. mayottensis strain 200901116, the complete polished genome sequences were generated using the PacBio single-molecule real-time (SMRT) sequencing method, which is a powerful approach for discovering extrachromosomal replicons. The strain 200901116 consists of two circular chromosomes (3,815,263 bp in chromosome 1 and 307,019 bp in chromosome 2) and two circular plasmids of 94,000 bp (p1_L200901116) and 53,283 bp (p2_L200901116) exhibiting homologies to the LE1-like and LE4-like prophages, respectively. The LE4-like plasmid, p2_L200901116, has a GC content of 39.9% and contains 86 predicted CDS, with 74 CDS on the sense strand. Phage-related encoded proteins include a bacteriophage resistance factor (PF05565 family), phage terminase large subunit GpA-like protein, phage tail protein, phage tail tape measure protein, phage late control protein D, and phage tail collar protein. Again, genes encoding capsid proteins or lytic enzymes were not identified (Supplementary Table S2). At the protein level, 16 out of 86 predicted p2_L200901116 products are 49.76 ± 3.67% similar (29.66 ± 3.33% identical) to LE4 proteins. Plasmid p2_L200901116 also exhibits a high syntheny with the genes encoding putative structural proteins (gp19, gp21, gp22, gp23, gp28, gp29, gp31, gp33, gp34, gp36, gp37, gp40, and gp41) found in LE3 and LE4, suggesting that the morphogenesis module is conserved between the two phages.

Clustering and heatmap of phage/prophage regions in Leptospira species. The colors represent, as indicated in the legend, the average similarity values for the 10 pairs of sequences having the highest bitcore values.

Circular genomic map and protein identity comparison between LE4, p2_L20090116 and lcp3. The circles represent from the outside to inside (1) LE4 CDS, and putative homologous loci with (2) p2_L200901116, (3) LE1 and (4) lcp3. The regions sharing bitscore values greater than or equal to 50 are in red while those sharing values inferior to 50 are in purple.
A genetic relationship between LE3/LE4 and lcp3 was also established; lcp3 is a 55-kb circular plasmid from the pathogen L. interrogans serovar Linhai strain 56609 which produced tailed phage particles after induction with mitomycin C10. This plasmid, which contains genes encoding 30 phage-related proteins may, therefore, be a prophage-inducible plasmid of a lytic phage. In addition, another lcp3-related plasmid was also detected in L. interrogans serovar Australis strain 5660710. These phages (LE3 and LE4) and prophages (p2_L200901116 and lcp3) have a similarly sized (48–55 kb) circular genome and they likely share a common ancestor of lytic tailed phages.
In an attempt to induce the LE4-like prophage in L. mayottensis strain 200901116, cells were treated with mitomycin C, hydrogen peroxide and heat shock and then fixed for electron microscopy. We were not able to detect any phage particles in culture supernatants, suggesting that the prophage induction may be sensitive to other stimuli such as signals encountered in vivo during host-pathogen interactions. Alternatively, p2_L200901116 is defective for phage growth as a consequence of DNA rearrangements or other factors.
Concluding Remarks
In summary, the present study suggests that Leptospira possess an unknown diversity of phages, of which most have no homologues with in public databases. LE3 and LE4 are genetically distinct from the temperate phage LE1. Leptospira genome sequencing revealed LE4-like prophages existing as circular plasmids in the pathogens L. interrogans and L. mayottensis. We suggest that LE4 is the founder of a new group of phages, to which LE3, lcp3, and p2_L200901116 also belong. This study is also the first to demonstrate that LPS is a receptor for leptophages and suggests that LPS structure dictates host succeptibility.
Prophages play an important role in the genetic evolution and diversification of bacteria, including the emergence of virulence26,27. Since prophages are ubiquitous in the genomes of pathogenic Leptospira strains11, it is likely that they have contributed to the adaptation of pathogens to their hosts. Further sequencing of phage genomes will advance several aspects of the field including detection methods for prophages in Leptospira genomes, development of genetic tools (such as phage-based DNA delivery systems), identification of novel genes, prediction of definitive or putative functions of hypothetical proteins, and better understanding of the phages contribution to bacterial evolution and virulence.
Methods
Bacterial strains and growth media
Leptospira biflexa serovar Patoc strain Patoc 1 was isolated from Parisian stream water28 and maintained in the collection of the National Reference Centre for Leptospirosis (Institut Pasteur, Paris, France). L. mayottensis strain 200901116 was isolated from a patient with leptospirosis in Mayotte, Indian Ocean25. L. meyeri strain 201601301 was isolated from stream water in Mayotte. Leptospira strains were grown at 30 °C in liquid Ellinghausen, McCullough, Johnson and Harris (EMJH) medium29.
Bacteriophages
Phages LE1, LE3 and LE4 were isolated from sewage water in Paris, France7. Titration of phages was done in EMJH 1.2% agar medium with an EMJH 0.6% agar overlay. For phage isolation, 400 mL of an exponential phase culture of L. biflexa strain Patoc 1 (DO420nm = 0.1) was infected with 106 PFU of phages (MOI = 0.001), then incubated at room temperature for 30 days. The phage lysate was centrifuged for 30 min at 7,000 g and the supernatant was supplemented with 8% PEG 8000 and 0.3 M NaCl and incubated overnight at 4 °C as previously described30. The phage solution was centrifuged for 30 min at 7,000 g. The supernatant was discarded and the pellet was re-suspended in 10 mL Difco™ Leptospira Medium Base EMJH. To remove contaminants, 20% chloroform was added, then eliminated by centrifugation for 30 min at 1700g, and the phage lysate was filtered through a 0.1 μm syringe filter. Phages were purified by isopycnic gradient centrifugation in 45% caesium chloride (38,000 rpm, 24 h, 4 °C; Beckman Coulter rotor SW41) as previously described31.
Phage absorption and one-step growth kinetics
For phage adsorption assays, L. biflexa strain Patoc 1 and LEPBIa2019 mutant cultures were infected with LE4 at MOI of 0.0001 at room temperature and at different times after infection (0 min, 30 min, 1 h, 2 h, 3 h and 6 h), the number of unadsorbed phages remaining in the supernatant following immediate filtration through a 0.22 µm filter was determined by plating. Briefly, 100 µL of the filtrate was incubated with 100 µL of a culture of 108 bacteria per mL for 30 min then phage titration was determined by the double-layer agar plate method7, expressed as plaque-forming units per milliliter (PFU/mL).
One step growth experiments were done as previously described32. Following an adsorption period of 30 min at a MOI of 10, samples were obtained at 2 hours intervals, and immediately filtrated and plated for phage titration determined by the double-layer agar plate method as described above. The burst size was determined relative to the initial number of phages (t = 0).
Isolation of phage-resistant mutants and lysogeny verification
A liquid culture of L. biflexa infected with LE4 (MOI = 0.001) was incubated for 30 days, and then the lysed culture, still showing several viable Leptospira as revealed by dark-field microscopy, was inoculated into fresh EMJH liquid medium. Colonies of the phage-resistant strain were isolated from an EMJH plate and the colonies were confirmed to resist phage infection by spot assays (described below). A total of 22 resistant clones were isolated in three independent experiments and all were tested by PCR assay using primers targeting LE4 sequence (LE4_F2 5′-ATGCGGAATGTCTTGAAGGC-3′ and LE4_R 5′-TGTCAAGGATGCGGTAGGTT-3′) to detect potential lysogeny. We then selected one phage-resistant mutant strain, called RLE4, for subsequent analysis.
For targeted mutagenesis, a spectinomycin resistance cassette replacing the coding sequence of LEPBIa2019 and flanked by 0.9 kb sequences homologous to the sequences flanking the target gene was synthesized by GeneArt (Life Technologies, Grand Island, NY, USA) and cloned in a pMK-RQ E. coli vector, which is not able to replicate in Leptospira. Allelic exchange was obtained by transforming the UV-pretreated suicide plasmid by electroporation as previously described9 with a Biorad Gene Pulser Xcell™.
Spot assays of both L. biflexa WT, RLE4, and LEPBIa2019 mutant strains were conducted as three independent replicates to determine the strain susceptibility to phage infection. One millilitre of an exponential phase culture (DO420nm = 0.1) was spread onto EMJH plates and, one hour later, drops containing 102, 103, or 104 PFU of LE1, LE3, or LE4 were spotted on lawns and then incubated at 30 °C for 4 days.
Electron Microscopy
Bacteriophages were allowed to adsorb onto a carbon-coated copper grid and negatively stained with 4% uranyl acetate33. Grids were observed under an FEI Tecnai T12 Transmission Electron Microscope (TEM) with an acceleration voltage of 120 kV and at a magnification of 49,000. To observe phage infection and host cell lysis, we used a MOI of 10 and a 2% glutaraldehyde fixation was performed prior negative staining; non-infected cells were used as controls.
Mass spectrometry
Mass spectrometry analysis was performed on LE3 and LE4 (titres of 109 PFU/mL and 1010 PFU/mL, respectively) purified by caesium chloride density gradient centrifugation. In order to get rid of BSA contained in the EMJH medium, a step of depletion was performed using a BSA Depletion Column according to the manufacturer protocol (IgY Kit, GenWay Biotech, San Diego, CA, USA). To remove contamination by PEG 8000 used during purification a 10 kDa filtration step was performed using an Amicon Ultra-0.5 ml Ultracel-10k (Millipore). After reduction and alkylation, proteins were digested by 0.5 μg of Sequencing-Grade Modified Trypsin (Promega, Madison, WI, USA) overnight at 37 °C according to the eFASP protocol34.
Tryptic peptides were analyzed on a Q Exactive Plus instrument (Thermo Fisher Scientific, Bremen) coupled with an EASY nLC 1200 chromatography system (Thermo Fisher Scientific). Five µL was loaded on an in-house packed 50 cm nano-HPLC column (75 μm inner diameter) with C18 resin (1.9 μm particles, 100 Å pore size, Reprosil-Pur Basic C18-HD resin, Dr. Maisch GmbH, Ammerbuch-Entringen, Germany) and equilibrated in 97% solvent A and 3% solvent B (ACN, 0.1% FA). Peptides were first eluted using a 3 to 22% gradient of solvent B for 112 min, then a 22 to 38% gradient of solvent B for 35 min and finally a 38 to 56% gradient of solvent B for 15 min all at 250 nL/min flow rate. The instrument method for the Q Exactive Plus was set up in the data dependent acquisition mode. After a survey scan in the Orbitrap (resolution 70,000), the 10 most intense precursor ions were selected for HCD fragmentation with a normalized collision energy set up to 27. Charge state screening was enabled, and precursors with unknown charge state or a charge state of 1 and >7 were excluded. Dynamic exclusion was enabled for 45 s. All data were searched using Andromeda with MaxQuant software35 version 1.5.3.8. Five amino acids were required as minimum peptide length and 1 unique peptide was required for protein identification. A false discovery rate (FDR) cutoff of 1% was applied at the peptide and protein levels.
Genome sequencing
Phages purified by a caesium chloride gradient were dialysed in Leptospira Medium Base EMJH (dialysis cassette 2000 MWCO, Thermo Fisher). Phage solutions were treated with DNase and RNase, and EDTA was used to terminate the reactions. Phage DNA was then extracted using the QIamp Minelute virus spin kit (Qiagen) and amplified with Repli-g mini kit (Qiagen). For whole genome sequencing of L. biflexa Patoc strains, DNA was extracted using a MagNA Pure 96 Instrument. Next-generation sequencing was performed by the Mutualized Platform for Microbiology (P2M) at Institut Pasteur, using the Nextera XT DNA Library Preparation kit (Illumina), the NextSeq 500 sequencing systems (Illumina), and the CLC Genomics Workbench 9 software (Qiagen) for analysis.
Genomic DNA of L. mayottensis strain 200901116 was extracted with the Genomic tip 100 g kit (Qiagen) from a 35 ml culture according to manufacturer protocols and the complete genome sequence was obtained using SMRT (Pacific Biosciences) technology. PacBio sequencing was performed at the Génome Québec Innovation Centre (McGill University, Montreal, Canada) using a Pacific BioScience RS II system. The sequencing reads were de novo assembled using HGAP36 through SMRT Analysis.
Bioinformatics analysis
Genome annotation was performed with the MaGe interface (https://www.genoscope.cns.fr/agc/mage)37. LE3 and LE4 sequences are available in this interface. Similarity searches were performed using BLASTp38 (with a BLASTp e-value of 1 × 10−3 as a cut-off) and conserved domains. ORF denomination in Table 1 and Supplementary Table S1 correspond to MaGe nomenclature.
We compared the Single-nucleotide polymorphisms (SNPs) of mutant RLE4 against the wild-type parental strain. Sequence reads were aligned with the annotated L. biflexa serovar Patoc strain Paris I genome (accession numbers CP000786, CP000787 and CP00078839) by using the Burrows-Wheeler Alignment tool (BWA mem 0.7.5a)40. SNP calling was done with the Genome Analysis Toolkit Unified Genotyper (GATK 2.7-2)41 by following Broad Institute best practices. Candidate SNPs were further filtered by requiring coverage of greater than half of the genome mean coverage and 95% read agreement to validate the call. SNPs, short indels, and coverage were visualized with SynTView42. The data are accessible through the following link: http://hub18.hosting.pasteur.fr/SynTView/flash/Leptospira/Leptospira_biflexa.html. This finding was confirmed by sequencing the PCR product of this region amplified with primers Pat2019-F (5′-GATGGAACATGATCCAACTA-3′) and Pat2019-R (5′-GAATTGAATGGTCTTGGCACA-3′).
Average similarity values were computed using the 10 hits having the highest bitscore values (blastp argument of DIAMOND43) for all combinations of phages presented in Fig. 5. Since some (pro)phages do not share 10 ORF, the average similarities values were multiplied by (n/10), where n is the number of shared ORF. The resulting square matrix was clustered using heatmap.2 from the statistical package R44. The map of LE4 and the tblastx results presented in Fig. 6 were visualized using circleator version 1.0.045.
Nucleotide sequence accession number
The LE1 bacteriophage genome accession number is BX571876. The nucleotide sequences of LE3 and LE4 bacteriophages have been deposited in the GenBank database as accession numbers MF974396 and MF974397, respectively. The sequence of Leptospira mayottensis strain 200901116 was deposited in GenBank under the accession number CP024871 (chromosome I), CP024872 (chromosome II), CP024873 (plasmid p1_200901116), and MF974398 (plasmid p2_200901116).
References
Doss, J., Culbertson, K., Hahn, D., Camacho, J. & Barekzi, N. A Review of Phage Therapy against Bacterial Pathogens of Aquatic and Terrestrial Organisms. Viruses 9, 50 (2017).
Koskella, B. & Brockhurst, M. A. Bacteria–phage coevolution as a driver of ecological and evolutionary processes in microbial communities. Fems Microbiol. Rev. 38, 916–931 (2014).
Klimenko, A. I., Matushkin, Y. G., Kolchanov, N. A. & Lashin, S. A. Bacteriophages affect evolution of bacterial communities in spatially distributed habitats: a simulation study. BMC Microbiol. 16, S10 (2016).
Picardeau, M. Virulence of the zoonotic agent of leptospirosis: still terra incognita? Nat. Rev. Microbiol. https://doi.org/10.1038/nrmicro.2017.5 (2017).
Costa, F. et al. Global Morbidity and Mortality of Leptospirosis: A Systematic Review. PLoS Negl. Trop. Dis. 9, e0003898 (2015).
Kropinski, A. M., Prangishvili, D. & Lavigne, R. Position paper: The creation of a rational scheme for the nomenclature of viruses of Bacteria and Archaea. Environ. Microbiol. 11, 2775–2777 (2009).
Saint Girons, I., Margarita, D., Amouriaux, P. & Baranton, G. First isolation of bacteriophages for a spirochaete: Potential genetic tools for Leptospira. Res. Microbiol. 141, 1131–1138 (1990).
Bourhy, P. et al. Complete Nucleotide Sequence of the LE1 Prophage from the Spirochete Leptospira biflexa and Characterization of Its Replication and Partition Functions. J. Bacteriol. 187, 3931–3940 (2005).
Saint Girons, I. et al. The LE1 Bacteriophage Replicates as a Plasmid within Leptospira biflexa: Construction of an L. biflexa-Escherichia coli Shuttle Vector. J. Bacteriol. 182, 5700–5705 (2000).
Zhu, W. et al. Identification of three extra-chromosomal replicons in Leptospira pathogenic strain and development of new shuttle vectors. BMC Genomics 16, 90 (2015).
Fouts, D. E. et al. What makes a bacterial species pathogenic?: Comparative genomic analysis of the genus Leptospira. PLoS Negl Trop Dis 10, e0004403 (2016).
Ricaldi, J. N. et al. Whole Genome Analysis of Leptospira licerasiae Provides Insight into Leptospiral Evolution and Pathogenicity. PLoS Negl. Trop. Dis. 6, e1853 (2012).
Qin, J.-H. et al. Identification of a Novel Prophage-Like Gene Cluster Actively Expressed in Both Virulent and Avirulent Strains of Leptospira interrogans Serovar Lai. Infect. Immun. 76, 2411–2419 (2008).
Huang, L. et al. Re-characterization of an extrachromosomal circular plasmid in the pathogenic Leptospira interrogans serovar Lai strain 56601. Acta Biochim. Biophys. Sin. 46, 605–611 (2014).
Bourhy, P. et al. A Genomic Island of the Pathogen Leptospira interrogans Serovar Lai Can Excise from Its Chromosome. Infect. Immun. 75, 677–683 (2007).
Satou, K. et al. Complete Genome Sequences of Low-Passage Virulent and High-Passage Avirulent Variants of Pathogenic Leptospira interrogans Serovar Manilae Strain UP-MMC-NIID, Originally Isolated from a Patient with Severe Leptospirosis, Determined Using PacBio Single-Molecule Real-Time Technology: Table 1. Genome Announc. 3, e00882–15 (2015).
Picardeau, M. Toolbox of Molecular Techniques for Studying Leptospira Spp. Curr. Top. Microbiol. Immunol. https://doi.org/10.1007/82_2017_45 (2017).
Ackermann, H.-W. Bacteriophage Electron Microscopy. Adv. Virus Res. 82, 1–32 (2012).
Manning, A. J. & Kuehn, M. J. Contribution of bacterial outer membrane vesicles to innate bacterial defense. BMC Microbiol. 11, 258 (2011).
Garcia-Doval, C. & Raaij, M. J. van. Bacteriophage Receptor Recognition and Nucleic Acid Transfer. In Structure and Physics of Viruses 489–518 https://doi.org/10.1007/978-94-007-6552-8_17 (Springer, Dordrecht, 2013).
Broeker, N. K. & Barbirz, S. Not a barrier but a key: How bacteriophages exploit host’s O-antigen as an essential receptor to initiate infection. Mol. Microbiol. 105, 353–357 (2017).
Knirel, Y. A. et al. Variations in O-Antigen Biosynthesis and O-Acetylation Associated with Altered Phage Sensitivity in Escherichia coli 4s. J. Bacteriol. 197, 905–912 (2015).
Sime-Ngando, T. Environmental bacteriophages: viruses of microbes in aquatic ecosystems. Front. Microbiol. 5 (2014).
Pappas, C. J., Benaroudj, N. & Picardeau, M. A Replicative Plasmid Vector Allows Efficient Complementation of Pathogenic Leptospira Strains. Appl. Environ. Microbiol. 81, 3176–3181 (2015).
Bourhy, P., Collet, L., Brisse, S. & Picardeau, M. Leptospira mayottensis sp. nov., a pathogenic species of the genus Leptospira isolated from humans. Int. J. Syst. Evol. Microbiol. 64, 4061–4067 (2014).
Casjens, S. Prophages and bacterial genomics: what have we learned so far? Mol. Microbiol. 49, 277–300 (2003).
Bille, E. et al. A virulence-associated filamentous bacteriophage of Neisseria meningitidis increases host-cell colonisation. PLOS Pathog. 13, e1006495 (2017).
Babudieri B. Cagliari-Sassari. Studio serologico del gruppo Semaranga-Patoc di Leptospira biflexa. 408–414 (1961).
Johnson, R. C. & Harris, V. G. Differentiation of Pathogenic and Saprophytic Leptospires I. Growth at Low Temperatures. J. Bacteriol. 94, 27–31 (1967).
Colombet, J. et al. Virioplankton ‘pegylation’: Use of PEG (polyethylene glycol) to concentrate and purify viruses in pelagic ecosystems. J. Microbiol. Methods 71, 212–219 (2007).
Mirande, M., Lazard, M. & Waller, J.-P. Small-scale purification of bacteriophage λ DNA by an airfuge centrifugation step in cesium chloride gradients. Gene Anal. Tech. 5, 80–82 (1988).
Kropinski, A. M. Practical Advice on the One-Step Growth Curve. In Bacteriophages 41–47 https://doi.org/10.1007/978-1-4939-7343-9_3 (Humana Press, New York, NY, 2018).
Børsheim, K. Y., Bratbak, G. & Heldal, M. Enumeration and biomass estimation of planktonic bacteria and viruses by transmission electron microscopy. Appl. Environ. Microbiol. 56, 352–356 (1990).
Erde, J., Loo, R. R. O. & Loo, J. A. Enhanced FASP (eFASP) to Increase Proteome Coverage and Sample Recovery for Quantitative Proteomic Experiments. J. Proteome Res. 13, 1885–1895 (2014).
Tyanova, S., Temu, T. & Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 11, 2301 (2016).
Chin, C.-S. et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569 (2013).
Vallenet, D. et al. MicroScope in 2017: an expanding and evolving integrated resource for community expertise of microbial genomes. Nucleic Acids Res. 45, D517–D528 (2017).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Picardeau, M. et al. Genome Sequence of the Saprophyte Leptospira biflexa Provides Insights into the Evolution of Leptospira and the Pathogenesis of Leptospirosis. PLoS ONE 3, e1607 (2008).
Li, H. & Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010).
McKenna, A. et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Lechat, P., Souche, E. & Moszer, I. SynTView — an interactive multi-view genome browser for next-generation comparative microorganism genomics. BMC Bioinformatics 14, 277 (2013).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
R Core Team. R: A language and environment for statistical computing. (2016). Available at, https://www.R-project.org/, (Accessed: 17th October 2017).
Crabtree, J. et al. Circleator: flexible circular visualization of genome-associated data with BioPerl and SVG. Bioinformatics 30, 3125–3127 (2014).
Acknowledgements
This work is part of a PhD thesis of O. S. who received financial support from “Université Paris Diderot” and “Sorbonne Paris Cité”. We thank Olivier Gorgette (Ultrapole, Institut Pasteur) and Soizick Lucas Staat (BMGE Unit, Institut Pasteur) for assistance in electron microscopy and phage purification, respectively. We thank Nathalie Armatys for technical support in control experiments. We thank Isabelle Saint Girons who layed the foundation for this work at the Institut Pasteur, and Robert Gaultney for proofreading the manuscript.
Author information
Authors and Affiliations
Contributions
O.S. designed and performed the experiments, analyzed the data and wrote the manuscript. A.T.V. analyzed the genomes and revised the manuscript. C.M. performed the mass spectrometry analysis. P.L. performed the SNP identification. J.C.R. designed the mass spectrometry analysis. F.J.V. revised the manuscript. M.P. assisted with experimental design and wrote the manuscript. P.B. conceived the study, assisted with experimental design and interpretation of data and edited manuscript. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schiettekatte, O., Vincent, A.T., Malosse, C. et al. Characterization of LE3 and LE4, the only lytic phages known to infect the spirochete Leptospira. Sci Rep 8, 11781 (2018). https://doi.org/10.1038/s41598-018-29983-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29983-6
This article is cited by
-
Leptospira interrogans Retains Direct Virulence After Long Starvation in Water
Current Microbiology (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
