
Abstract
Bi2O2Se is an emerging semiconducting, air-stable layered material (Nat. Nanotechnol. 2017, 12, 530; Nano Lett. 2017, 17, 3021), potentially exceeding MoS2 and phosphorene in electron mobility and rivalling typical Van der Waals stacked layered materials in the next-generation high-speed and low-power electronics. Holding the promise of functional versatility, it is arousing rapidly growing interest from various disciplines, including optoelectronics, thermoelectronics and piezoelectronics. In this work, we comprehensively study the electrical properties of the native point defects in Bi2O2Se, as an essential step toward understanding the fundamentals of this material. The defect landscapes dependent on both Fermi energy and the chemical potentials of atomic constituents are investigated. Along with the bulk defect analysis, a complementary inspection of the surface properties, within the simple context of charge neutrality level model, elucidates the observed n-type characteristics of Bi2O2Se based FETs. This work provides important guide to engineer the defects of Bi2O2Se for desired properties, which is key to the successful application of this emerging layered material27.
Similar content being viewed by others


Introduction
The richness of exotic physical properties of layered materials, such as graphene1,2,3, transition-metal dichalcogenides4,5, black phosphorous6, indium selenide7, has given rise to diverse intriguing applications, including electronic logic, memory devices8,9,10 and optoelectronic devices11,12,13. The extraordinariness of layered materials has also captivated the broad spintronics2 and piezoelectronics14 communities. Apart from the unprecedented device functionalities, another natural reason to adopt layered materials is the room for device scaling to low dimension. Hitherto, however, no single layered material stands out as being desirable in all technological aspects. For example, graphene is gapless and therefore unsuitable for conventional transistors, black phosphorous is not air-stable. Thus, immense interest has been sparked in exploring a wider range of layered materials.
Recently, semiconducting Bi2O2Se layered material has been synthesized, exhibiting thickness-dependent band gap and air-stability15,16. What’s more, it is anticipated to have lower in-plane electron effective mass than those of MoS2 and black phosphorous, which has been evidenced by the ultrahigh electron Hall mobility and quantum oscillation at low temperature15. Field-effect-transistors (FETs) based on Bi2O2Se show encouraging performance and substantial room for further optimization15, making Bi2O2Se a promising candidate for future high-speed and low-power electronic applications. The combination of its exceptional optical and electrical properties has also been exploited in integrated photodetectors of high photoresponsitivity at selective wavelength, holding the promise for next-generation optoelectronic systems17. Preceding the showcase in modern electronics, Bi2O2Se was a traditional thermoelectric material18. The keen interest in the functional versatility of Bi2O2Se has been propelled by the theoretical prediction of its larger piezoelectricity and ferroelectricity, under in-plane strain, than those of monolayer MoS219, which opens up prospects for energy conversion devices, sensors and non-volatile ferroelectric memories.
Although Bi2O2Se is gaining increasing interest from various disciplines, understanding the fundamentals of Bi2O2Se as a semiconductor, such as the native point defects, is still premature. Knowing the behaviors of native point defects is essential to the successful application of any semiconductors. These defects, which are naturally present in certain amount, control directly or indirectly the electrical and optical properties of the materials, such as the electrical conductivity. The defect concentration is governed primarily by defect formation energies, especially during the prolonged anneals at elevated temperatures. Thus the formation energies of individual native defects are quantities of central importance. In this letter, we comprehensively investigate the native point defects of Bi2O2Se by first-principles calculations. The results provide an important guide to engineer Bi2O2Se for desired properties and design functional Bi2O2Se devices.
Results and Discussion
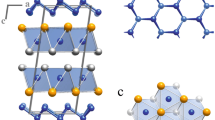
Our calculations are based on density functional theory within the generalized gradient approximation,20 using the Cambridge Sequential Total Energy Package21. 90 atoms’ Bi2O2Se supercell is used as the host of various native point defects, where the lattice constants are fixed to the calculated values. Cutoff energy of the plane wave basis set is 680 eV. All atoms are relaxed in each optimization cycle until atomic forces on each atom are smaller than 0.01 eV Å−1 and the energy variation between subsequent iterations falls below 5 × 10−6 eV. Total energies are evaluated on 3 × 3 × 3 Monkhorst−Pack k-meshes. Unlike other layered materials which have individual atomic layers stacked by van der Waals interactions, Bi2O2Se lacks a well-defined van der Waals gap but displays out-of-plane electrostatic interactions between planar covalently bonded oxide layer (Bi2O2) and Se square array, as shown in Fig. 1a. The calculated band structure and atomic projected density of states (PDOSs) are shown in Fig. 1b. Indirect band gap of 0.76 eV with conduction band minimum (CBM) near Γ point is in good agreement with the value of 0.80 eV measured by angle-resolved photoemission spectroscopy15. The electronic states near the CBM and the valence band maximum (VBM) originate mainly from the Bi and Se/O p-orbital bands, respectively.

(a) Atomic structure of Bi2O2Se, (b) band structure of Bi2O2Se, (c) PDOSs of Bi2O2Se.
Bi2O2Se is a ternary semiconductor with ample defect configurations. We consider ten of them in this work, including vacancies, interstitials and antisites in the relevant charge states. The formation energy \({{\rm{\Delta }}{\rm{H}}}_{{\rm{f}}}({\rm{\alpha }},{\rm{q}})\) of defect α in charge state q depends on the chemical potentials μ of the atomic constituents as well as the electron chemical potential, namely, Fermi energy εF. In Bi2O2Se,
where \({\rm{E}}({\rm{\alpha }},{\rm{q}})\) is the total energy of the supercell containing a type α defect and charge q, \({\rm{E}}({{\rm{Bi}}}_{2}{{\rm{O}}}_{2}{\rm{Se}})\) is the total energy of the defect free supercell, n’s and q are the numbers of the atoms and electrons, respectively, that transferred from the defect free supercell to the reservoirs in forming the defect cell. CFreysoldt is the charge state and cell size correction to the defect-formation energy22. According to Freysoldt et al., the correction consists of three contributions: a lattice term, a self-interaction term and a potential alignment term22. The lattice term accounts for the electrostatic interaction of the defect charge in the supercell with its array of periodic images in the remaining crystal. We use Gaussian defect charge distribution. The lattice energy includes the self-interaction term of the defect charge with its own potential, which must be removed from the correction term. The potential alignment term allows for a meaningful comparison of the formation energies of different charged defects as the charged defect in the supercell will introduce a constant shift of the electrostatic potential and the valence band maximum compared to the ideal host system. As the dielectric anisotropy in layered material systems could be strong, we generalize the Freysoldt scheme to account for the anisotropy. This is achieved by using the dielectric tensor for the calculation of Coulomb interaction potential in the reciprocal space. The dielectric tensor is computed from density-functional perturbation theory. A correction for filling the CBM and emptying the VBM has also been considered23. Freysoldt correction leads to well-converged defect formation energies (Fig. S1). The chemical potentials are allowed to vary over a restricted range determined by equilibrium thermodynamics24: εF is bound between the VBM and CBM of Bi2O2Se, and μ’s are bound by the values that (i) will cause precipitation of solid elemental Bi in the trigonal phase, molecular O and solid elemental Se in the trigonal phase, i.e.,
(ii) maintain the stable Bi2O2Se compound, i.e.,
(iii) will cause the formation of solid binaries Bi2O3 and Bi2Se3 in the monoclinic and trigonal phases, respectively, i.e.,
The calculated range of atomic chemical potentials for stable Bi2O2Se is shown on the two-dimensional “μO vs μSe” plane in Fig. 2. The formation of Bi2O3 and Bi2Se3 from their component elements is exothermic by 4.0 eV and 1.2 eV per Bi, respectively. The formation of Bi2O2Se from its component binary oxides is exothermic by 0.05 eV per Bi.

Calculated range of atomic chemical potentials for stable Bi2O2Se. Selected (Se-poor, Bi-rich) and (Se-rich, Bi-poor) conditions are labelled by red point A and B, respectively.
We first consider anion deficiency related defects, including O and Se vacancies (Ov, Sev), Bi interstitial (Biin) and Bi antisites (BiO, BiSe). The defect formation energies as a function of Fermi energy εF are shown in Fig. 3, with values of the atomic chemical potentials μO and μSe assigned at two representative points on the “μO vs μSe” plane, respectively. The kinks in the curves for a given defect indicate transitions between different charge states. For both Ov and Sev, they occur exclusively in the positively charged state (charge states from 0 to 2+ are considered) and therefore act as shallow donors. The ξ(2+/1+) transition levels for Ov and Sev occur at 0.61 eV and 0.64 eV above the VBM, respectively. The atomic structures of Ov1+ and Sev1+ are shown in Fig. 4a,b. PDOSs (Fig. S2ab) of Ov° and Sev° provide alternative way of understanding the shallow donor effect of Ov and Sev, where we see that electrons are readily provided to the conduction band by thermal excitation at steady state. Sev remains to be the most stable defect throughout the band gap under the (Se-poor, Bi-rich) condition (Fig. 3a). Ov has slightly higher formation energy. The unintentional n-type doping of Bi2O2Se is therefore likely to be due to the existence of Sev and Ov. Under the (Se-rich, Bi-poor) condition, their formation energies increase and they dominate only in the p-type Bi2O2Se, counteracting the p-type conductivity.


Atomic structures of (a) Ov1+ and (b) Sev1+.
We then consider Biin at the center of the Se square, which is found to be energetically favorable compared with other interstitial sites. Biin occurs exclusively in the positively charged state (charge states from 3− to 3+ are considered), thus acting as a shallow donor. A ξ(3+/2+) transition level occurs at 0.19 eV above the VBM. However, because Biin has quite high formation energy in n-type Bi2O2Se even under the (Se-poor, Bi-rich) condition, it is not likely to be present to drive unintentional conductivity of Bi2O2Se. The atomic structure of Biin2+ is shown in Fig. 5a. It can be seen that the interstitial Bi atom repels the two Bi atoms right below and above in the Bi2O2 layers to the centers of the O squares.

Atomic structures of (a) Biin2+ and (b) neutral BiSe.
Next, we consider two kinds of Bi antisites. For BiSe, a ξ(1+/0) transition level occurs at 0.30 eV above the VBM (charge states from 1− to 5+ are considered), indicating that it is a deep donor center. It is the third most stable defect next to Sev and Ov in p-Bi2O2Se under the (Se-poor, Bi-rich) condition, compensating the p-type conductivity. The formation energy of BiSe rapidly increases with anion chemical potentials and BiSe becomes less likely to exist under the (Se-rich, Bi-poor) condition. The atomic structure of neutral BiSe is shown in Fig. 5b.
For BiO, transition levels ξ(4+/2+) and ξ(2+/0) occur at 0.33 eV and 0.50 eV above the VBM, respectively (charge states from 1− to 5+ are considered). Therefore, it is a deep donor center and it compensates the p-type conductivity of Bi2O2Se. However, it has quite high formation energy even in the (Se-poor, Bi-rich) condition, so it is not likely to exist. It is obvious that BiO is a negative-U defect, with transition level ξ(4+/3+) higher than ξ(3+/2+), and ξ(2+/1+) higher than ξ(1+/0). Negative-U behavior has been typically related to unusually large local lattice relaxations that stabilize particular charge states. Here, substitutional Bi in the 4+ charged state undergoes displacement ~1 Å vertical to the Bi2O2 plane (Fig. 6), whereas it remains almost onsite in the case of BiO3+ and BiO2+. At the same time, there is ~0.2 Å difference in the displacement of a neighboring Bi atom vertical to the Bi2O2 plane between BiO° and BiO1+/ BiO2+, whereas the latter two are comparable (not shown).

Atomic structures of (a) BiO4+ and (b) neutral BiO2+.
Next, we investigate cation deficiency related native point defects, including O and Se interstitials (Oin, Sein), Bi vacancies (Biv) and O/Se antisites (OBi, SeBi). For Oin right above a Se ion, which is found to be energetically favorable, it repels the Se ion out of the Se square plane and forms seleninyl ion SeO−2+q (charge states q from 2− to 2+ are considered). The atomic structure of Oin° is shown in Fig. 7a. Oin remains neutral throughout the band gap, rendering on average −1 intermediate oxidation state for Se and O atoms in the seleninyl ion. Its formation energy is relatively high under the (Se-poor, Bi-rich) condition compared with Ov and Sev, but is reduced under the (Se-rich, Bi-poor) condition, becoming the most favorable defect in the Fermi energy range between 0.13 eV and 0.58 eV above the VBM. For Sein at the edge center of the Se square, which is found to be energetically favorable, it repels the two neighboring Se ions at the vertices, forming triselenide anion Se3−4+q (charge states q from 2− to 4+ are considered). The atomic structure of Sein° is shown in Fig. 7b. Sein assumes positively charged state as long as the Fermi level is below the transition level ξ(1+/0) at 0.26 eV above the VBM, acting as an acceptor compensating center in p-type Bi2O2Se. On average, each Se atom in the triselenide anion acquires intermediate oxidation state between −2 and 0. It has higher formation energy than Oin does. PDOSs of Oin° (Sein0) (Fig. 7c,d) show that the seleninyl ion SeO−2 (triselenide anion Se3−4) has filled antibonding frontier orbital states below (above) the VBM. It is well known that chalcogen elements (S, Se, Te) have strong tendency to form polychalcogenide anion XN−Q (X: chalcogen element) of variable chain length N, with average oxidation state assumed by each atom intermediate between −2 and 0. This rationalizes the favorable nonnegative charge states for Oin and Sein.

Atomic structures of charge neutral (a) Oin and (b) Sein, PDOSs of charge neutral (c) Oin and (d) Sein. The vertical dash lines indicate the VBM and CBM of the host system, drawn from the PDOSs of the atoms in bulk which serve as references.
Next, we consider Biv. It is a shallow acceptor which occurs exclusively in the negative charge state, with transition levels ξ(1−/2−) and ξ(2−/3−) occur at 0.28 eV and 0.66 eV above the VBM, respectively (charge states from 3- to 0 are considered). PDOSs (Fig. S3) of Biv0 provide alternative way of understanding the shallow acceptor effect of Biv, where we see that it is ready to accept electrons near the VBM by thermal excitation at steady state, leaving holes in the valence band. The formation energy of Biv under the (Se-poor, Bi-rich) condition is relatively high compared with those of Sev and Ov. Under the (Se-rich, Bi-poor) condition, however, the formation energy of Biv rapidly decreases and Biv becomes the dominant defect in n-type Bi2O2Se, compensating the prevalent conductivity. The atomic structure of Biv3− is shown in Fig. 8a.

Atomic structure of (a) Biv3−, (b) OBi2− and (c) SeBi1−.
Finally, we consider OBi and SeBi. OBi occurs exclusively in the negative charge state (charge states from 5− to 1+ are considered) and acts as a shallow acceptor. The transition level ξ(1−/2−) occur at 0.28 eV above the VBM. The atomic structure of OBi2− is shown in Fig. 8b. The substitutional O atom undergoes large displacement to the Se square layer, locating in the center of two neighboring Se atoms. The formation energy of OBi is high even under the (Se-rich, Bi-poor) condition. Thus, it is not likely to exist.
For SeBi, the transition level ξ(0/1−) occurs at 0.31 eV above the VBM. Thus, it compensates donors. The formation energy of SeBi under the (Se-poor, Bi-rich) condition is quite high but rapidly decreases with increasing anion chemical potentials. Under the (Se-rich, Bi-poor) condition, SeBi becomes the second most likely compensating center next to Biv in n-type Bi2O2Se. The atomic structure of SeBi1− in Fig. 8c shows that the substitutional Se atom displaces toward the center of the underlying Se square.
Here to, we have studied the electrical properties of ten types of native point defects. As previously pointed out, the electrical conductivity of material can be significantly affected by its native point defects. In Bi2O2Se FETs, whose channels remain conducting at Vg = 0, the total resistance decreases with increasing gate bias15, which is a clear signature of n-type characteristics. Figure 3 implies the possibility of (Se-poor, Bi-rich) fabrication condition for the Bi2O2Se FETs since Sev and Ov, which are shallow donors, are the most likely defects under this condition.
The conductivity of Bi2O2Se can alternatively be understood within the context of charge neutrality level (CNL) model25. The CNL model is useful because it is simple and gives good chemical trends, while requiring no specified details of surface chemical bonding which are outside of the scope of this work. The CNL is the demarcation between the surface states that are predominantly donor-like (valence band states) and acceptor-like (conduction band states), namely, at CNL they have equal densities. Mathematically, the CNL is the branch point of the imaginary bulk band structure of the semiconductor. It is calculated as the zero of the Greens function of the band structure averaged over the Brillouin zone:
where δ is a small number to be used if the CNL lies inside a band. It can also be expressed as a sum over special points of the Brillouin zone (such as the Monkhorst–Pack grid)26:
The CNL is then a weighted average of the valence and conduction band DOS:
In this definition, the CNL is an intrinsic property of the bulk semiconductor; it does not depend on the interface, or interface bonding, or whatever it is attached to.
According to equation (7), the CNL of Bi2O2Se is calculated to be ~0.7 eV above the VBM, very close to the CBM. In the absence of gate bias, the Fermi level at the surface of Bi2O2Se is aligned with the CNL to ensure charge neutrality, resulting in surface electron accumulation. This explains why Bi2O2Se based FETs are in the ON state when Vg = 0 and show n-type behavior.
Conclusion
In summary, we have systematically studied all anion and cation deficiency related native point defects of Bi2O2Se in all relevant charge states. The abounding defect behaviors resulting from the ternary elemental compositions and the unique stacking structure are analyzed. Defect landscape is found to vary with Fermi energy and the chemical potentials of the atomic constituents. Our results suggest the possibility of (Se-poor, Bi-rich) fabrication condition of the previously reported Bi2O2Se FETs15. Under this condition, Sev and Ov are the dominant defects and they act as shallow donors, accounting for the unintentional n-type conductivity of Bi2O2Se. Alternatively, the n-type characteristics of Bi2O2Se FETs can also be understood in the context of the CNL model. The CNL of Bi2O2Se is computed to be close to the CBM, resulting in surface electron accumulation. This work provides important guide to engineer the defects of Bi2O2Se for desired properties, which is key to the successful application of this emerging layered material.
References
Castro Neto, A. H., Guinea, F., Peres, N. M. R., Novoselov, K. S. & Geim, A. K. The Electronic Properties of Graphene. Rev. Mod. Phys. 81, 109–162 (2009).
Han, W., Kawakami, R. K., Gmitra, M. & Fabian, J. Graphene Spintronics. Nat. Nanotechnol. 9, 794–807 (2014).
Bonaccorso, F., Sun, Z., Hasan, T. & Ferrari, A. C. Graphene Photonics and Optoelectronics. Nat. Photonics 4, 611–622 (2010).
Wang, Q. H., Kalantar-Zadeh, K., Kis, A., Coleman, J. N. & Strano, M. S. Electronics and Optoelectronics of Two-Dimensional Transition Metal Dichalcogenides. Nat. Nanotechnol. 7, 699–712 (2012).
Xu, X., Yao, W., Xiao, D. & Heinz, T. F. Spin and Pseudospins in Layered Transition Metal Dichalcogenides. Nat. Phys. 10, 343–350 (2014).
Xia, F., Wang, H. & Jia, Y. Rediscovering Black Phosphorus as an Anisotropic Layered Material for Optoelectronics and Electronics. Nat. Commun. 5(1–6), 4458 (2014).
Bandurin, D. A. et al. High Electron Mobility, Quantum Hall Effect and Anomalous Optical Response in Atomically Thin InSe. Nat. Nanotechnol. 12, 223–228 (2016).
Radisavljevic, B., Radenovic, A., Brivio, J., Giacometti, V. & Kis, A. Single-Layer MoS2 Transistors. Nat. Nanotechnol. 6, 147–150 (2011).
Li, L. et al. Black Phosphorus Field-Effect Transistors. Nat. Nanotechnol. 9, 372–377 (2014).
Bessonov, A. A. et al. Layered Memristive and Memcapacitive Switches for Printable Electronics. Nat. Mater. 14, 199–204 (2015).
Wu, J. et al. Organic Light-Emitting Diodes on Solution-Processed Graphene Transparent Electrodes. ACS Nano 4, 43–48 (2009).
Liu, Z. et al. Organic Photovoltaic Devices Based on a Novel Acceptor Material: Graphene. Adv. Mater. 20, 3924–3930 (2008).
Lopez-Sanchez, O., Lembke, D., Kayci, M., Radenovic, A. & Kis, A. Ultrasensitive Photodetectors Based on Monolayer MoS2. Nat. Nanotechnol. 8, 497–501 (2013).
Wu, W. et al. Piezoelectricity of Single-Atomic-Layer MoS2 for Energy Conversion and Piezotronics. Nature 514, 470–474 (2014).
Wu, J. et al. High Electron Mobility and Quantum Oscillations in Non-Encapsulated Ultrathin Semiconducting Bi2O2Se. Nat. Nanotechnol. 12, 530–534 (2017).
Wu, J. et al. Controlled Synthesis of High-Mobility Atomically Thin Bismuth Oxyselenide Crystals. Nano Lett. 17, 3021–3026 (2017).
Wu, J. et al. Chemical Patterning of High-Mobility Semiconducting 2D Bi2O2Se Crystals for Integrated Optoelectronic Devices. Adv. Mater. 29(1-6), 1704060 (2017).
Ruleova, P. et al. Thermoelectric Properties of Bi2O2Se. Mater. Chem. Phys. 119, 299–302 (2010).
Wu, M. & Zeng, X. C. Bismuth Oxychalcogenides: A New Class of Ferroelectric/Ferroelastic Materials with Ultra High Mobility. Nano Lett. 17, 6309–6314 (2017).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Clark, S. J. et al. First Principles Methods Using CASTEP. Z. Kristallogr. 220, 567–570 (2017).
Freysoldt, C., Neugebauer, J. & Van de Walle, C. G. Fully Ab Initio Finite-Size Corrections for Charged-Defect Supercell Calculations. Phys. Rev. Lett. 102(1–4), 016402 (2009).
Freysoldt, C. et al. First-Principles Calculations for Point Defects in Solids. Rev. Mod. Phys. 86, 253–305 (2014).
Zhang, S. B. & Northrup, J. E. Chemical Potential Dependence of Defect Formation Energies in GaAs: Application to Ga Self-Diffusion. Phys. Rev. Lett. 67, 2339–2342 (1991).
Tersoff, J. Schottky Barrier Heights and the Continuum of Gap States. Phys. Rev. Lett. 52, 465–468 (1984).
Cardona, M. & Christensen, N. E. Acoustic Deformation Potentials and Heterostructure Band Offsets in Semiconductors. Phys. Rev. B 35, 6182–6194 (1987).
Zhang, W., Lin, J., Xu, W., Fu, H. & Yang, G. SCStore: Managing Scientific Computing Packages for Hybrid System with Containers. Tsinghua Science and Technology 22, 675–681 (2017).
Acknowledgements
H. Li thanks Beijing Natural Science Foundation (No. 4164087) and National Natural Science Foundation (No. 61704096) for financial support. R. Gillen thanks Deutsche Forschungsgemeinschaft (DFG) within the Cluster of Excellence Engineering of Advanced Materials (project EXC 315) (Bridge Funding) for financial support. L.P. Shi thanks National Natural Science foundation (No. 61603209, 61475080, 61327902), SuZhou-Tsinghua innovation leading program (2016SZ0102) and Beijing Innovation Centre for Future Chip for financial support. J. Robertson thanks EPSRC for financial support. Computational resources are provided by high performance computing service of Tsinghua National Laboratory for Information Science and Technology and high performance computing service of University of Cambridge.
Author information
Authors and Affiliations
Contributions
H. Li conceived this work. H. Li, X.X and Y. Zhang performed DFT calculations. H. Li wrote the manuscript. All authors discussed the results and implications and commented on the manuscript at all stages.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, H., Xu, X., Zhang, Y. et al. Native point defects of semiconducting layered Bi2O2Se. Sci Rep 8, 10920 (2018). https://doi.org/10.1038/s41598-018-29385-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-29385-8
This article is cited by
-
Highly insulating phase of Bi2O2Se thin films with high electronic performance
Nano Research (2023)
-
Huge permittivity and premature metallicity in Bi2O2Se single crystals
Science China Physics, Mechanics & Astronomy (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
